
Recent Chemical and Pharmacological Developments on 14-Oxygenated-N-methylmorphinan-6-ones



Abstract
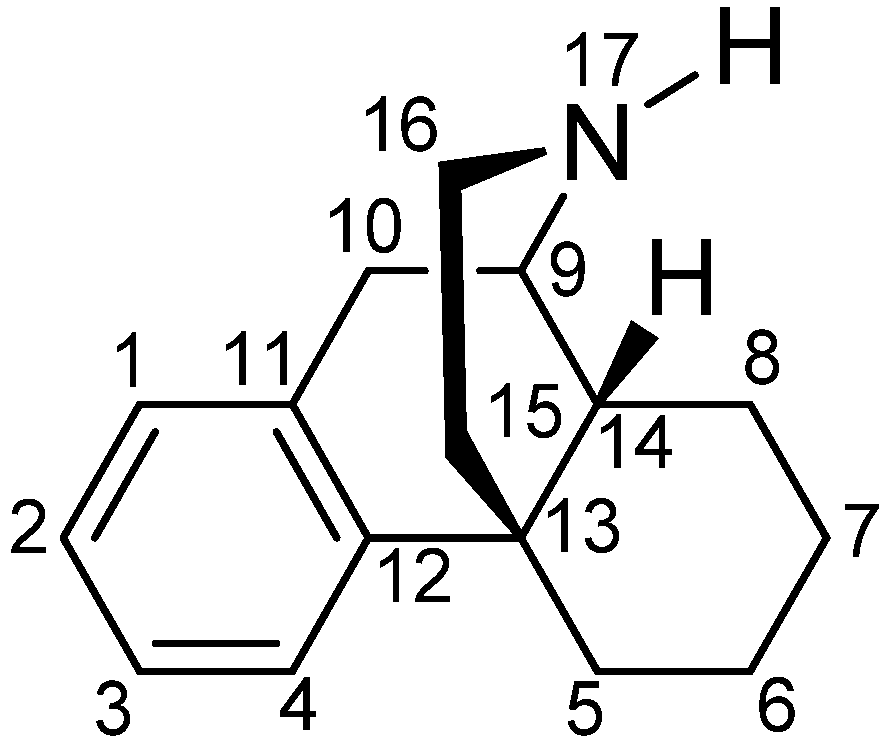
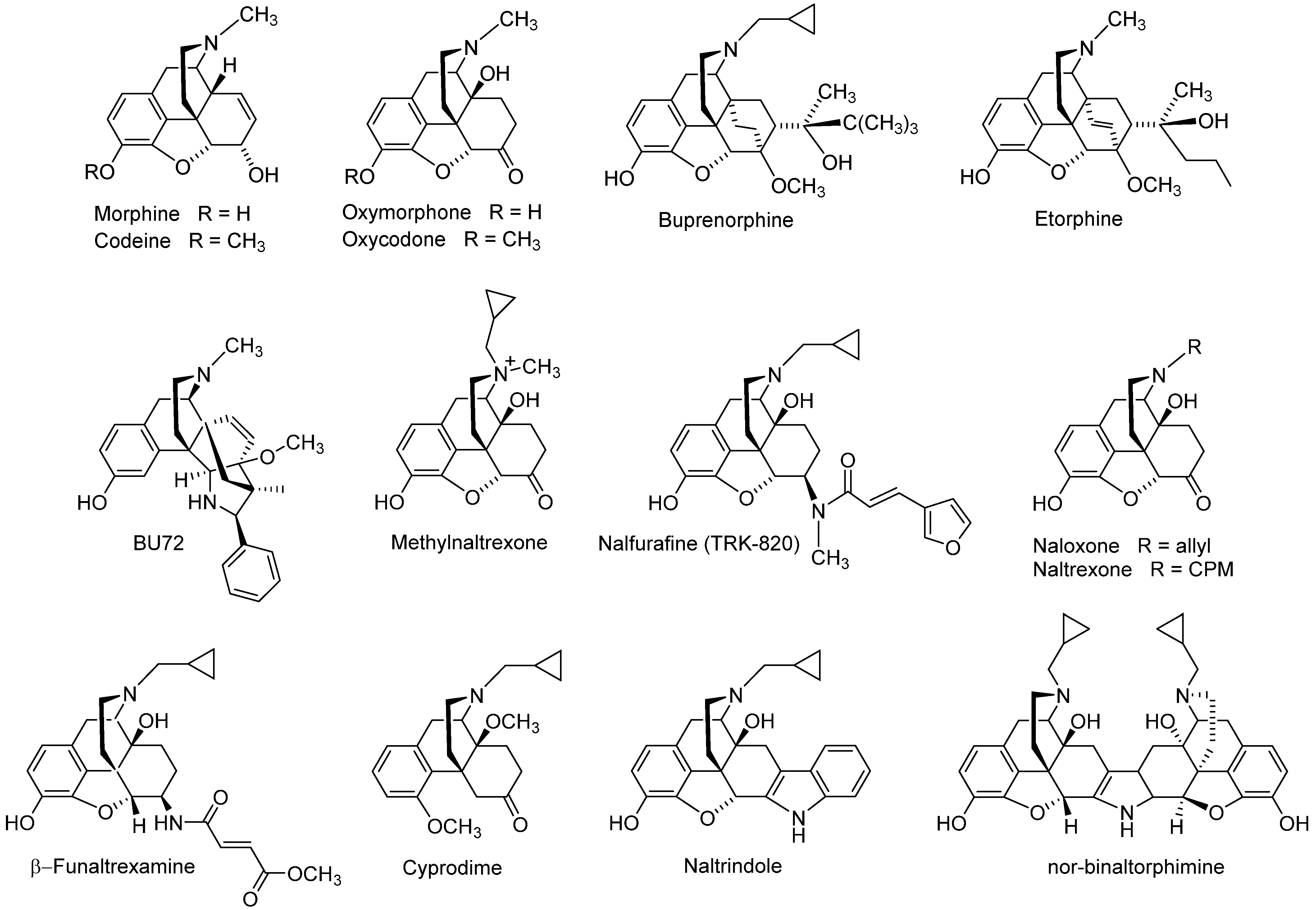
1. Introduction
2. Modifications in Position 6 of 14-Oxygenated-N-methylmorphinan-6-ones: Design, Synthesis and SAR Studies
2.1. Deletion of the Carbonyl Group in Position 6 via Wolff–Kishner Reduction
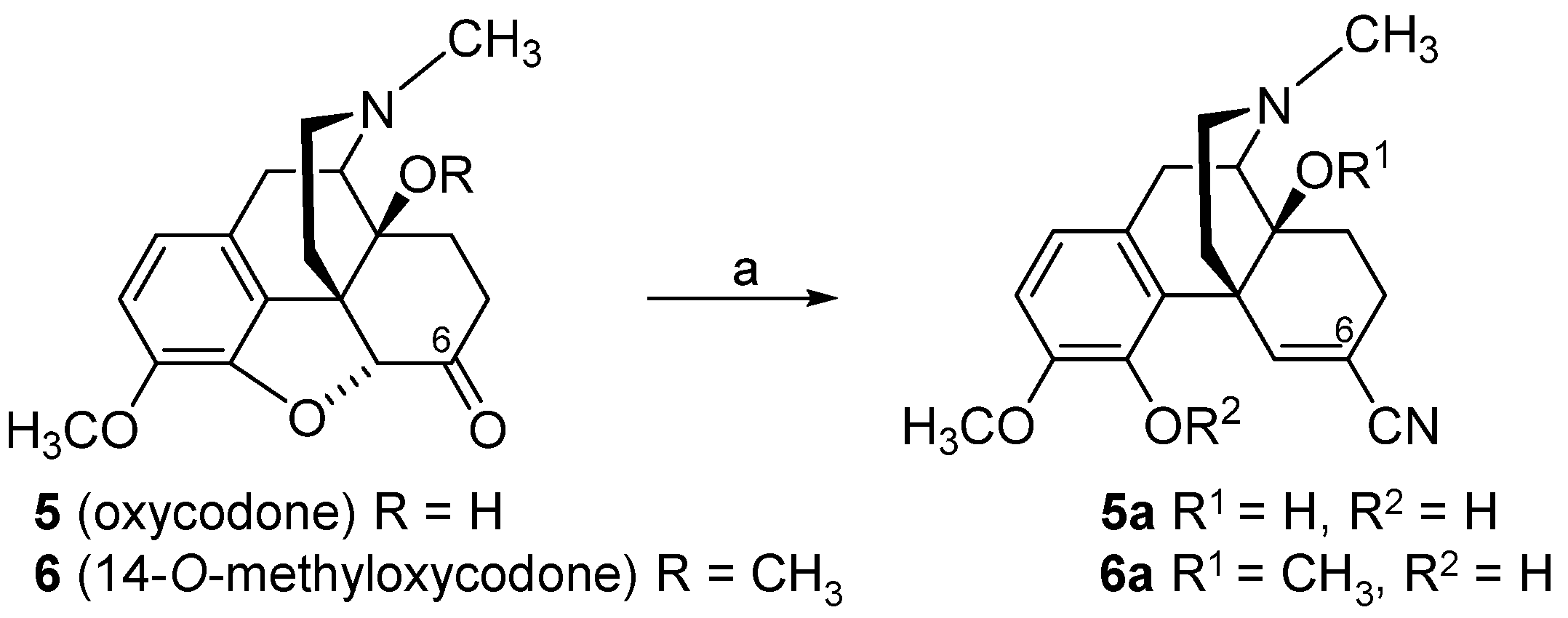
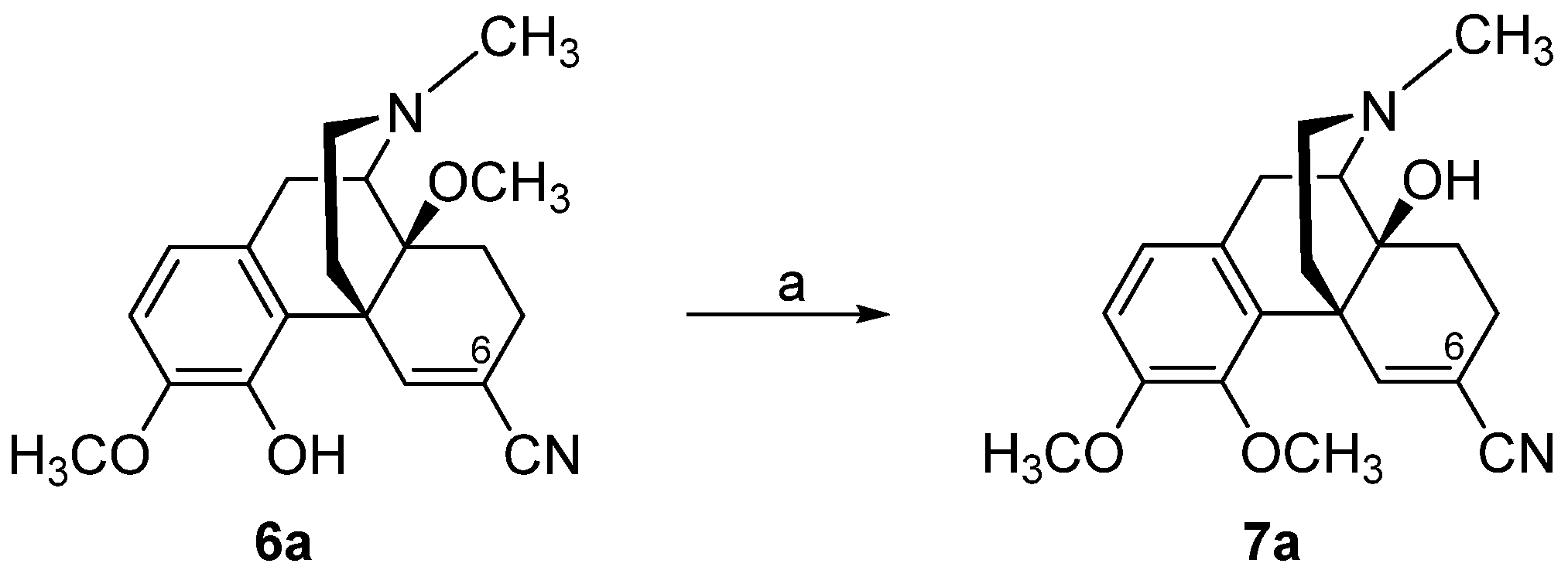
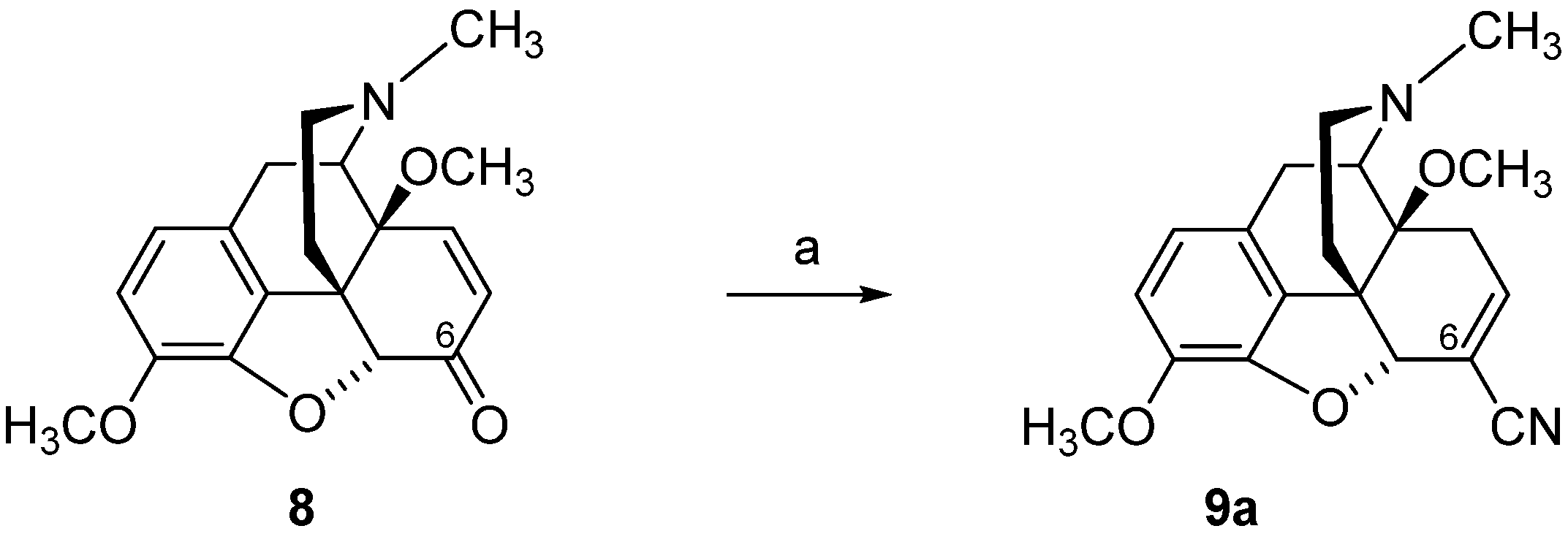
2.2. Introduction of Acrylonitrile Substructures in Position 6
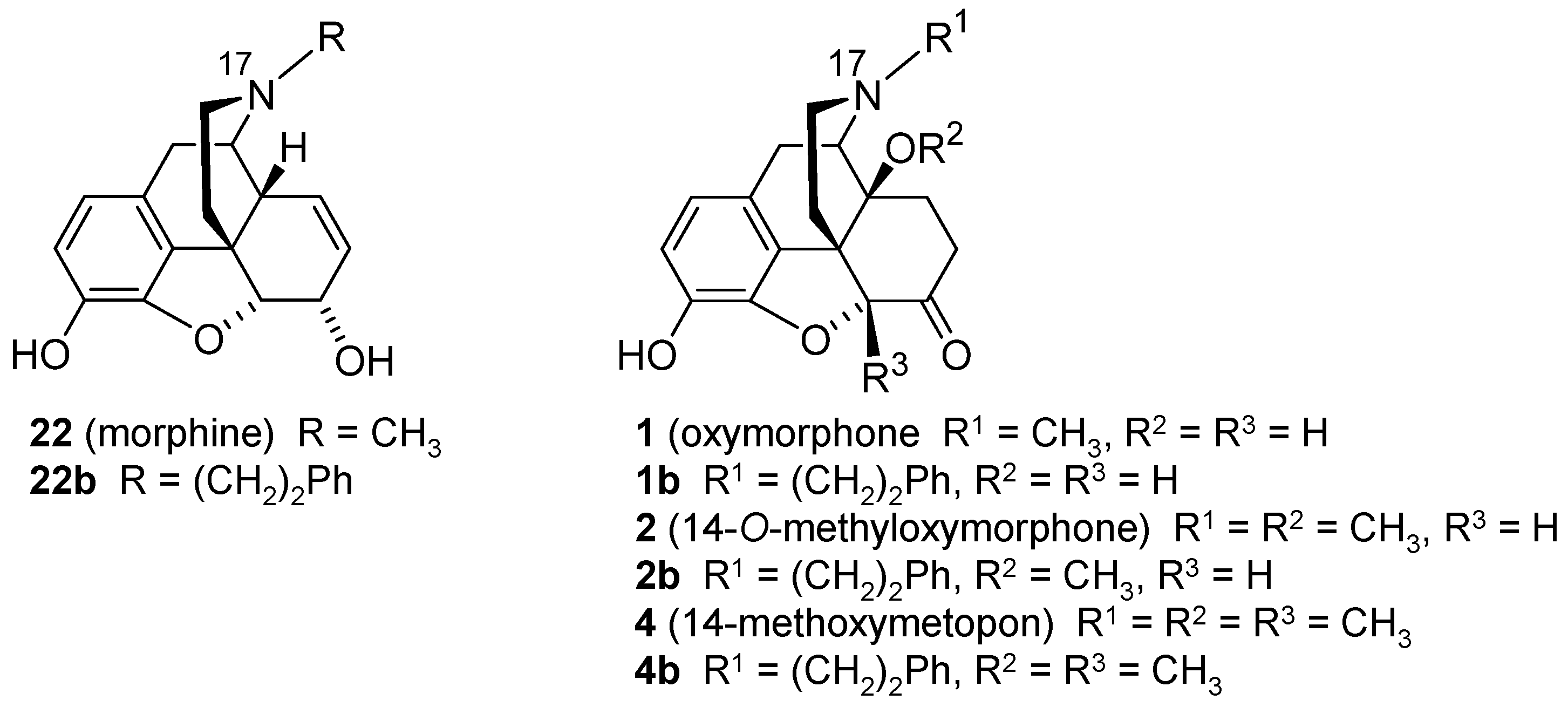
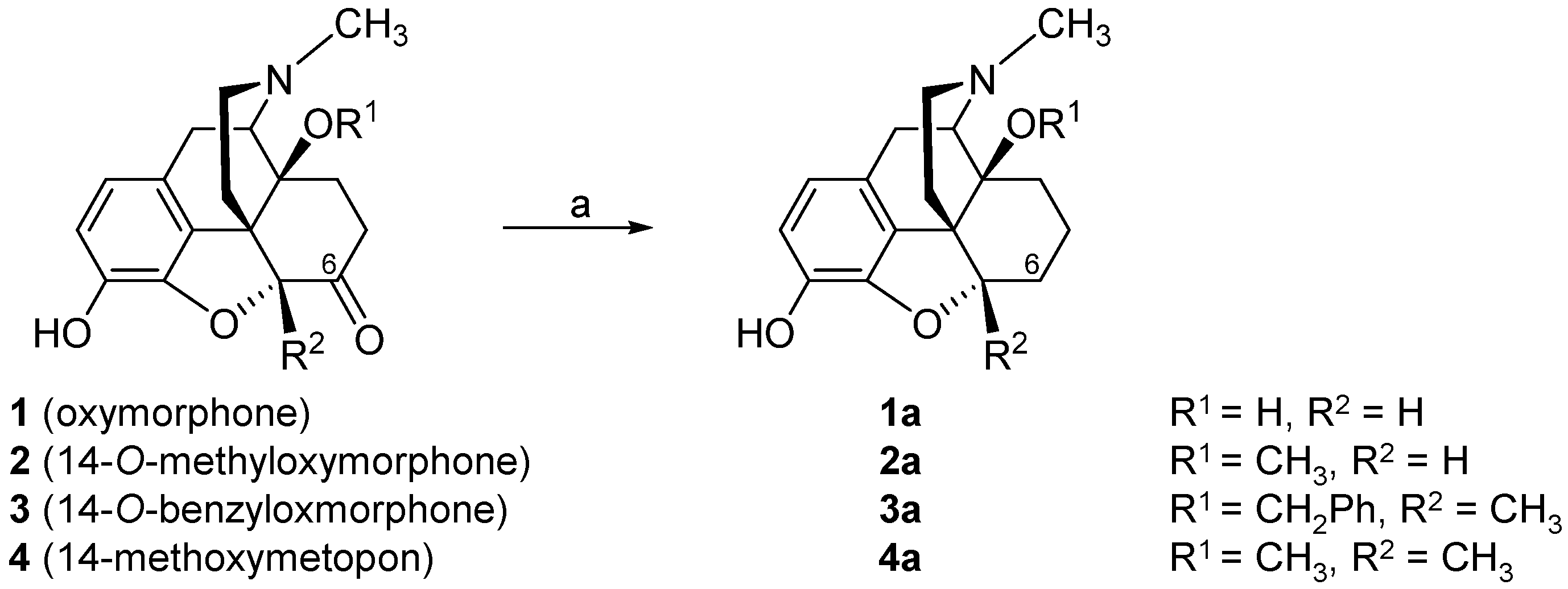
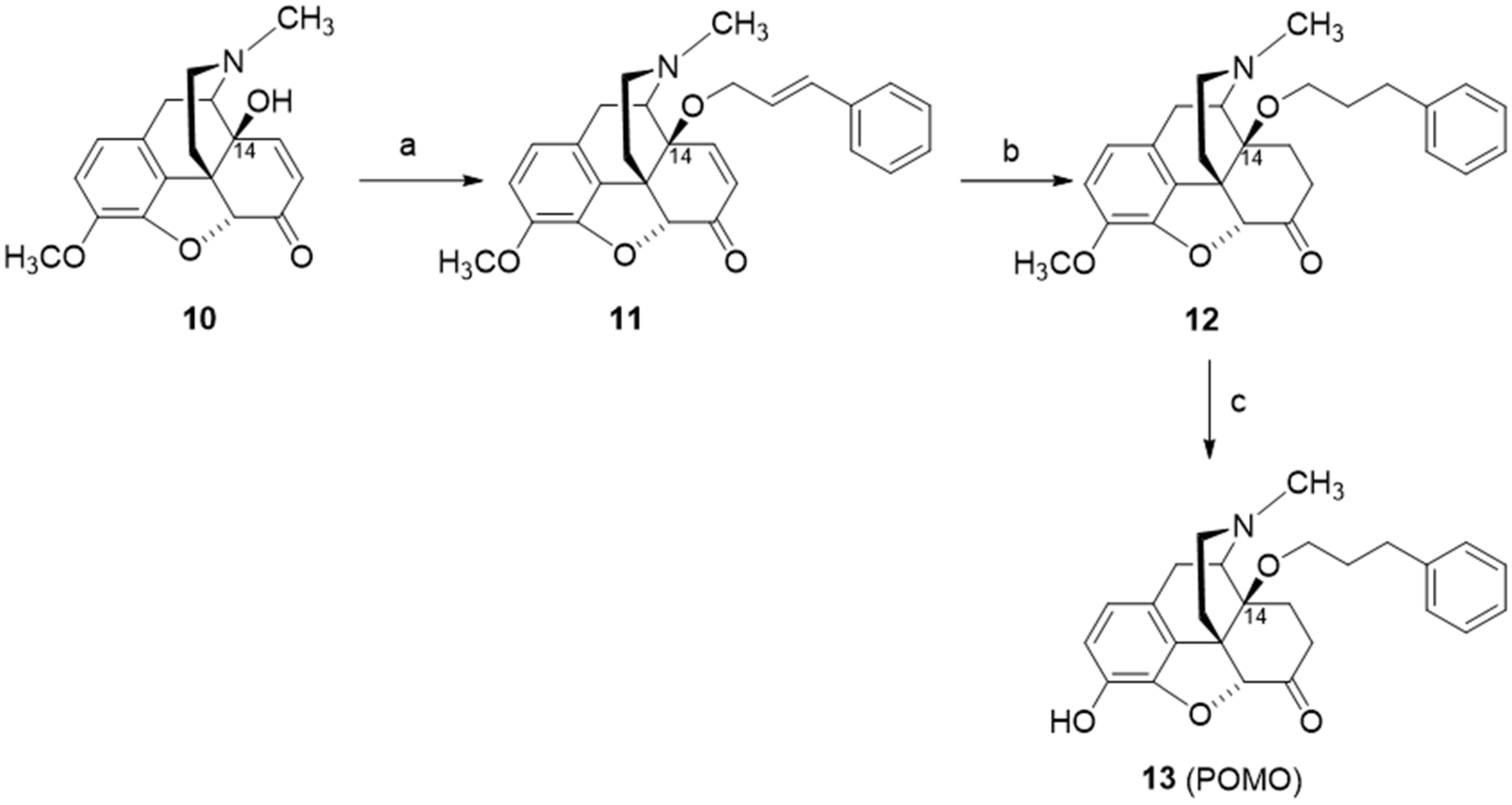
3. Modifications in Position 14 of 14-Oxygenated-N-methylmorphinan-6-ones: Design, Synthesis and SAR Studies
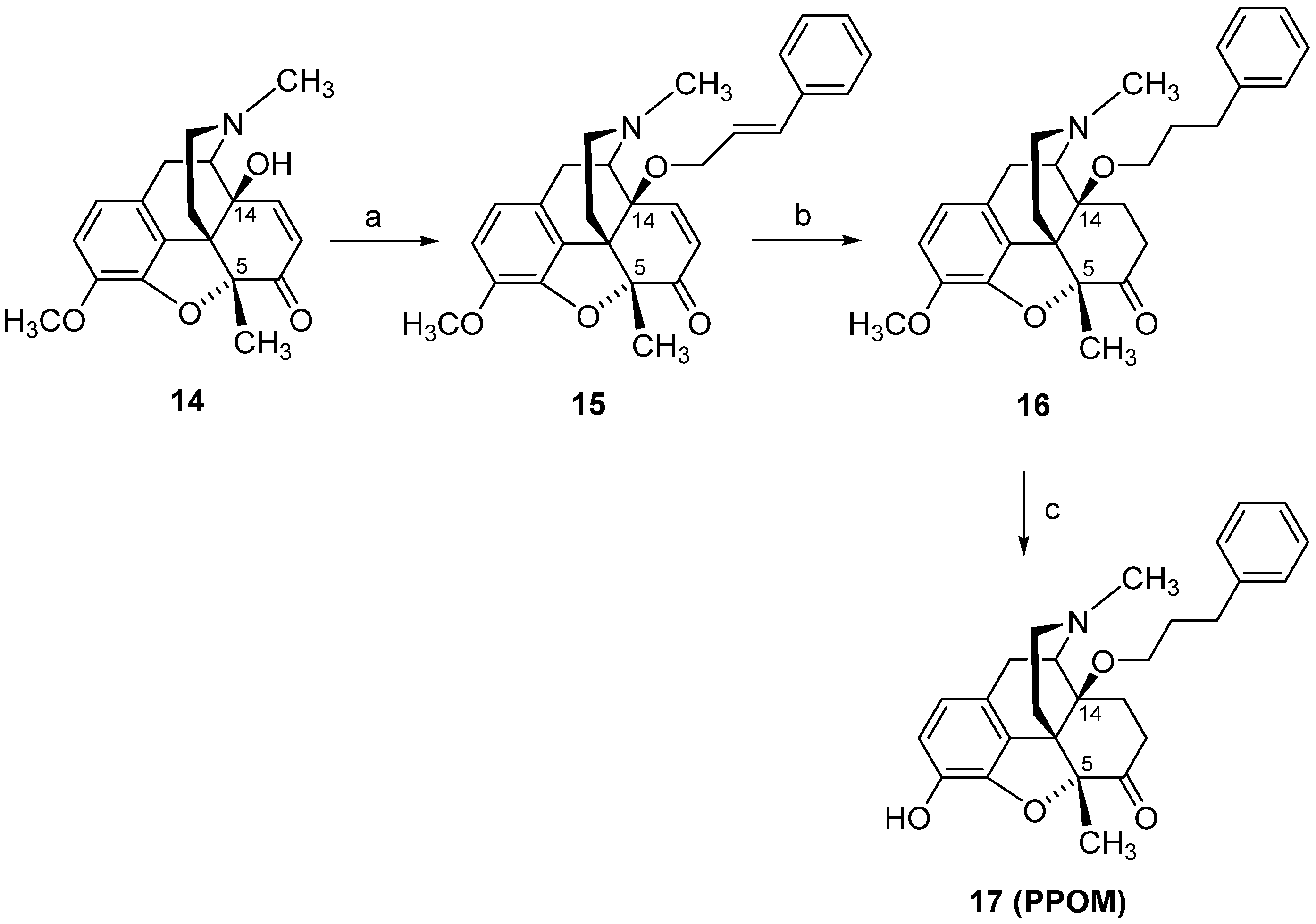
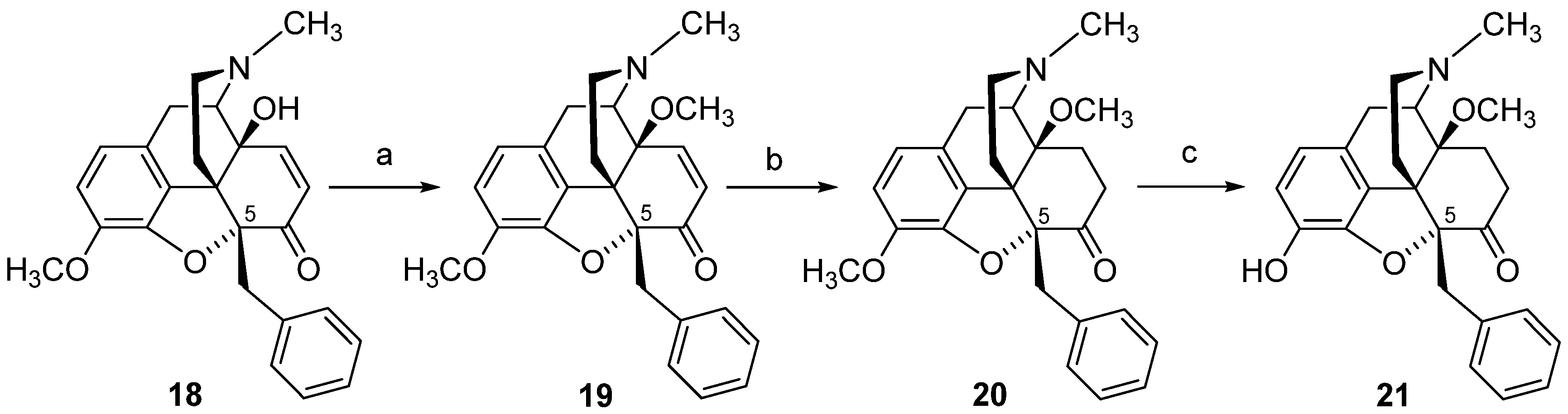
4. Modifications in Position 5 of 14-Oxygenated-N-methylmorphinan-6-ones: Design, Synthesis and SAR Studies
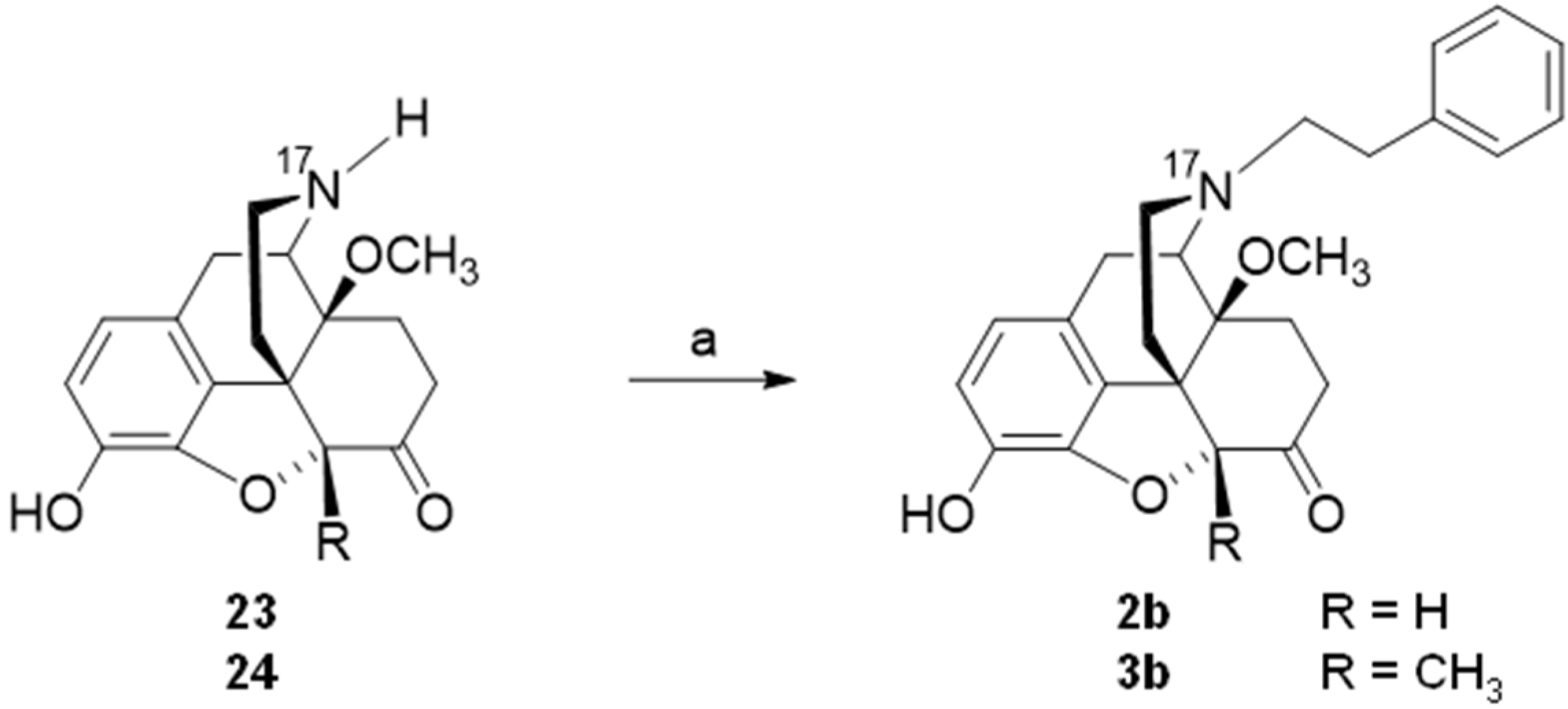
5. Modifications in Position 17 of N-Methylmorphinan-6-ones: Design, Synthesis and SAR Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kapur, B.M.; Lala, P.K.; Shaw, J.L. Pharmacogenetics of chronic pain management. Clin. Biochem. 2014, 47, 1169–1187. [Google Scholar] [CrossRef]
- Volkow, N.; Benveniste, H.; McLellan, A.T. Use and misuse of opioids in chronic pain. Annu. Rev. Med. 2018, 69, 451–465. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Roberson, D.P.; Bean, B.P.; Woolf, C.J. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 2017, 16, 545–564. [Google Scholar] [CrossRef]
- Stein, C.; Kopf, A. Pain therapy—Are there new options on the horizon? Best Pract. Res. Clin. Rheumatol. 2019, 33, 101420. [Google Scholar] [CrossRef]
- Miller, L.R.; Cano, A. Comorbid chronic pain and depression: Who is at risk? J. Pain 2009, 10, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, E.; Porreca, F. Reward and motivation in pain and pain relief. Nat. Neurosci. 2014, 17, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W. Mu opioid pharmacology: 40 years to the promised land. Adv. Pharmacol. 2018, 82, 261–291. [Google Scholar]
- Sobczak, Ł.; Goryński, K. Pharmacological aspects of over-the-counter opioid drugs misuse. Molecules 2020, 25, 3905. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Blanco, C. The changing opioid crisis: Development, challenges and opportunities. Mol. Psychiatry 2021, 26, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Obeng, S.; Hiranita, T.; León, F.; McMahon, L.R.; McCurdy, C.R. Novel approaches, drug candidates, and targets in pain drug discovery. J. Med. Chem. 2021, 64, 6523–6548. [Google Scholar] [CrossRef]
- Rapaka, R.S.; Sadée, W. Drug Addiction: From Basic Research to Therapy; Springer: New York, NY, USA, 2008. [Google Scholar]
- Nagase, H. Chemistry of Opioids; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Pasternak, G.W. The Opiate Receptors; Humana Press: Totowa, NJ, USA, 2011. [Google Scholar]
- Albert-Vartanian, A.; Boyd, M.R.; Hall, A.L.; Morgado, S.J.; Nguyen, E.; Nguyen, V.P.; Patel, S.P.; Russo, L.J.; Shao, A.J.; Raffa, R.B. Will peripherally restricted κ opioid receptor agonists (pKORAs) relieve pain with less opioid adverse effects and abuse potential? J. Clin. Pharm. Ther. 2016, 41, 371–382. [Google Scholar] [CrossRef]
- Jutkiewicz, E.M. Delta Opioid Receptor Pharmacology and Therapeutic Applications. In Handbook of Experimantal Pharmacology; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar]
