
Bile-Acid-Appended Triazolyl Aryl Ketones: Design, Synthesis, In Vitro Anticancer Activity and Pharmacokinetics in Rats

Abstract
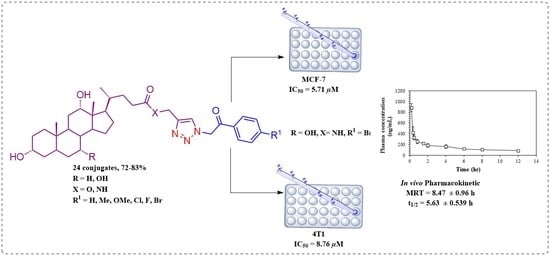
:
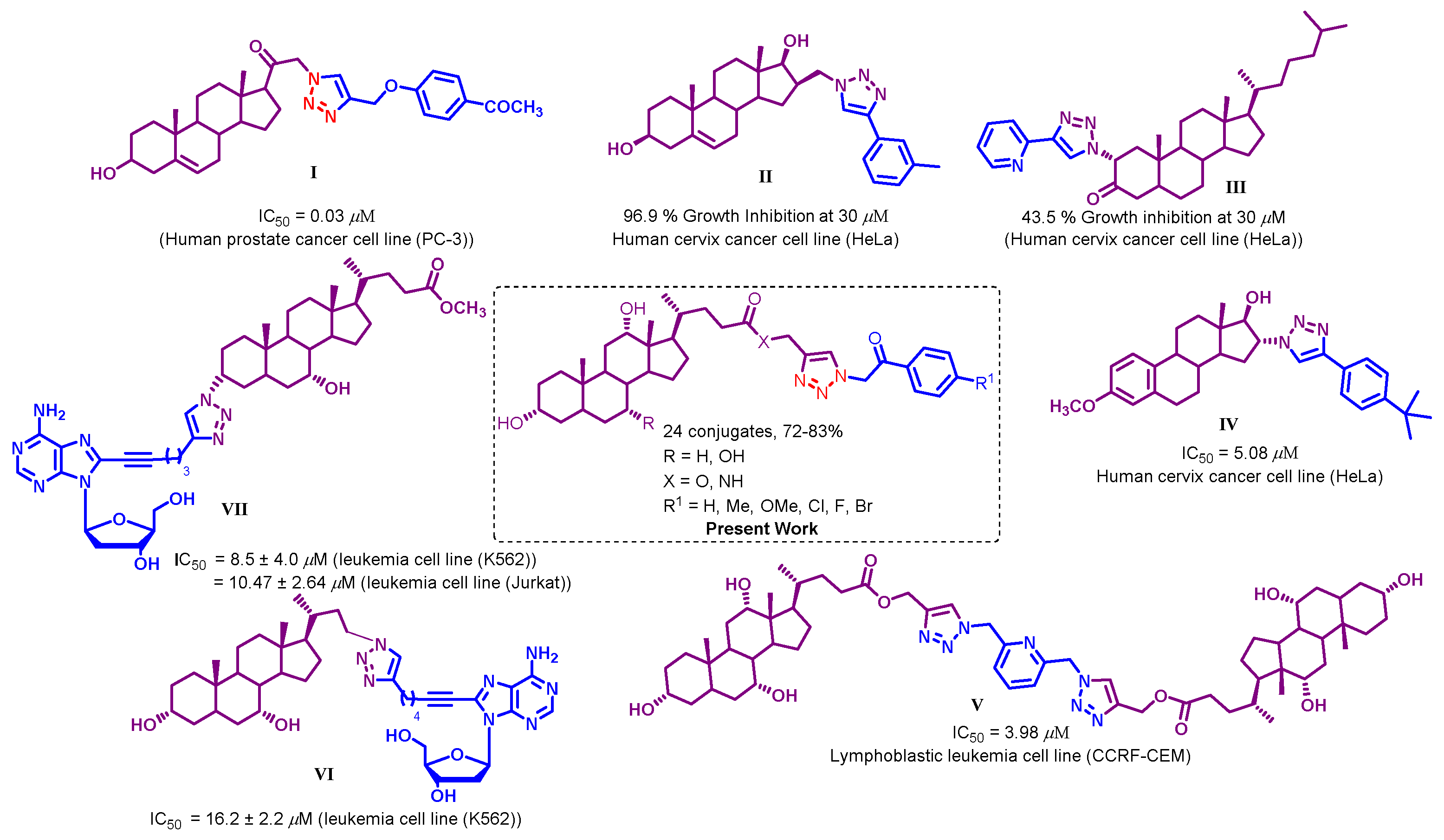
1. Introduction
2. Results and Discussion
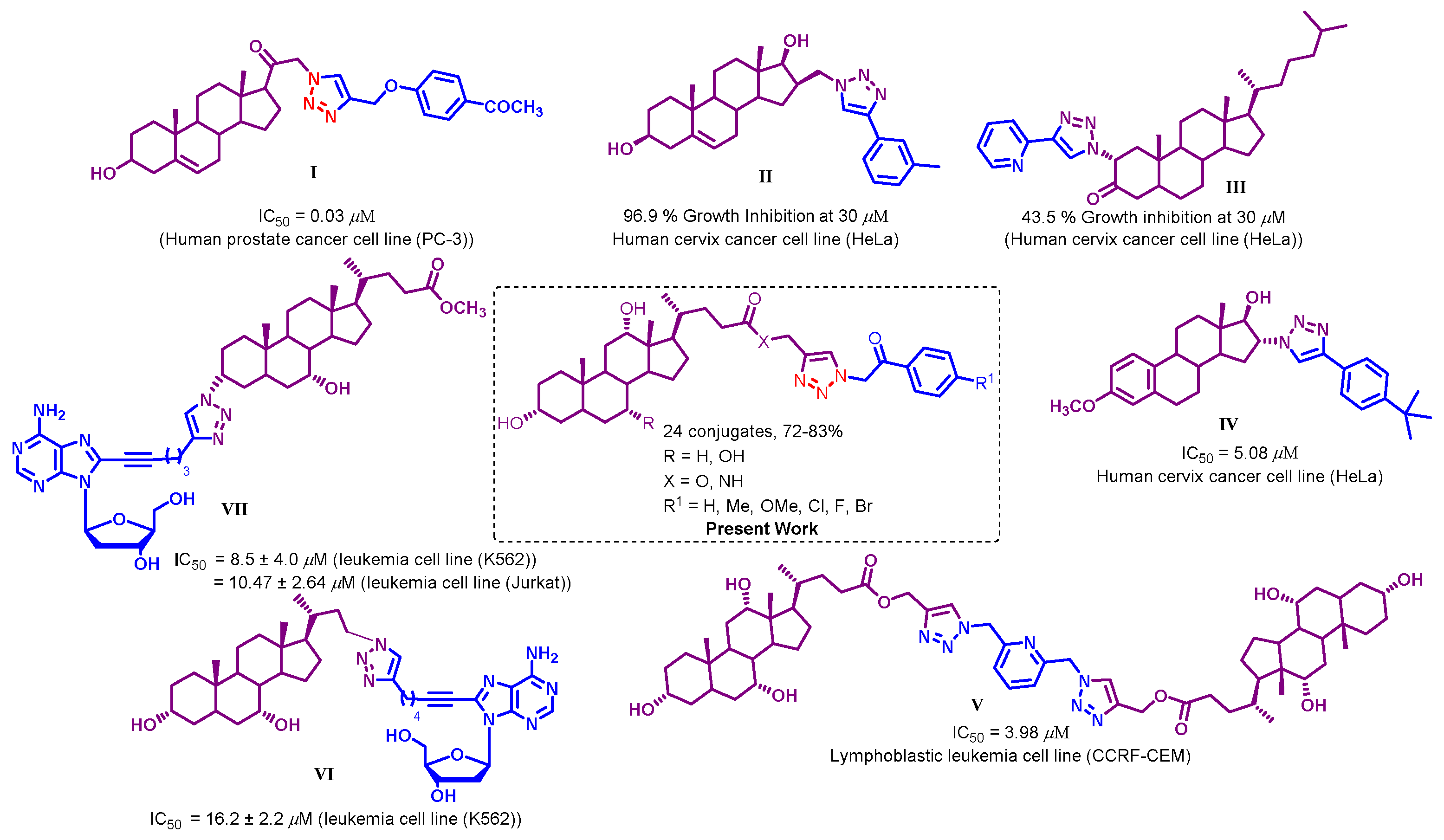
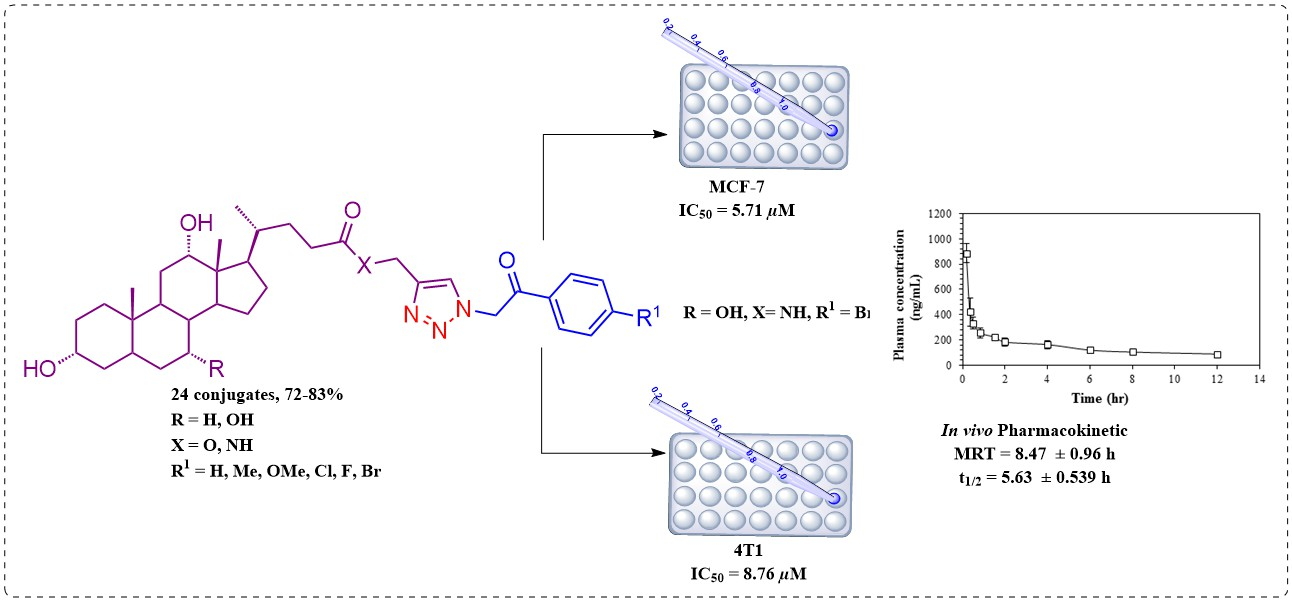
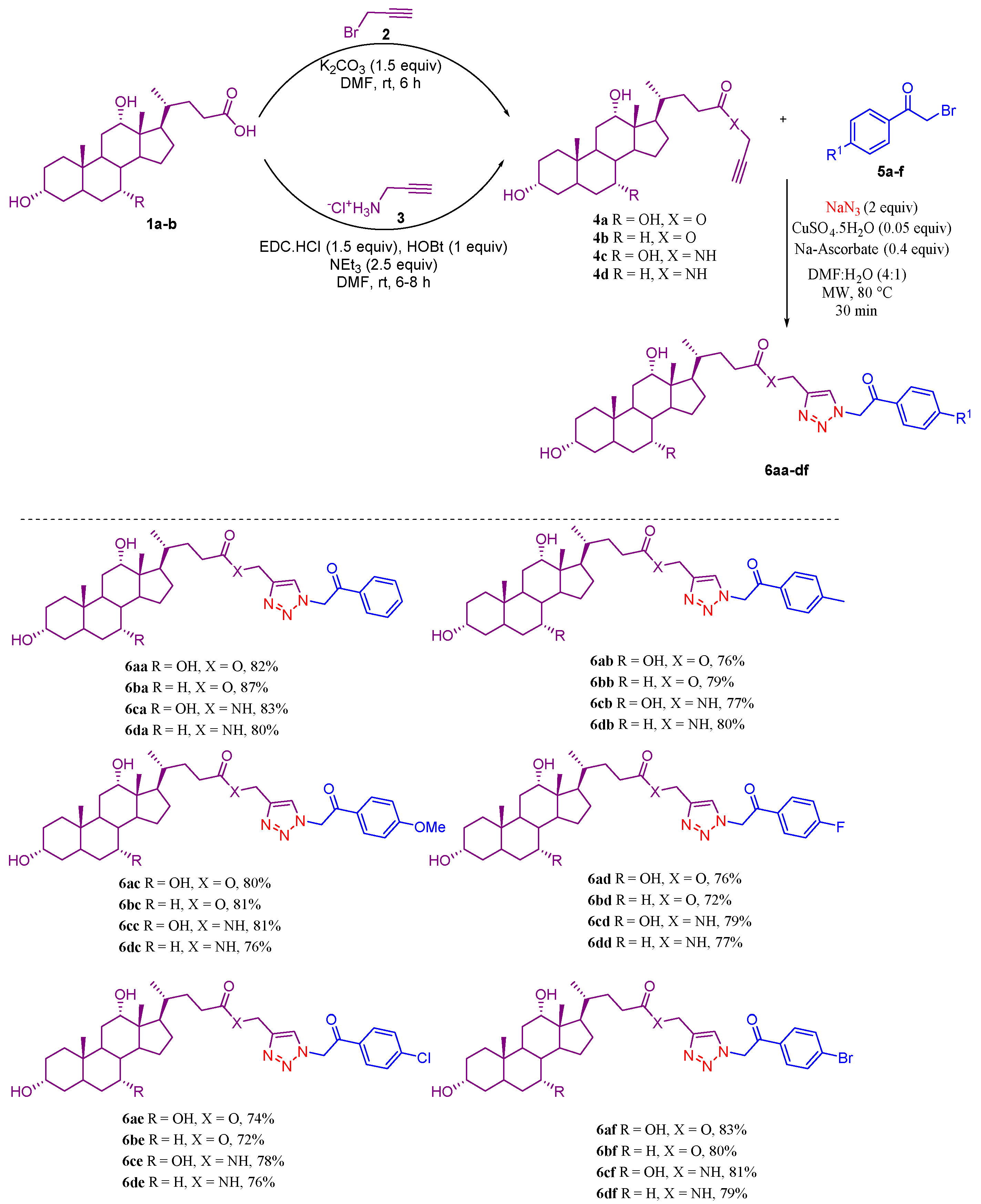
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cytotoxic Activity
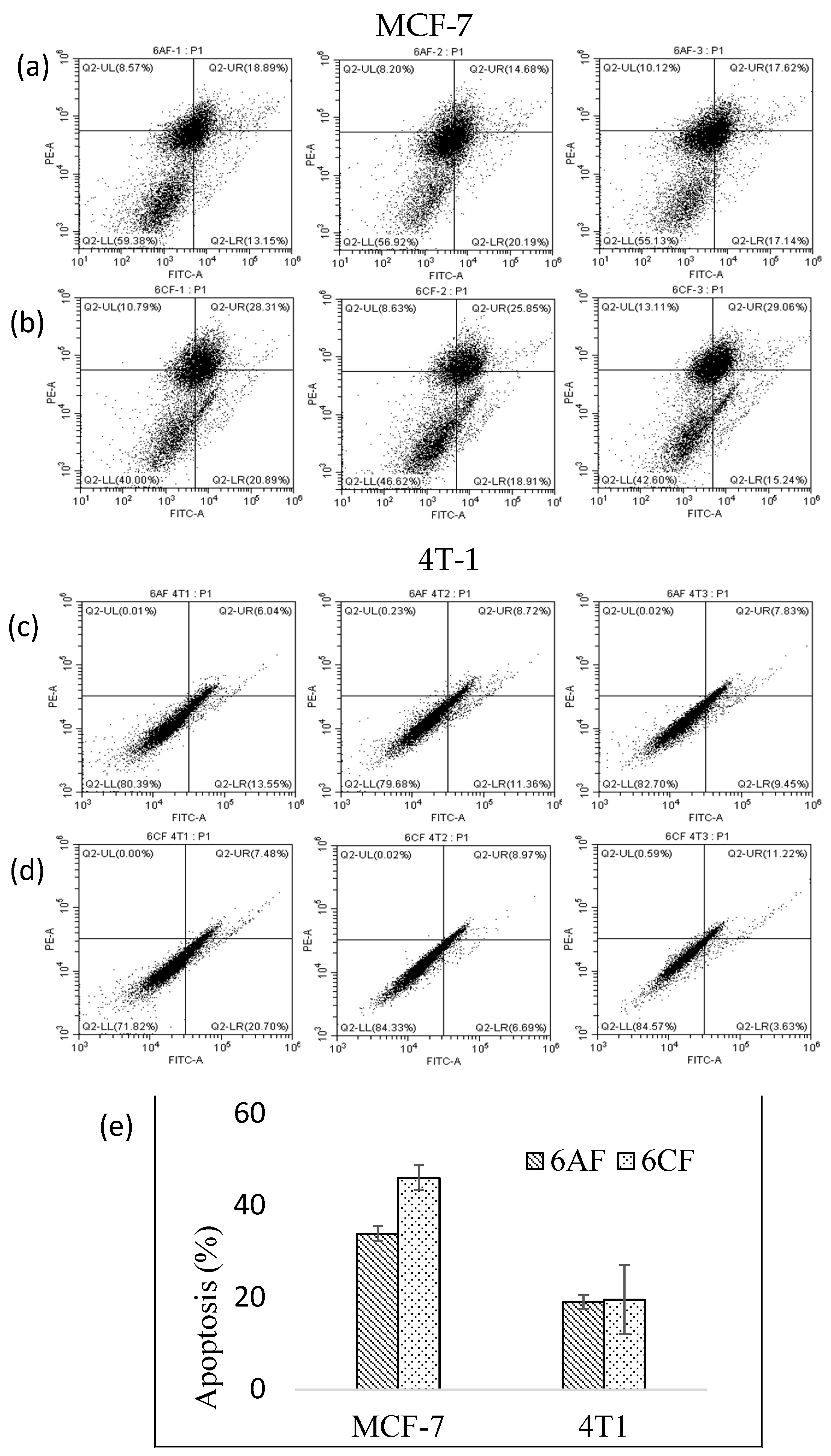
2.2.2. Apoptotic Study
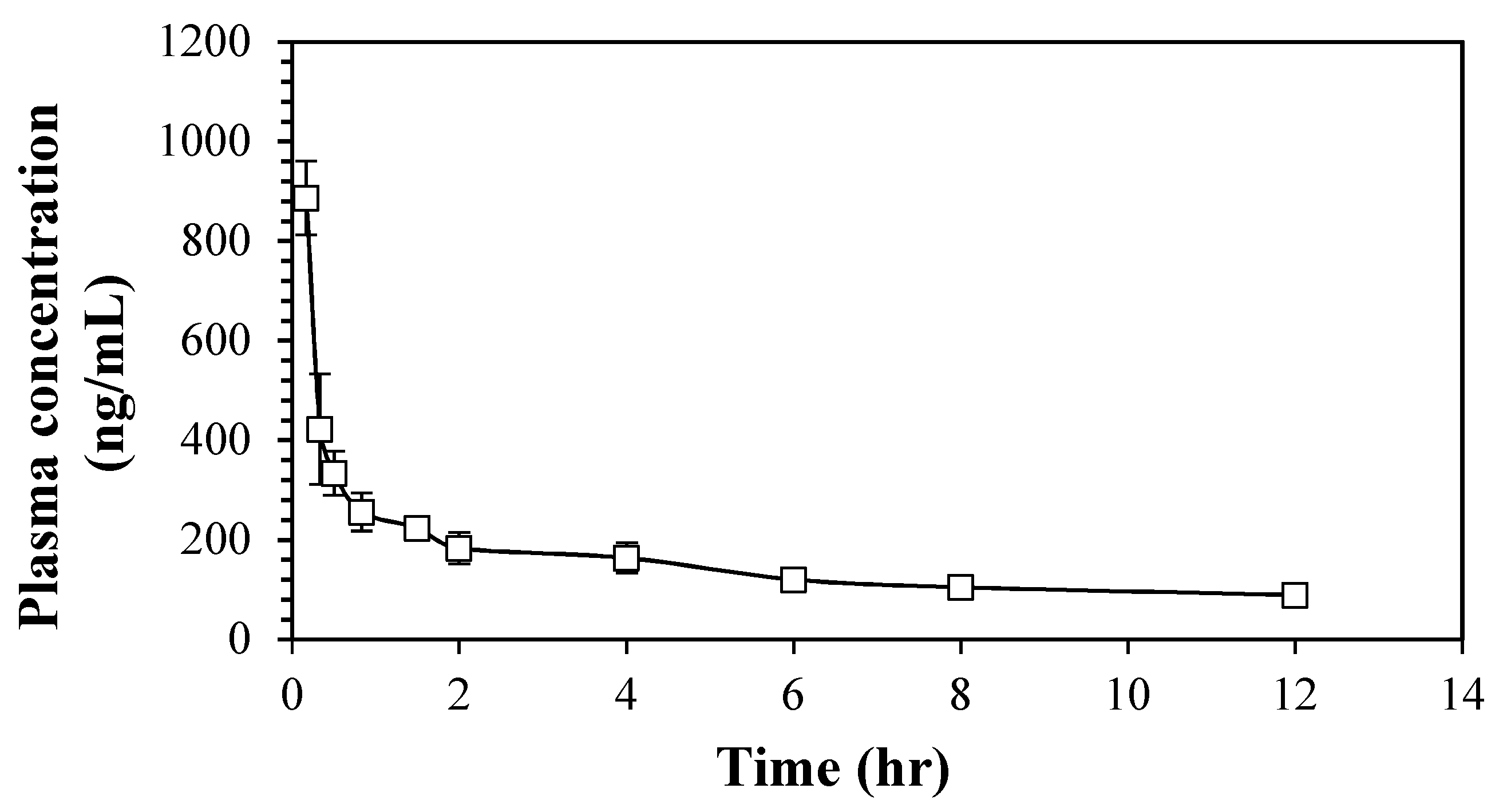
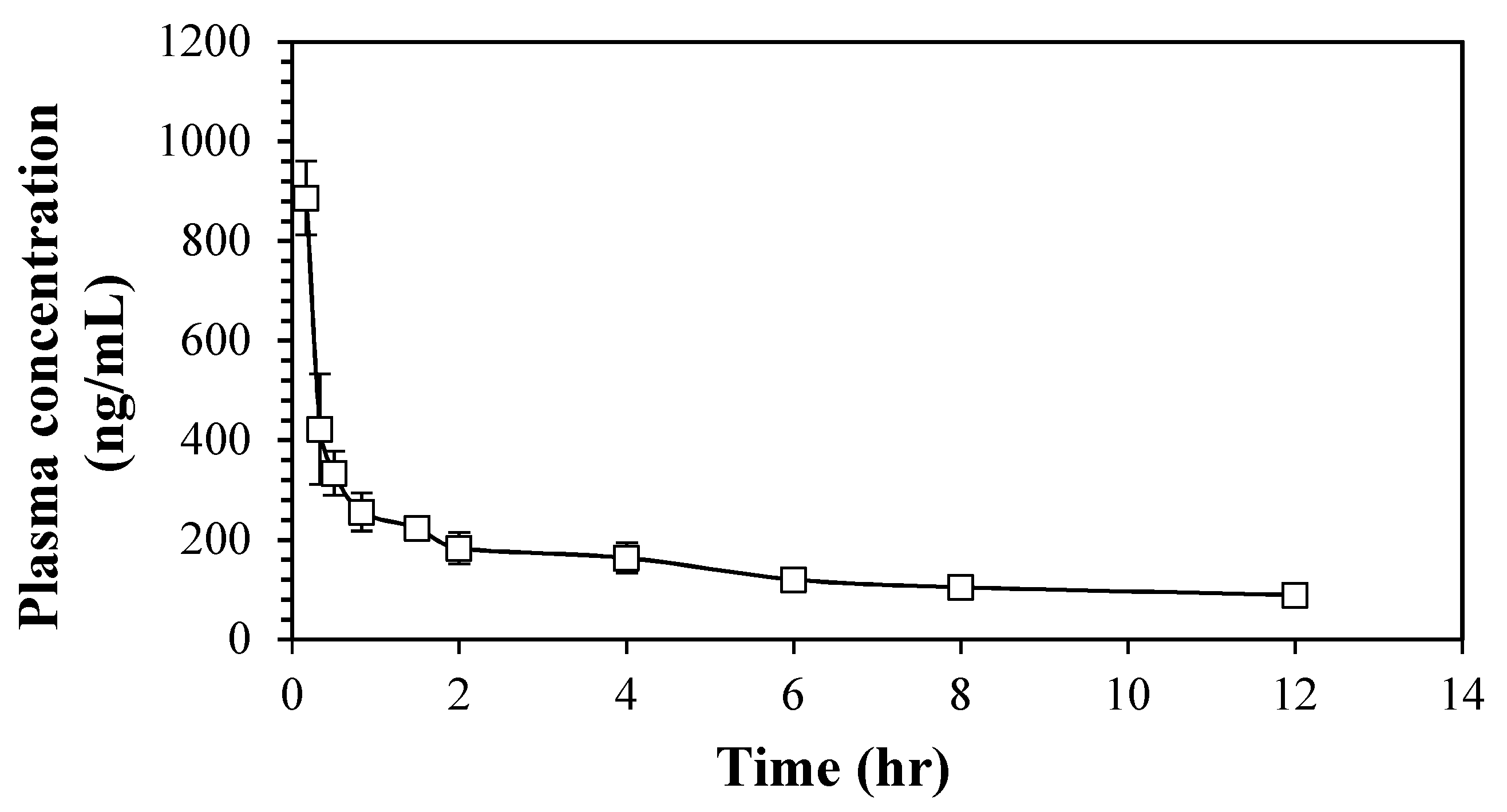
2.2.3. Pharmacokinetic Study of 6cf
3. Materials and Methods
3.1. General Procedure for the Synthesis of CA and DCA Propargyl Amides (4c,d)
3.2. General Procedure for the Synthesis of Bile-Acid-Appended Triazolyl Aryl Ketones (6aa–df)
3.3. Biological Assay
3.3.1. Cell Culture and MTT Assay
3.3.2. Apoptotic Study
3.3.3. Pharmacokinetic Study of 6cf
3.3.4. Determination of 6cf in Rat Plasma
3.3.5. Liquid Chromatographic Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bach, P.B.; Jett, J.R.; Pastorino, U.; Tockman, M.S.; Swensen, S.J.; Begg, C.B. Computed tomography screening and lung cancer outcomes. Jama 2007, 297, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.B. Mechanism-based target identification and drug discovery in cancer research. Science 2000, 287, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arve, L.; Voigt, T.; Waldmann, H. Charting biological and chemical space: PSSC and SCONP as guiding principles for the development of compound collections based on natural product scaffolds. Qsar Comb. Sci. 2006, 25, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Gali, R.; Banothu, J.; Porika, M.; Velpula, R.; Hnamte, S.; Bavantula, R.; Abbagani, S.; Busi, S. Indolylmethylene benzo [h] thiazolo [2,3-b] quinazolinones: Synthesis, characterization and evaluation of anticancer and antimicrobial activities. Bioorg. Med. Chem. Lett. 2014, 24, 4239–4242. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T. Molecular portraits of breast cancer: Tumour subtypes as distinct disease entities. Eur. J. Cancer 2004, 40, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. Ca-Cancer J. Clin. 2011, 61, 212–236. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Roberts Jr, T.G. Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65. [Google Scholar] [CrossRef]