- Machelska, H.; Celik, M.Ö. Advances in achieving opioid analgesia without side effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Caló, G. The Nociceptin/Orphanin FQ Peptide Receptor. In Handbook of Experimantal Pharmacology; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Cunningham, C.W.; Elballa, W.M.; Vold, S.U. Bifunctional opioid receptor ligands as novel analgesics. Neuropharmacology 2019, 151, 195–207. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased opioid ligands. Molecules 2020, 25, 4257. [Google Scholar]
- Paton, K.F.; Atigari, D.V.; Kaska, S.; Prisinzano, T.; Kivell, B.M. Strategies for developing κ opioid receptor agonists for the treatment of pain with fewer side effects. J. Pharmacol. Exp. Ther. 2020, 375, 332–348. [Google Scholar] [CrossRef]
- Spetea, M.; Schmidhammer, H. Opioids and their receptors: Present and emerging concepts in opioid drug discovery. Molecules 2020, 25, 5658. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and exogenous opioids in pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the nanobody-stabilized active state of the κ opioid receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.M.L.; Filizola, M. Insights from molecular dynamics simulations of a number of G-protein coupled receptor targets for the treatment of pain and opioid use disorders. Front. Mol. Neurosci. 2019, 12, 207. [Google Scholar] [CrossRef]
- Manglik, A. Molecular basis of opioid action: From structures to new leads. Biol. Psychiatry 2020, 87, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef]
- Devereaux, A.L.; Mercer, S.L.; Cunningham, C.W. DARK classics in chemical neuroscience: Morphine. ACS Chem. Neurosci. 2018, 9, 2395–2407. [Google Scholar] [CrossRef]
- Gulland, J.; Robinson, R. Constitution of codeine and thebaine. Mem. Proc. Manch. Lit. Philos. Soc. 1925, 69, 79–86. [Google Scholar]
- Gates, M.; Tschudi, G. The synthesis of morphine. J. Am. Chem. Soc. 1956, 78, 1380–1393. [Google Scholar] [CrossRef]
- Mella-Raipan, J.; Javier Romero-Parra, J.; Recabarren-Gajardo, G. DARK classics in chemical neuroscience: Heroin and desomorphine. ACS Chem. Neurosci. 2020, 11, 3905–3927. [Google Scholar] [CrossRef]
- Casy, A.F.; Parfitt, R.T. Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Takemori, A.E.; Portoghese, P.S. Selective naltrexone-derived opioid receptors antagonists. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 239–269. [Google Scholar] [CrossRef] [PubMed]
- Schmidhammer, H. Opioid receptor antagonists. Prog. Med. Chem. 1998, 35, 83–132. [Google Scholar] [PubMed]
- Metcalf, M.D.; Coop, A. Kappa opioid antagonists: Past successes and future prospects. AAPS J. 2005, 7, E704–E722. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.S.; Hruby, V.J. Analgesics. In Synthesis of Essential Drugs; Elsevier: Amsterdam, The Netherlands, 2006; pp. 19–55. [Google Scholar]
- Fürst, S.; Hosztafi, S. The chemical and pharmacological importance of morphine analogues. Acta Physiol. Hung. 2008, 95, 3–44. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Spetea, M. Synthesis of 14-alkoxymorphinans and their pharmacological activities. Top. Curr. Chem. 2011, 299, 63–91. [Google Scholar] [PubMed]
- Lewis, J.W.; Husbands, S.M. 14-Amino-4,5-epoxymorphinan derivatives and their pharmacological actions. Top. Curr. Chem. 2011, 299, 93–119. [Google Scholar] [PubMed]
- Stavitskaya, L.; Coop, A. Most recent developments and modifications of 14-alkylamino and 14-alkoxy-4,5-epoxymorphinan derivatives. Mini Rev. Med. Chem. 2011, 11, 1002–1008. [Google Scholar] [CrossRef]
- Spetea, M.; Schmidhammer, H. Recent advances in the development of 14-alkoxy substituted morphinans as potent and safer opioid analgesics. Curr. Med. Chem. 2012, 19, 2442–2457. [Google Scholar] [CrossRef]
- Spetea, M.; Asim, M.F.; Wolber, G.; Schmidhammer, H. The µ opioid receptor and ligands acting at the µ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed]
- Mazak, K.; Noszal, B.; Hosztafi, S. Physicochemical and pharmacological characterization of permanently charged opioids. Curr. Med. Chem. 2017, 24, 3633–3648. [Google Scholar] [CrossRef] [PubMed]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef]
- Imam, M.Z.; Kuo, A.; Ghassabian, S.; Smith, M.T. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255. [Google Scholar] [CrossRef]
- Nagase, H.; Hayakawa, J.; Kawamura, K.; Kawai, K.; Takezawa, Y.; Matsuura, H.; Tajima, C.; Endo, T. Discovery of a structurally novel opioid kappa-agonist derived from 4,5-epoxymorphinan. Chem. Pharm. Bull. 1998, 46, 366–369. [Google Scholar] [CrossRef]
- Nagase, H.; Fuji, H. Opioids in preclinical and clinical trials. Top. Curr. Chem. 2011, 299, 29–62. [Google Scholar]
- Cowan, A.; Kehner, G.B.; Inan, S. Targeting itch with ligands selective for κ opioid receptors. Handb. Exp. Pharmacol. 2015, 226, 291–314. [Google Scholar]
- Shigeki, I. Nalfurafine hydrochloride to treat pruritus: A review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 249–255. [Google Scholar]
- Goodman, A.J.; Le Bourdonnec, B.; Dolle, R.E. Mu opioid receptor antagonists: Recent developments. Chem. Med. Chem. 2007, 2, 1552–1557. [Google Scholar] [CrossRef]
- Carroll, F.I.; Carlezon, W.A., Jr. Development of κ opioid receptor antagonists. J. Med. Chem. 2013, 56, 2178–2195. [Google Scholar] [CrossRef]
- Bidlack, J.M. Mixed κ/μ partial opioid agonists as potential treatments for cocaine dependence. Adv. Pharmacol. 2014, 69, 387–418. [Google Scholar]
- Moss, J. Identifying and treating opioid side effects: The development of methylnaltrexone. Anesthesiology 2019, 130, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Fürst, S.; Hosztafi, S.; Friedmann, T. Structure-activity relationships of synthetic and semisynthetic opioid agonists and antagonists. Curr. Med. Chem. 1995, 1, 423–440. [Google Scholar]
- Cami-Kobeci, G.; Neal, A.P.; Bradbury, F.A.; Purington, L.C.; Aceto, M.D.; Harris, L.S.; Lewis, J.W.; Traynor, J.R.; Husbands, S.M. Mixed kappa/mu opioid receptor agonists: The 6 beta-naltrexamines. J. Med. Chem. 2009, 52, 1546–1552. [Google Scholar] [CrossRef]
- Li, G.; Aschenbach, L.C.; Chen, J.; Cassidy, M.P.; Stevens, D.L.; Gabra, B.H.; Selley, D.E.; Dewey, W.L.; Westkaemper, R.B.; Zhang, Y. Design, synthesis, and biological evaluation of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives as mu opioid receptor selective antagonists. J. Med. Chem. 2009, 52, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Subrath, J.; Le Rouzic, V.; Polikar, L.; Burgman, M.; Nagakura, K.; Ocampo, J.; Haselton, N.; Pasternak, A.R.; Grinnell, S.; et al. Synthesis and evaluation of aryl-naloxamide opiate analgesics targeting truncated exon 11-associated μ opioid receptor (MOR-1) splice variants. J. Med. Chem. 2012, 55, 6352–6362. [Google Scholar] [CrossRef] [PubMed]
- Schmidhammer, H.; Spetea, M.; Windisch, P.; Schütz, J.; Riba, P.; Al-Khrasani, M.; Fürst, S. Functionalization of the carbonyl group in position 6 of morphinan-6-ones. Development of novel 6-amino and 6-guanidino substituted 14-alkoxymorphinans. Curr. Pharm. Des. 2013, 19, 7391–7399. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascuta, M.; Ben Haddou, T.; Guerrieri, E.; Noha, S.M.; Schläfer, L.; Schmidhammer, H.; Spetea, M. Synthesis, pharmacology, and molecular docking studies on 6-desoxo-N-methylmorphinans as potent μ-opioid receptor agonists. J. Med. Chem. 2017, 60, 9407–9412. [Google Scholar] [CrossRef] [PubMed]
- Kishner, N. Wolff–Kishner reduction; Huang–Minlon modification. J. Russ. Phys. Chem. Soc. 1911, 43, 582–595. [Google Scholar]