- Rosen, H.; Abribat, T. The rise and rise of drug delivery. Nat. Rev. Drug Discov. 2005, 4, 381. [Google Scholar] [CrossRef]
- Kim, N.-D.; Im, E.-O.; Choi, Y.-H.; Yoo, Y.-H. Synthetic bile acids: Novel mediators of apoptosis. Bmb Rep. 2002, 35, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Carey, E.J.; Lindor, K.D. Chemoprevention of colorectal cancer with ursodeoxycholic acid: Cons. Clin. Res. Hepatol. Gastroenterol. 2012, 36, S61–S64. [Google Scholar] [CrossRef]
- Serfaty, L.; Bissonnette, M.; Poupon, R. Ursodeoxycholic acid and chemoprevention of colorectal cancer. Gastroenterol. Clin. Biol. 2010, 34, 516–522. [Google Scholar] [CrossRef]
- Serfaty, L. Chemoprevention of colorectal cancer with ursodeoxycholic acid: Pro. Clin. Res. Hepatol. Gastroenterol. 2012, 36, S53–S60. [Google Scholar] [CrossRef]
- Tatsumura, T.; Sato, H.; Yamamoto, K.; Ueyama, T. Ursodeoxycholic acid prevents gastrointestinal disorders caused by anticancer drugs. Jpn. J. Surg. 1981, 11, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Zhao, X.; Liu, W.; Deng, J.; Tan, X.; Qiu, L. Anticancer effect of ursodeoxycholic acid in human oral squamous carcinoma HSC-3 cells through the caspases. Nutrients 2015, 7, 3200–3218. [Google Scholar] [CrossRef] [Green Version]
- Dyakova, L.; Culita, D.-C.; Marinescu, G.; Alexandrov, M.; Kalfin, R.; Patron, L.; Alexandrova, R. Metal Zn (II), Cu (II), Ni (II) complexes of ursodeoxycholic acid as putative anticancer agents. Biotechnol. Biotechnol. Equip. 2014, 28, 543–551. [Google Scholar] [CrossRef]
- Marchesi, E.; Chinaglia, N.; Capobianco, M.L.; Marchetti, P.; Huang, T.-E.; Weng, H.-C.; Guh, J.-H.; Hsu, L.-C.; Perrone, D.; NavacchiaKomori, M.L. Dihydroartemisinin–bile acid hybridization as an effective approach to enhance dihydroartemisinin anticancer activity. ChemMedChem 2019, 14, 779–787. [Google Scholar] [CrossRef]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Frank, E.; Molnar, J.; Zupko, I.; Kadar, Z.; Wolfling, J. Synthesis of novel steroidal 17α-triazolyl derivatives via Cu (I)-catalyzed azide-alkyne cycloaddition, and an evaluation of their cytotoxic activity in vitro. Steroids 2011, 76, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Aly, M.R.E.S.; Saad, H.A.; Mohamed, M.A.M. Click reaction based synthesis, antimicrobial, and cytotoxic activities of new 1,2,3-triazoles. Bioorg. Med. Chem. Lett. 2015, 25, 2824–2830. [Google Scholar] [CrossRef]
- Aly, M.R.E.S.; Saad, H.A.; Abdel-Hafez, S.H. Synthesis, antimicrobial and cytotoxicity evaluation of new cholesterol congeners. Beilstein J. Org. Chem. 2015, 11, 1922–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertino, M.; Lopez, C.; Theoduloz, C. Schmeda-Hirschmann, G. 1,2,3-Triazole-substituted oleanolic acid derivatives: Synthesis and antiproliferative activity. Molecules 2013, 18, 7661–7674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Luan, W.; Wang, S.; Cui, S.; Li, F.; Liu, Y.; Liu, Y.; Cheng, M. A library of 1,2,3-triazole-substituted oleanolic acid derivatives as anticancer agents: Design, synthesis, and biological evaluation. Org. Biomol. Chem. 2015, 13, 1507–1514. [Google Scholar] [CrossRef]
- Bębenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Latocha, M.; Boryczka, S. Novel triazoles of 3-acetylbetulin and betulone as anticancer agents. Med. Chem. Res. 2018, 27, 2051–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeed, R.; Sangwan, P.L.; Chinthakindi, P.K.; Khan, I.; Dangroo, N.A.; Thota, N.; Hamid, A.; Sharma, P.R.; Saxena, A.K.; Koul, S. Synthesis of 3-O-propargylated betulinic acid and its 1, 2, 3-triazoles as potential apoptotic agents. Eur. J. Med. Chem. 2013, 63, 782–792. [Google Scholar] [CrossRef]
- Sidova, V.; Zoufaly, P.; Pokorny, J.; Dzubak, P.; Hajduch, M.; Popa, I.; Urban, M. Cytotoxic conjugates of betulinic acid and substituted triazoles prepared by Huisgen Cycloaddition from 30-azidoderivatives. PLoS ONE 2017, 12, e0171621. [Google Scholar] [CrossRef] [Green Version]