- Wolff, L. Methode zum Ersatz des Sauerstoffatoms der Ketone und Aldehyde durch Wasserstoff. Liebigs Ann. Chem. 1912, 394, 86–108. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H. Structure of the µ-opioid receptor–G i protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Hahn, E.F. Long-acting opiate agonists and antagonists: 14-Hydroxydihydromorphinone hydrazones. J. Med. Chem. 1980, 23, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Tóth, G.; Benyhe, S.; Hosztafi, S.; Borsodi, A. Synthesis and binding of [3H]-oxymorphazone to rat brain membranes. Life Sci. 1987, 40, 1579–1588. [Google Scholar] [CrossRef]
- Krizsan, D.; Varga, E.; Hosztafi, S.; Benyhe, S.; Szücs, M.; Borsodi, A. Irreversible blockade of the high and low affinity [3H]-naloxone binding sites by C-6 derivatives of morphinane-6-ones. Life Sci. 1991, 48, 439–451. [Google Scholar] [CrossRef]
- Monory, K.; Greiner, E.; Sartania, N.; Sallai, L.; Pouille, Y.; Schmidhammer, H.; Hanoune, J.; Borsodi, A. Opioid binding profiles of new hydrazone, oxime, carbazone and semicarbazone derivatives of 14-alkoxymorphinans. Life Sci. 1999, 22, 2011–2220. [Google Scholar] [CrossRef]
- Gergely, A.; Gyimesi-Forras, K.; Horvath, P.; Hosztafi, S.; Kökösi, J.; Nagy, P.I.; Szasz, G.; Szentesi, A. 6-Oxo-morphinane oximes: Pharmacology, chemistry and analytical application. Curr. Med. Chem. 2004, 11, 2555–2564. [Google Scholar] [CrossRef]
- Lacko, E.; Varadi, A.; Rapavi, R.; Zador, F.; Riba, P.; Benyhe, S.; Borsodi, A.; Hosztafi, S.; Timar, J.; Noszal, B.; et al. A novel µ-opioid receptor ligand with high in vitro and in vivo agonist efficacy. Curr. Med. Chem. 2012, 19, 4699–4707. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Rief, S.B.; Haddou, T.B.; Fink, M.; Kristeva, E.; Mittendorfer, H.; Haas, S.; Hummer, N.; Follia, V.; Guerrieri, E.; et al. Synthesis, biological, and structural explorations of new zwitterionic derivatives of 14-O-methyloxymorphone, as potent μ/δ opioid agonists and peripherally selective antinociceptives. J. Med. Chem. 2019, 62, 641–653. [Google Scholar] [CrossRef]
- Zádor, F.; Mohammadzadeh, A.; Balogh, M.; Zádori, Z.S.; Király, K.; Barsi, S.; Galambos, A.R.; László, S.B.; Hutka, B.; Váradi, A.; et al. Comparisons of in vivo and in vitro opioid effects of newly synthesized 14-methoxycodeine-6-O-sulfate and codeine-6-O-sulfate. Molecules 2020, 25, 1370. [Google Scholar] [CrossRef]
- Fürst, Z.; Borsodi, A.; Friedmann, T.; Hosztafi, S. 6-Substituted oxycodone derivatives have strong antinociceptive effects and block irreversibly the low affinity [3H]-naloxone binding sites in rat brain. Pharmacol. Res. 1992, 25, 31–32. [Google Scholar] [CrossRef]
- Grinnell, S.G.; Majumdar, S.; Narayan, A.; Le Rouzic, V.; Ansonoff, M.; Pintar, J.E.; Pasternak, G.W. Pharmacologic characterization in the rat of a potent analgesic lacking respiratory depression, IBNtxA. J. Pharmacol. Exp. Ther. 2014, 350, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Greiner, E.; Schottenberger, H.; Wurst, K.; Schmidhammer, H. Novel class of morphinans with acrylonitrile incorporated substructures as key intermediates for non-oxygen-bridged opioid ligands. J. Am. Chem. Soc. 2001, 123, 3840–3841. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Greiner, E.; Aceto, M.D.; Harris, L.S.; Coop, A.; Schmidhammer, H. Effect of a 6-cyano substituent in 14-oxygenated N-methylmorphinans on opioid receptor binding and antinociceptive potency. J. Med. Chem. 2005, 48, 5052–5055. [Google Scholar] [CrossRef]
- Schütz, J.; Windisch, P.; Kristeva, E.; Wurst, K.; Ongania, K.H.; Horvath, U.E.; Schottenberger, H.; Laus, G.; Schmidhammer, H. Mechanistic diversity of the van Leusen reaction applied to 6-ketomorphinans and synthetic potential of the resulting acrylonitrile substructures. J. Org. Chem. 2005, 70, 5323–5326. [Google Scholar] [CrossRef]