- Csuk, R.; Barthel, A.; Kluge, R.; Strohl, D. Synthesis, cytotoxicity and liposome preparation of 28-acetylenic betulin derivatives. Bioorg. Med. Chem. 2010, 18, 7252–7259. [Google Scholar] [CrossRef]
- Csuk, R.; Barthel, A.; Sczepek, R.; Siewert, B.; Schwarz, S. Synthesis, encapsulation and antitumor activity of new betulin derivatives. Arch. Pharm. 2011, 344, 37–49. [Google Scholar] [CrossRef]
- Jurášek, M.; Džubák, P.; Sedlák, D.; Dvořáková, H.; Hajdúch, M.; Bartůněk, P.; Drašar, P. Preparation, preliminary screening of new types of steroid conjugates and their activities on steroid receptors. Steroids 2013, 78, 356–361. [Google Scholar] [CrossRef]
- Mohamed, Z.; El-Koussi, N.A.; Mahfouz, N.M.; Youssef, A.F.; Jaleel, G.A.A.; Shouman, S.A. Cu (I) catalyzed alkyne-azide 1, 3-dipolar cycloaddition (CuAAC): Synthesis of 17α-[1-(substituted phenyl)-1,2,3-triazol-4-yl]-19-nor-testosterone-17β-yl acetates targeting progestational and antipro-liferative activities. Eur. J. Med. Chem. 2015, 97, 75–82. [Google Scholar] [CrossRef]
- Kadar, Z.; Kovacs, D.; Frank, E.; Schneider, G.; Huber, J.; Zupko, I.; Bartok, T.; Wolfling, J. Synthesis and in vitro antiproliferative activity of novel androst-5-ene triazolyl and tetrazolyl derivatives. Molecules 2011, 16, 4786–4806. [Google Scholar] [CrossRef] [PubMed]
- Molnar, J.; Frank, E.; Minorics, R.; Kadar, Z.; Ocsovszki, I.; Schonecker, B.; Wolfling, J.; Zupko, I. A click approach to novel D-ring-substituted 16α-triazolylestrone derivatives and characterization of their antiproliferative properties. PLoS ONE 2015, 10, e0118104. [Google Scholar] [CrossRef] [Green Version]
- Kadar, Z.; Molnar, J.; Schneider, G.; Zupko, I.; Frank, E. A facile ‘click’approach to novel 15β-triazolyl-5α-androstane derivatives, and an evaluation of their antiproliferative activities in vitro. Bioorg. Med. Chem. 2012, 20, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Kadar, Z.; Baji, A.; Zupko, I.; Bartok, T.; Wolfling, J.; Frank, E. Efficient approach to novel 1α-triazolyl-5α-androstane derivatives as potent antiproliferative agents. Org. Biomol. Chem. 2011, 9, 8051–8057. [Google Scholar] [CrossRef] [PubMed]
- Kadar, Z.; Frank, E.; Schneider, G.; Molnar, J.; Zupko, I.; Koti, J.; Schonecker, B.; Wolfling, J. Efficient synthesis of novel A-ring-substituted 1, 2, 3-triazolylcholestane derivatives via catalytic azide-alkyne cycloaddition. Arkivoc 2012, 3, 279–296. [Google Scholar] [CrossRef]
- Solum, E.J.; Vik, A.; Hansen, T.V. Synthesis, cytotoxic effects and tubulin polymerization inhibition of 1,4-disubstituted 1,2,3-triazole analogs of 2-methoxyestradiol. Steroids 2014, 87, 46–53. [Google Scholar] [CrossRef]
- Banday, A.H.; Shameem, S.A.; Gupta, B.; Kumar, H.S. D-ring substituted 1,2,3-triazolyl 20-keto pregnenanes as potential anticancer agents: Synthesis and biological evaluation. Steroids 2010, 75, 801–804. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Perrone, D. Bile acid conjugates with anticancer activity: Most recent research. Molecules 2021, 26, 25. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Mari, L.; Chinaglia, N.; Gallerani, E.; Gavioli, R.; Capobianco, M.L.; Perrone, D. Rational Design of Nucleoside–Bile Acid Conjugates Incorporating a Triazole Moiety for Anticancer Evaluation and SAR Exploration. Molecules 2017, 22, 1710. [Google Scholar] [CrossRef] [Green Version]
- Perrone, D.; Bortolini, O.; Fogagnolo, M.; Marchesi, E.; Mari, L.; Massarenti, C.; Navacchia, M.L.; Sforza, F.; Varani, K.; Capobianco, M.L. Synthesis and in vitro cytotoxicity of deoxyadenosine–bile acid conjugates linked with 1,2,3-triazole. New J. Chem. 2013, 37, 3559–3567. [Google Scholar] [CrossRef]
- Agarwal, D.S.; Krishna, V.S.; Sriram, D.; Yogeeswari, P.; Sakhuja, R. Clickable conjugates of bile acids and nucleosides: Synthesis, characterization, in vitro anticancer and antituberculosis studies. Steroids 2018, 139, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.S.; Singh, R.P.; Lohitesh, K.; Jha, P.N.; Chowdhury, R.; Sakhuja, R. Synthesis and evaluation of bile acid amides of α-cyanostilbenes as anticancer agents. Mol. Divers. 