- Oldenziel, O.H.; van Leusen, D.; van Leusen, A.M. Chemistry of sulfonylmethyl isocyanides. 13. A general one-step synthesis of nitriles from ketones using tosylmethyl isocyanide. Introduction of a one-carbon unit. J. Org. Chem. 1977, 42, 3114–3118. [Google Scholar] [CrossRef]
- Ben Haddou, T.; Malfacini, D.; Calo, G.; Aceto, M.D.; Harris, L.S.; Traynor, J.R.; Coop, A.; Schmidhammer, H.; Spetea, M. Exploring pharmacological activities and signaling of morphinans substituted in position 6 as potent agonists interacting with the μ opioid receptor. Mol. Pain 2014, 10, 48. [Google Scholar]
- Ananthan, S.; Saini, S.K.; Dersch, C.M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-Alkoxy- and 14-acyloxypyridomorphinans: μ agonist/δ antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Polgar, W.E.; Cami-Kobeci, G.; Thomas, M.P.; Khroyan, T.V.; Toll, L.; Husbands, S.M. Synthesis, biological evaluation, and SAR studies of 14β-phenylacetyl substituted 17-cyclopropylmethyl-7,8-dihydronoroxymorphinones derivatives: Ligands with mixed NOP and opioid receptor profile. Front. Psychiatry 2018, 9, 430. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Lei, W.; Ray, A.; Saini, S.K.; Zhang, S.; Molnar, G.; Barlow, D.; Karlage, K.L.; Bilsky, E.J.; Houseknecht, K.L.; et al. Synthesis and structure-activity relationships of 5′-aryl-14-alkoxypyridomorphinans: Identification of a μ opioid receptor agonist/δ opioid receptor antagonist ligand with systemic antinociceptive activity and diminished opioid side effects. J. Med. Chem. 2020, 63, 7663–7694. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Aeppli, L.; Atwell, L.; Fritsch, F.; Jacobson, A.E.; Nebuchla, M.; Sperk, G. Synthesis and biological evaluation of 14-alkoxymorphinans. 1. Highly potent opioid agonists in the series of (−)-14-methoxy-N-methylmorphinan-6-ones. J. Med. Chem. 1984, 27, 1575–1579. [Google Scholar] [CrossRef]
- Lattanzi, R.; Spetea, M.; Schüllner, F.; Rief, S.B.; Krassnig, R.; Negri, L.; Schmidhammer, H. Synthesis and biological evaluation of 14-alkoxymorphinans. 22. Influence of the 14-alkoxy group and the substitution in position 5 in 14-alkoxymorphinan-6-ones on in vitro and in vivo activities. J. Med. Chem. 2005, 48, 3372–3378. [Google Scholar] [CrossRef]
- Spetea, M.; Bohotin, C.R.; Asim, M.F.; Stübegger, K.; Schmidhammer, H. In vitro and in vivo pharmacological profile of the 5-benzyl analogue of 14-methoxymetopon, a novel µ opioid analgesic with reduced propensity to alter motor function. Eur. J. Pharm. Sci. 2010, 41, 125–135. [Google Scholar] [CrossRef]
- Lattanzi, R.; Rief, S.; Schmidhammer, H.; Negri, L.; Spetea, M. In vitro and in vivo pharmacological activities of 14-O-phenylpropyloxymorphone, a potent mixed mu/delta/kappa-opioid receptor agonist with reduced constipation in mice. Front. Pharmacol. 2018, 9, 1002. [Google Scholar] [CrossRef]
- Noha, S.M.; Schmidhammer, H.; Spetea, M. Molecular docking, molecular dynamics, and structure–activity relationship explorations of 14-oxygenated N-methylmorphinan-6-ones as potent μ-opioid receptor agonists. ACS Chem. Neurosci. 2017, 8, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Obeng, S.; Wang, H.; Jali, A.; Stevens, D.L.; Akbarali, H.I.; Dewey, W.L.; Selley, D.E.; Zhang, Y. Structure–activity relationship studies of 6α- and 6β-indolylacetamidonaltrexamine derivatives as bitopic mu opioid receptor modulators and elaboration of the “message-address concept” to comprehend their functional conversion. ACS Chem. Neurosci. 