2018, 22, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.S.; Anantaraju, H.S.; Sriram, D.; Yogeeswari, P.; Nanjegowda, S.H.; Mallu, P.; Sakhuja, R. Synthesis, characterization and biological evaluation of bile acid-aromatic/heteroaromatic amides linked via amino acids as anti-cancer agents. Steroids 2016, 107, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Vatmurge, N.S.; Hazra, B.G.; Pore, V.S.; Shirazi, F.; Deshpande, M.V.; Kadreppa, S.; Chattopadhyay, S.; Gonnade, R.G. Synthesis and biological evaluation of bile acid dimers linked with 1, 2, 3-triazole and bis-β-lactam. Org. Biomol. Chem. 2008, 6, 3823–3830. [Google Scholar] [CrossRef]
- Jarrahpour, A.; Fathi, J.; Mimouni, M.; Hadda, T.; Sheikh, J.; Chohan, Z.; Parvez, A. Petra, osiris and molinspiration (POM) together as a successful support in drug design: Antibacterial activity and biopharmaceutical characterization of some azo Schiff bases. Med. Chem. Res. 2012, 21, 1984–1990. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Gündüz, M.G.; Uğur, S.B.; Güney, F.; Özkul, C.; Krishna, V.S.; Kaya, S.; Sriram, D.; Doğan, S.D. 1,3-Disubstitutedureaderivatives: Synthesis, antimicrobial activity evaluation and in silico studies. Bioorg. Chem. 2020, 102, 2020. [Google Scholar] [CrossRef]
- Bartzatt, R.; Donigan, L. Applying pattern recognition methods to analyze the molecular properties of a homologous series of nitrogen mustard agents. AAPS PharmSciTech 2006, 7, 35. [Google Scholar] [CrossRef]
- Hao, T.; Ling, Y.; Wu, M.; Shen, Y.; Gao, Y.; Liang, S.; Gao, Y.; Qian, S. Enhanced oral bioavailability of docetaxel in rats combined with myricetin: In situ and in vivo evidences. Eur. J. Pharm. Sci. 2017, 101, 71–79. [Google Scholar] [CrossRef]
- Sharma, S.; Mazumdar, S.; Italiya, K.S.; Date, T.; Mahato, R.I.; Mittal, A.; Chitkara, D. Cholesterol and Morpholine Grafted Cationic Amphiphilic Copolymers for miRNA-34a Delivery. Mol. Pharm. 2018, 15, 2391–2402. [Google Scholar] [CrossRef]
- Italiya, K.S.; Mazumdar, S.; Sharma, S.; Chitkara, D.; Mahato, R.I.; Mittal, A. Self-assembling lisofylline-fatty acid conjugate for effective treatment of diabetes mellitus. Nanomedicine 2019, 15, 175–187. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
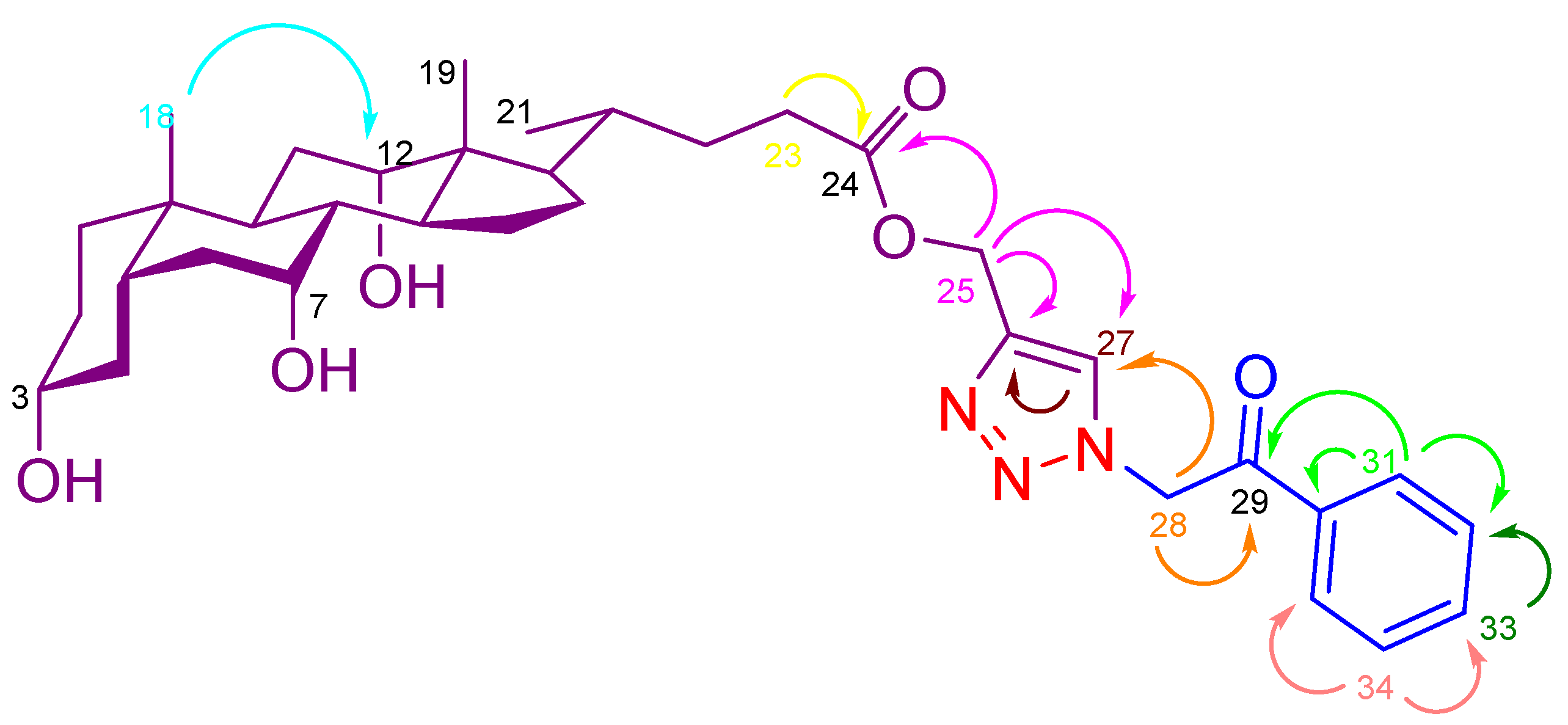
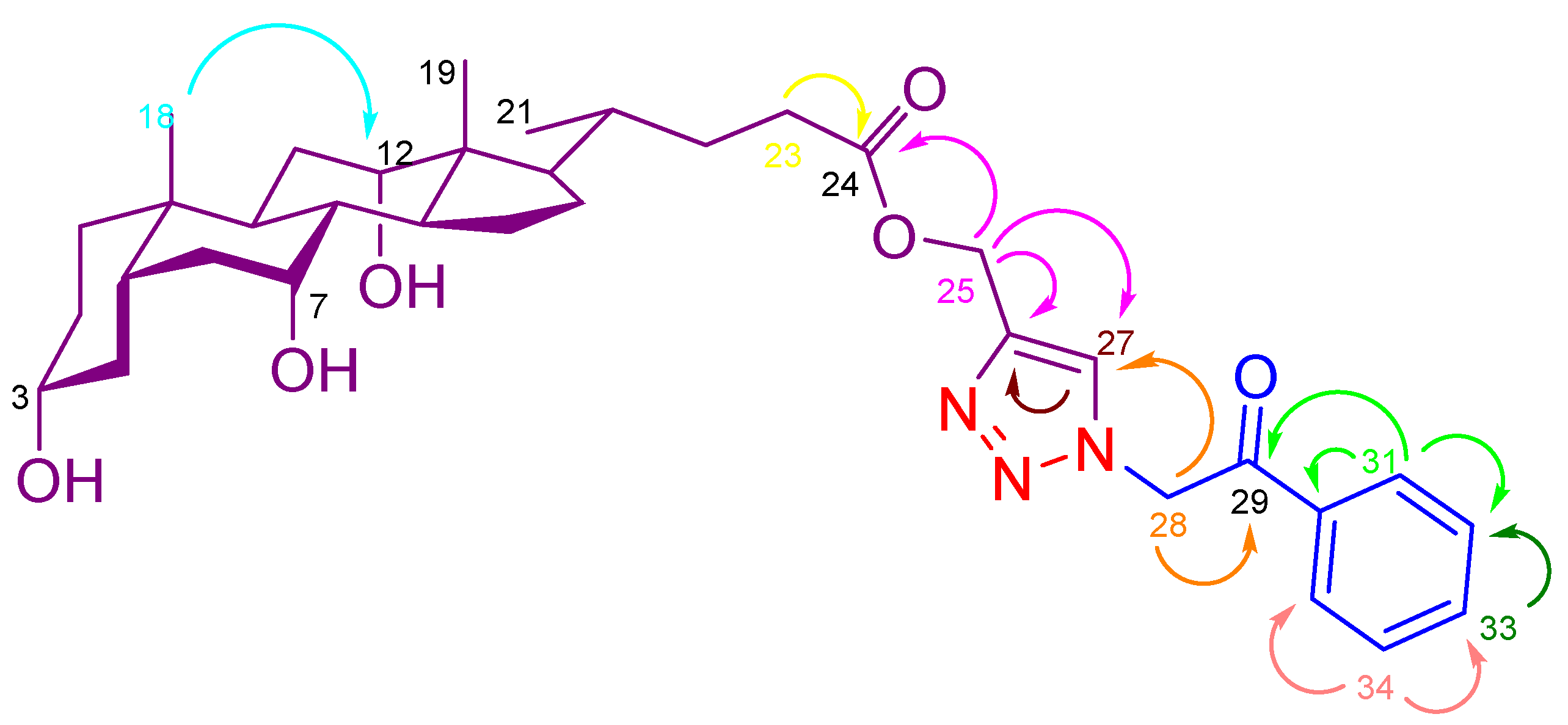