2018, 10, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascuta, M.; Bermudez, M.; Haddou, T.B.; Guerrieri, E.; Schläfer, L.; Ritsch, A.; Hosztafi, S.; Lantero, A.; Kreutz, C.; Massotte, D.; et al. N-Phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones turns selective µ opioid receptor ligands into dual µ/δ opioid receptor agonists. Sci. Rep. 2020, 10, 5653. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Schüllner, F.; Moisa, R.C.; Berzetei-Gurske, I.P.; Schraml, B.; Dörfler, C.; Aceto, M.D.; Harris, L.S.; Coop, A.; Schmidhammer, H. Synthesis and biological evaluation of 14-alkoxymorphinans. 21. Novel 4-alkoxy and 14-phenylpropoxy derivatives of the mu opioid receptor antagonist cyprodime. J. Med. Chem. 2004, 47, 3242–3247. [Google Scholar] [CrossRef]
- Azzam, A.A.; Mcdonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Wtorek, K.; Piekielna-Ciesielska, J.; Janecki, T.; Janecka, A. The search for opioid analgesics with limited tolerance liability. Peptides 2020, 130, 170331. [Google Scholar] [CrossRef]
- Lei, W.; Vekariya, R.H.; Ananthan, S.; Streicher, J.M. A novel mu-delta opioid agonist demonstrates enhanced efficacy with reduced tolerance and dependence in mouse neuropathic pain models. J. Pain 2020, 21, 146–160. [Google Scholar] [CrossRef]
- Dumitrascuta, M.; Bermudez, M.; Trovato, O.; De Neve, J.; Ballet, S.; Wolber, G.; Spetea, M. Antinociceptive efficacy of the µ-opioid/nociceptin peptide-based hybrid KGNOP1 in inflammatory pain without rewarding effects in mice: An experimental assessment and molecular docking. Molecules 2021, 26, 3267. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Spetea, M. Development of 5-substituted N-methylmorphinan-6-ones as potent opioid analgesics with improved side-effect profile. Int. J. Med. Chem. 2012, 2012, 208039. [Google Scholar]
- Schmidhammer, H.; Schratz, A.; Mitterdorfer, J. Synthesis and biological evaluation of 14-alkoxymorphinans. 8. 14-Methoxymetopon, an extremely potent opioid agonist. Helv. Chim. Acta 1990, 73, 1784–1787. [Google Scholar] [CrossRef]
- Schütz, J.; Spetea, M.; Koch, M.; Aceto, M.D.; Harris, L.S.; Coop, A.; Schmidhammer, H. Synthesis and biological evaluation of 14-alkoxymorphinans. 20. 14-Phenylpropoxymetopon: An extremely powerful analgesic. J. Med. Chem. 2003, 46, 4182–4187. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Deeter, J.B.; Jones, N.D.; Leander, J.D.; Schoepp, D.D.; Swartzendruber, J.K. Synthesis, Structure elucidation, and pharmacological evaluation of 5-methyl-oxymorphone (=4,5α-Epoxy-3,14-dihydroxy-5,17-dimethylmorphinan-6-one). Helv. Chim. Acta 1988, 71, 1801–1804. [Google Scholar] [CrossRef]
- Gates, M.; Boden, R.M.; Sundararaman, P. Derivatives of the thebaine anion. 2. 5-Methylmorphine, 5-methylcodeine, 5-methylheroin, and some related compounds. J. Org. Chem. 1989, 54, 972–974. [Google Scholar] [CrossRef]
- Sterken, J.; Troubleyn, J.; Gasthuys, F.; Maes, V.; Diltoer, M.; Verborgh, C. Intentional overdose of large animal immobilon. Eur. J. Emerg. Med. 2004, 11, 298–301. [Google Scholar] [CrossRef]
- Weijlard, J.; Erickson, A.E. N-Allylnormorphine. J. Am. Chem. Soc. 1942, 64, 869–870. [Google Scholar] [CrossRef]
- Foldes, F.F.; Lunn, J.N.; Moore, J.; Brown, I.M. N-Allylnoroxymorphone: A new potent narcotic antagonist. Am. J. Med. Sci. 1963, 245, 23–30. [Google Scholar] [CrossRef]
- Anton, R.F. Clinical use of opioid antagonists in the treatment of alcohol dependence. In Opioid Receptors and Antagonists: From Bench to Clinic; Dean, R., Bilsky, E.J., Negus, S.S., Eds.; Humana Press: New York, NY, USA, 2008; pp. 371–386. [Google Scholar]
- Clark, R.L.; Pessolano, A.A.; Weijlard, J.; Pfister, K. N-Substituted epoxymorphinans. J. Am. Chem. Soc. 1953, 75, 4963–4967. [Google Scholar] [CrossRef]
- Small, L.F.; Eddy, N.B.; Ager, J.H.; May, E.L. An improved synthesis of N-phenethylnormorphine and analogs. J. Org. Chem. 1958, 23, 1387–1388. [Google Scholar] [CrossRef]
- Hosztafi, S.; Simon, C.M.; Makleit, S. Synthesis of N-demethyl-N-substituted14-hydroxycodeine and morphine derivatives. Synth. Commun. 1992, 22, 2527–2541. [Google Scholar] [CrossRef]
- May, E.L.; Eddy, N.B. Structures related to morphine. XII.1 (±)-20-Hydroxy-5,9-dimethyl-2-phenethyl-6,7- benzomorphan (NIH 7519) and its optical forms. J. Org. Chem. 1959, 24, 1435–1437. [Google Scholar] [CrossRef]
- Seki, I.; Takagi, H.; Kobayashi, S. Pharmacological studies on morphine derivatives. 3. On the chemical structure-activity relationships of 14-hydroxymorphine derivatives. Yakugaku Zasshi J. Pharm. Soc. Jpn. 1964, 84, 280–286. [Google Scholar] [CrossRef][Green Version]
- Winter, C.A.; Orahovats, P.D.; Lehman, E.G. Analgesic activity and morphine antagonism of compounds related to nalorphine. Arch. Int. Pharmacodyn. Ther. 1957, 110, 186–202. [Google Scholar]
- Loew, G.W.; Berkowitz, D.S. Quantum chemical studies of n-substituent variation in the oxymorphone series of opiate narcotics. J. Med. Chem. 1978, 21, 101–106. [Google Scholar] [CrossRef]
- Ben Haddou, T.; Béni, S.; Hosztafi, S.; Malfacini, D.; Calo, G.; Schmidhammer, H.; Spetea, M. Pharmacological investigations of n-substituent variation in morphine and oxymorphone: Opioid receptor binding, signaling and antinociceptive activity. PLoS ONE 2014, 9, e99231. [Google Scholar]
- Hashimoto, A.; Jacobson, A.E.; Rothman, R.B.; Dersch, C.M.; George, C.; Flippen-Anderson, J.L.; Rice, K.C. Probes for narcotic receptor mediated phenomena. Part 28: New opioid antagonists from enantiomeric analogues of 5-(3-hydroxyphenyl)-N-phenylethylmorphan. Bioorg. Med. Chem. 2002, 10, 3319–3329. [Google Scholar] [CrossRef]
- Zezula, J.; Singer, L.; Przybył, A.K.; Hashimoto, A.; Dersch, C.M.; Rothman, R.B.; Deschamps, J.; Lee, Y.S.; Jacobson, A.E.; Rice, K.C. Synthesis and pharmacological effects of the enantiomers of the N-phenethyl analogues of the ortho and para e- and f-oxide-bridged phenylmorphans. Org. Biomol. Chem. 2008, 6, 2868–2883. [Google Scholar] [CrossRef]
- Wang, M.; Irvin, T.C.; Herdman, C.A.; Hanna, R.D.; Hassan, S.A.; Lee, Y.-S.; Kaska, S.; Crowley, R.S.; Prisinzano, T.E.; Withey, S.L.; et al. The intriguing effects of substituents in the N-phenethyl moiety of norhydromorphone: A bifunctional opioid from a set of “tail wags dog” experiments. Molecules 2020, 25, 2640. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
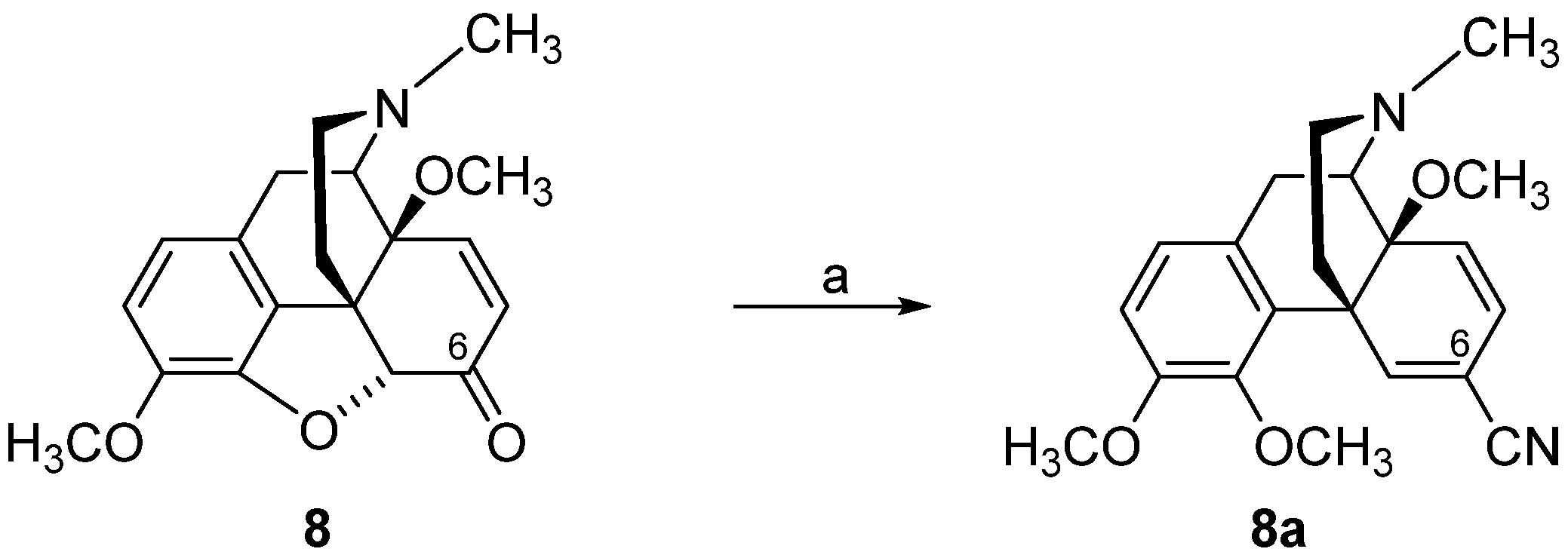{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Affinity (Ki, nM) a | MOP Agonist Activity b | Antinociception c | |||||
|---|---|---|---|---|---|---|---|
| MOP | DOP | KOP | Ki Ratios MOP/DOP/KOP | EC50 (nM) | % stim. | ED50 (µg/kg, s.c.) | |
| 1 | 1.79 | 70.0 | 25.3 | 1/39/14 | 7.80 | 91.5 | 382 |
| 1a | 1.65 | 201 | 21.6 | 1/1122/13 | 8.99 | 104 | 383 |
| 2 | 0.32 | 8.80 | 10.1 | 1/28/32 | 1.45 | 96.4 | 14.2 |
| 2a | 0.24 | 8.64 | 3.99 | 1/36/17 | 1.37 | 105 | 17.3 |
| 3 | 0.45 | 1.05 | 1.27 | 1/2.3/1.8 | 0.32 | 101 | 2.38 |
| 3a | 0.048 | 0.89 | 0.85 | 1/21/18 | 0.13 | 103 | 2.41 |
| 4 | 0.25 | 19.8 | 15.1 | 1/79/60 | 3.28 | 99.2 | 27.1 |
| 4a | 0.34 | 12.9 | 7.92 | 1/38/23 | 2.95 | 104 | 25.1 |
| Binding Affinity (Ki, nM) a | MOP Agonist Activity b | |||||
|---|---|---|---|---|---|---|
| MOP | DOP | KOP | Ki Ratios MOP/DOP/KOP | EC50 (nM) | % stim. | |
| 5 | 43.6 | 1087 | 2658 | 1/25/61 | 500 | 92 |
| 5a | 31.7 | 498 | 1648 | 1/16/52 | 273 | 98 |
| 6 | 35.3 | 116 | 454 | 1/3/13 | 325 | 137 |
| 6a | 5.38 | 197 | 378 | 1/37/70 | 26.2 | 85 |