| S. No. | Labeling | 1H NMR | 13C NMR |
|---|---|---|---|
| 1. | 18 | 0.57 (s, 3H) | 12.8 |
| 2. | 19 | 0.80 (s, 3H) | 23.1 |
| 3. | 21 | 0.92 (d, J = 6.2 Hz, 3H) | 17.4 |
| 4. | 3 | 3.17 (d, J = 5.1 Hz, 1H) | 70.9 |
| 5. | 7 | 3.61 (brs, 1H) | 66.7 |
| 6. | 12 | 3.78 (d, J = 3.8 Hz, 1H) | 71.5 |
| 7. | OH | 4.33 (d, J = 4.3 Hz, 1H) | - |
| 8. | OH | 4.12 (d, J = 3.8 Hz, 1H) | - |
| 9. | OH | 4.01 (d, J = 3.3 Hz, 1H) | - |
| 10. | 25 (COO-CH2) | 5.18 (s, 2H) | 57.5 |
| 11. | 28 (N-CH2) | 6.20 (s, 2H) | 56.3 |
| 12. | 27 (Triazole-H) | 8.11 (s, 1H) | 126.9 |
| 13. | 24 (COO) | - | 173.6 |
| 14. | 29 (CO) | - | 192.5 |
| Com. No. | % Cell Viability at 10 µM | milog P d | TPSA e | % ABS f | Drug-Likeness Score | ||
|---|---|---|---|---|---|---|---|
| MCF-7 a | 4T1 b | HEK 293 c | |||||
| 6aa | 77.32 | 74.32 | 86.08 | 4.54 | 134.78 | 62.50 | 0.75 |
| 6ba | 91.90 | 97.81 | 88.92 | 5.45 | 114.55 | 69.48 | 0.65 |
| 6ca | 92.06 | 85.16 | 90.13 | 3.89 | 137.57 | 61.53 | 0.69 |
| 6da | 91.20 | 99.56 | 97.86 | 4.81 | 117.34 | 68.51 | 0.60 |
| 6ab | 57.35 | 72.93 | 87.63 | 4.99 | 134.78 | 62.50 | 0.82 |
| 6bb | 67.60 | 85.69 | 89.17 | 5.90 | 114.55 | 69.48 | 0.72 |
| 6cb | 61.86 | 96.74 | 86.00 | 4.34 | 137.57 | 61.53 | 0.75 |
| 6db | 78.69 | 53.67 | 89.43 | 5.26 | 117.34 | 68.51 | 0.66 |
| 6ac | 55.59 | 80.02 | 87.88 | 4.59 | 144.01 | 59.31 | 0.98 |
| 6bc | 69.62 | 68.40 | 90.77 | 5.51 | 123.78 | 66.29 | 0.89 |
| 6cc | 95.36 | 56.79 | 96.75 | 3.95 | 146.81 | 58.35 | 0.96 |
| 6dc | 68.52 | 64.92 | 86.53 | 4.87 | 126.58 | 65.32 | 0.88 |
| 6ad | 66.48 | 47.70 | 86.79 | 4.70 | 134.78 | 62.50 | 1.07 |
| 6bd | 79.62 | 52.35 | 89.37 | 5.62 | 114.55 | 69.48 | 0.97 |
| 6cd | 88.72 | 70.65 | 97.51 | 4.06 | 137.57 | 61.53 | 1.01 |
| 6dd | 73.76 | 89.90 | 81.61 | 4.97 | 117.34 | 68.51 | 0.92 |
| 6ae | 79.62 | 58.92 | 96.21 | 5.21 | 134.78 | 62.50 | 1.14 |
| 6be | 68.52 | 76.01 | 88.37 | 6.13 | 114.55 | 69.48 | 1.05 |
| 6ce | 69.16 | 50.59 | 92.69 | 4.57 | 137.57 | 61.53 | 1.09 |
| 6de | 86.85 | 62.89 | 90.45 | 5.49 | 117.34 | 68.50 | 1.01 |
| 6af | 26.52 | 55.91 | 88.20 | 5.34 | 134.78 | 62.50 | 0.94 |
| 6bf | 44.43 | 60.86 | 86.82 | 6.26 | 114.55 | 69.48 | 0.85 |
| 6cf | 37.53 | 49.27 | 87.82 | 4.70 | 137.57 | 61.53 | 0.88 |
| 6df | 86.45 | 48.38 | 93.46 | 5.62 | 117.34 | 68.51 | 0.79 |
| DTX g | 46.47 | 56.88 | - | - | - | - | - |
| Compound | IC50 (µM) | |
|---|---|---|
| MCF-7 a | 4T1 b | |
| 6af | 2.61 ± 0.70 | 12.84 ± 1.80 |
| 6bf | 18.26 ± 1.48 | 9.68 ± 1.59 |
| 6cf | 5.71 ± 1.00 | 8.76 ± 1.29 |
| DTX | 9.46 ± 0.98 | 13.85 ± 1.07 |
| Parameters | 6cf (Mean ± SEM) |
|---|---|
| Initial Concentration, C0 (ng/mL) | 1752.69 ± 66.52 |
| Half-Life, t1/2 (h) | 5.63 ± 0.539 |
| Elimination Rate Constant, Ke (1/h) | 0.13 ± 0.01 |
| Area Under the Curve (0 to 12 h), AUC0–last (ng.h/mL) | 1995.306 ± 87.43 |
| Area Under the Curve (0 to infinity), AUC0–∞ (ng.h/mL) | 2690.50 ± 113.20 |
| Area Under the First Moment Curve (0 to 12 h), AUMC0–last (ng.h/mL) | 8155.94 ± 311.78 |
| Area Under the First Moment Curve (0 to infinity), AUMC0–∞ (ng.h/mL) | 22276.39 ± 2023.334 |
| Mean Residence Time, MRT (h) | 8.47 ± 0.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, D.S.; Mazumdar, S.; Italiya, K.S.; Chitkara, D.; Sakhuja, R. Bile-Acid-Appended Triazolyl Aryl Ketones: Design, Synthesis, In Vitro Anticancer Activity and Pharmacokinetics in Rats. Molecules 2021, 26, 5741. https://doi.org/10.3390/molecules26195741
Agarwal DS, Mazumdar S, Italiya KS, Chitkara D, Sakhuja R. Bile-Acid-Appended Triazolyl Aryl Ketones: Design, Synthesis, In Vitro Anticancer Activity and Pharmacokinetics in Rats. Molecules. 2021; 26(19):5741. https://doi.org/10.3390/molecules26195741
Chicago/Turabian StyleAgarwal, Devesh S., Samrat Mazumdar, Kishan S. Italiya, Deepak Chitkara, and Rajeev Sakhuja. 2021. "Bile-Acid-Appended Triazolyl Aryl Ketones: Design, Synthesis, In Vitro Anticancer Activity and Pharmacokinetics in Rats" Molecules 26, no. 19: 5741. https://doi.org/10.3390/molecules26195741
APA StyleAgarwal, D. S., Mazumdar, S., Italiya, K. S., Chitkara, D., & Sakhuja, R. (2021). Bile-Acid-Appended Triazolyl Aryl Ketones: Design, Synthesis, In Vitro Anticancer Activity and Pharmacokinetics in Rats. Molecules, 26(19), 5741. https://doi.org/10.3390/molecules26195741