| 7a | 2.44 | 107 | 364 | 1/44/149 | 42.5 | 97 |
| 8a | 0.54 | 30.3 | 200 | 1/56/370 | 1.64 | 133 |
| 9a | 7.39 | 239 | 194 | 1/32/26 | 25.1 | 121 |
| ED50 (mg/kg, s.c.) a | |||
|---|---|---|---|
| Hot-Plate Test | Tail-Flick Test | PPQ Abdominal Stretching Test | |
| 5 | 1.37 | 0.94 | 0.38 |
| 5a | 0.50 | 1.88 | 0.18 |
| 6 | 1.02 | 0.80 | 0.22 |
| 6a | 0.25 | 0.21 | 0.11 |
| 7a | 0.15 | 0.12 | 0.026 |
| 8a | 0.080 | 0.040 | 0.0023 |
| 9a | 0.089 | 0.12 | 0.003 |
| Binding Affinity (Ki, nM) a | Agonist Activity (EC50, nM; %stim) b | ||||||
|---|---|---|---|---|---|---|---|
| MOP | DOP | KOP | Ki ratios MOP/DOP/KOP | MOP | DOP | KOP | |
| 2 | 0.10 | 4.80 | 10.2 | 1/48/102 | 1.62; 97 | 43.8; 106 | 144; 65 |
| 3 | 0.12 | 2.14 | 1.18 | 1/18/10 | 0.32; 101 | - c | - |
| 13 | 0.073 | 0.13 | 0.30 | 1/1.8/4.1 | 0.082; 100 | 0.28; 91 | 0.38; 39 |
| Antinociception a (AD50 nmol/kg, s.c.) | Gastrointestinal Transit b (EC50 nmol/kg, s.c.) | |
|---|---|---|
| 2 | 53 | 37 |
| 3 | 9.6 | 27 c |
| 13 | 0.70 | 1.70 |
| Morphine | 6690 | 3800 7622 c |
| Binding Affinity (Ki, nM) a | Agonist Activity (EC50, nM; %stim) b | ||||||
|---|---|---|---|---|---|---|---|
| MOP | DOP | KOP | Ki Ratios MOP/DOP/KOP | MOP | DOP | KOP | |
| 2 | 0.10 | 4.8 | 10.2 | 1/48/102 | 1.21; 95 23.7; 103 d | 38.5; 102 | 135; 65.6 |
| 4 | 0.15 | 13.3 | 25.2 | 1/89/168 | 2.66; 99 63.0; 99 d | 36.8; 100 | 181; 68.9 |
| 17 | 0.20 | 0.14 | 0.40 | 1/0.7/2 | - c | - | - |
| 21 | 0.31 | 13.1 | 22.8 | 1/42/73 | 1.86; 95 e 13.8; 85 d | 31.7; 126 e | 116; 59.0 e |
| ED50 (µg/kg, s.c.) a | |||
|---|---|---|---|
| Hot-Plate Test | Tail-Flick Test | PPQ Abdominal Stretching Test | |
| 2 | 17 | 14 | - b |
| 4 | 30 | 30 | 9.0 |
| 17 | 0.10 | 0.08 | 0.16 |
| 21 | 53 | 43 | - |
| Morphine | 850 | 1920 | 400 |
| Binding Affiniy Ki (nM) a | ||||
|---|---|---|---|---|
| MOP | DOP | KOP | Ki Ratios MOP/DOP/KOP | |
| Morphine (22) | 3.35 | 195 | 96.4 | 1/58/29 |
| 22b | 0.25 | 24.5 | 93.5 | 1/98/374 |
| Oxymorphone (1) | 1.41 | 79.1 | 32.6 | 1/56/23 |
| 1b | 0.12 | 10.7 | 42.2 | 1/89/352 |
| 2 | 0.27 | 9.08 | 10.3 | 1/34/38 |
| 2b | 0.19 | 1.81 | 15.8 | 1/9.5/83 |
| 4 | 0.25 | 18.6 | 12.8 | 1/74/51 |
| 4b | 0.24 | 1.45 | 35.3 | 1/6.0/147 |
| In Vitro Agonist Activity a | Antinocieption b | ||||||
|---|---|---|---|---|---|---|---|
| MOP | DOP | KOP | ED50 mg/kg, s.c. | ||||
| EC50 (nM) | % stim. | EC50 (nM) | % stim. | EC50 (nM) | % stim. | ||
| >Morphine (22) | 34.4 | 89 | 668 | 109 | 710 | 76 | 3.06 |
| 22b | 10.3 | 113 | 712 | 138 | 1049 | 19 | 0.11 |
| Oxymorphone (1) | 7.80 | 92 | 259 | 87 | 463 | 48 | 0.35 |
| 1b | 2.67 | 97 | 131 | 101 | 225 | 7.5 | 0.15 |
| 2 | 1.21 | 95 | 38.5 | 102 | 135 | 65.9 | 0.014 |
| 2b | 1.26 | 98 | 9.34 | 107 | 144 | 35.4 | 0.014 |
| 4 | 2.66 | 99 | 36.8 | 100 | 181 | 68.9 | 0.024 |
| 4b | 1.86 | 102 | 9.54 | 103 | 334 | 51.3 | 0.024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spetea, M.; Schmidhammer, H. Recent Chemical and Pharmacological Developments on 14-Oxygenated-N-methylmorphinan-6-ones. Molecules 2021, 26, 5677. https://doi.org/10.3390/molecules26185677
Spetea M, Schmidhammer H. Recent Chemical and Pharmacological Developments on 14-Oxygenated-N-methylmorphinan-6-ones. Molecules. 2021; 26(18):5677. https://doi.org/10.3390/molecules26185677
Chicago/Turabian StyleSpetea, Mariana, and Helmut Schmidhammer. 2021. "Recent Chemical and Pharmacological Developments on 14-Oxygenated-N-methylmorphinan-6-ones" Molecules 26, no. 18: 5677. https://doi.org/10.3390/molecules26185677
APA StyleSpetea, M., & Schmidhammer, H. (2021). Recent Chemical and Pharmacological Developments on 14-Oxygenated-N-methylmorphinan-6-ones. Molecules, 26(18), 5677. https://doi.org/10.3390/molecules26185677