
Isolation, Characterization, Complete Structural Assignment, and Anticancer Activities of the Methoxylated Flavonoids from Rhamnus disperma Roots

 ,
, 
 and
and
Abstract
:1. Introduction
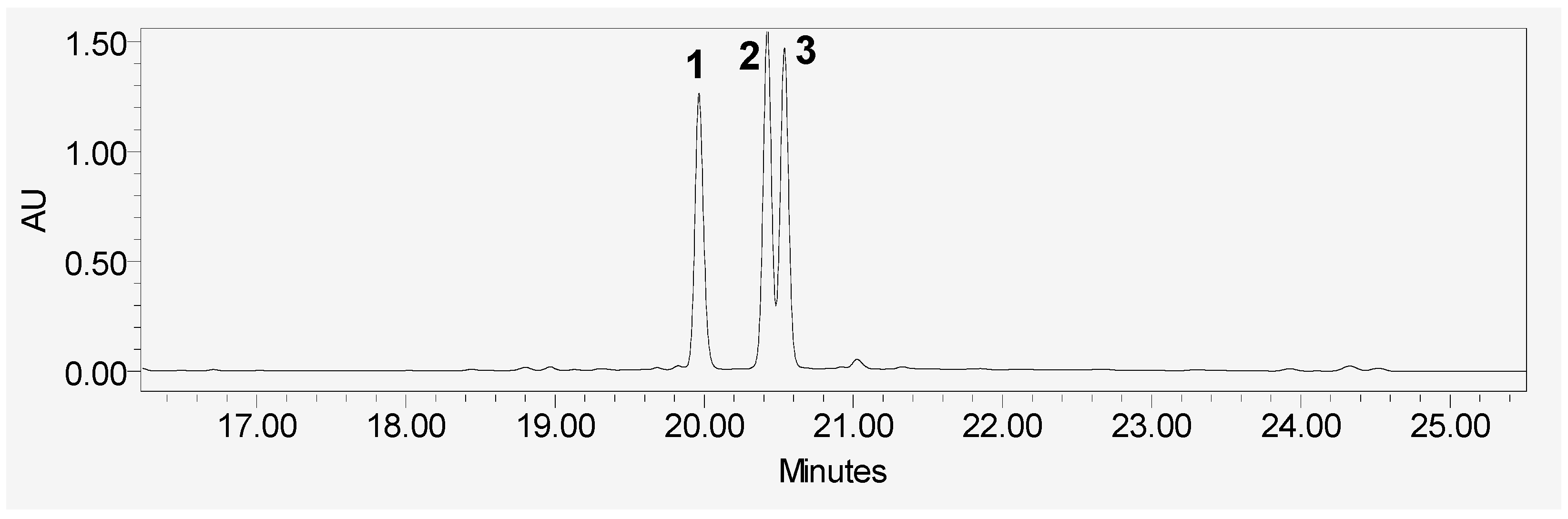
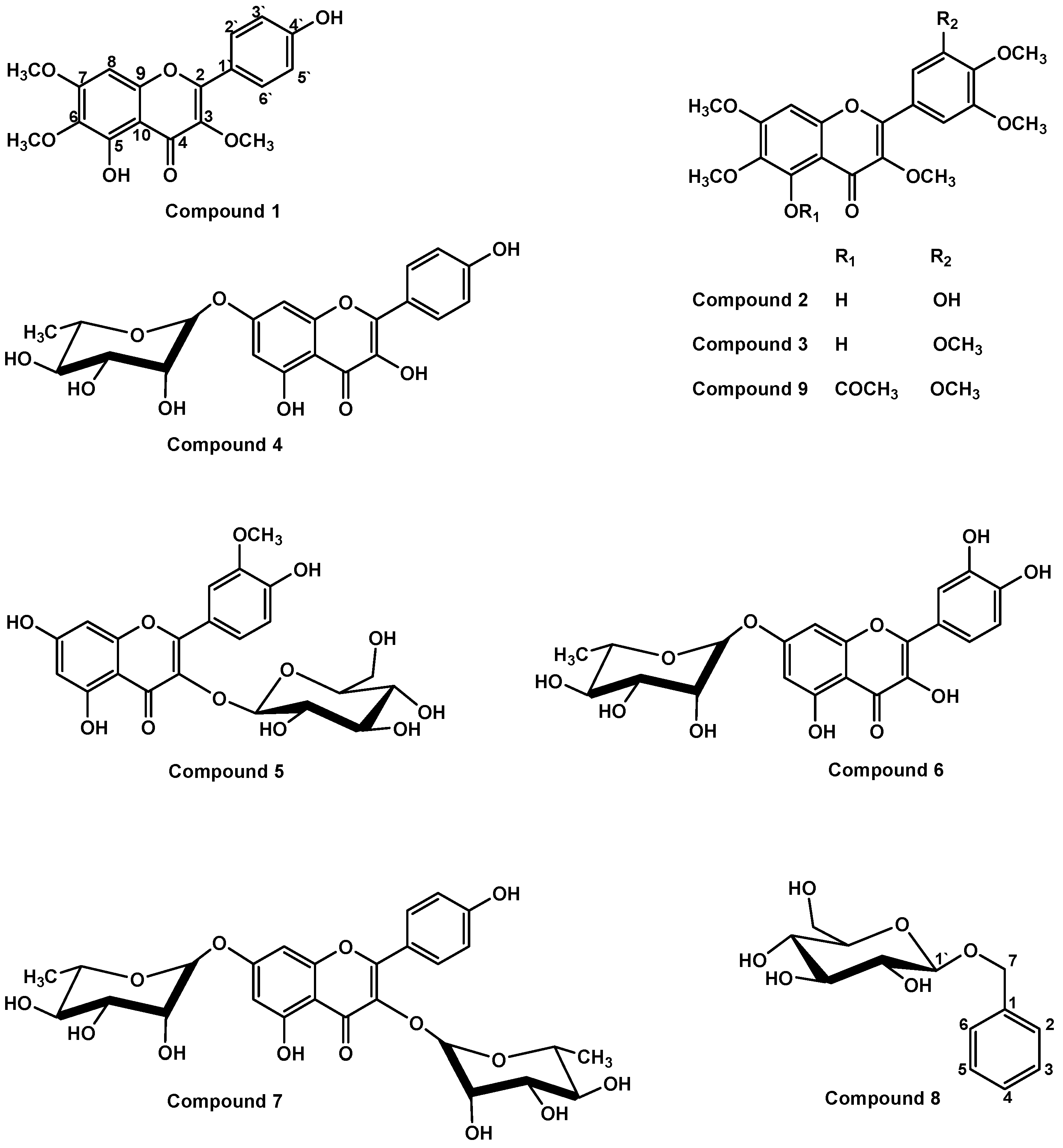
2. Results and Discussion
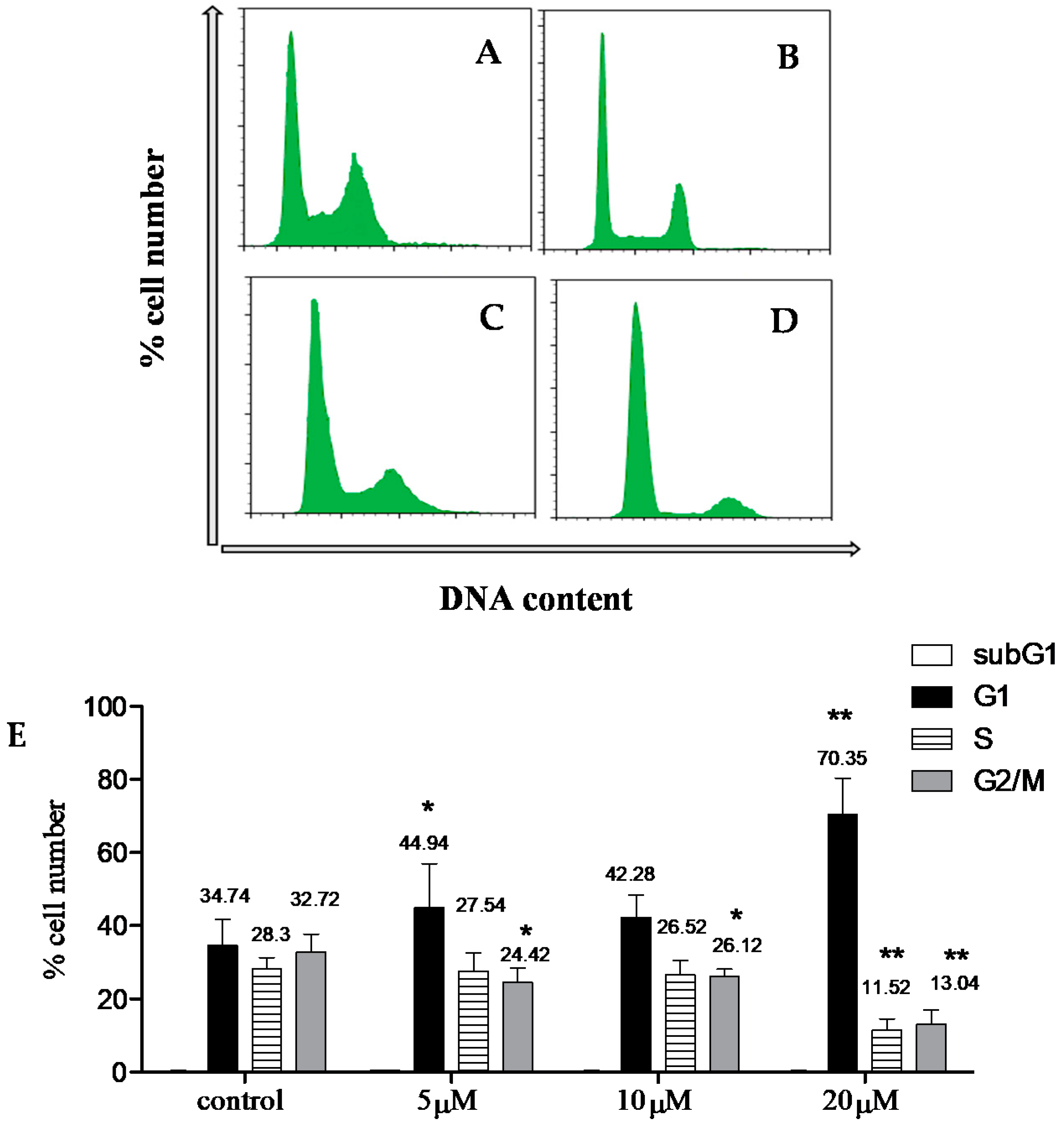
Cytotoxicity Assay
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectroscopic Analysis of the Isolated Compounds
3.5. Acetylation of Compound 3
3.6. Acid Hydrolysis of Compounds 4–8
3.7. Cytotoxicity Assay
3.8. Annexin VFITC/PI Apoptosis Assay
3.9. Cell Cycle Analysis
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Boulos, L. Flora of Egypt: With 409 Line Drawings and 94 Colour Photographs; Geraniaceae-Boraginaceae; Al Hadara Publishing: Cairo, Egypt, 2000. [Google Scholar]
- Marna, E. Cultivated Plants; Academic Press Publishing: London, UK; New York, NY, USA, 1961. [Google Scholar]
- Collenette, S. Wildflowers of Saudi Arabia; National Commission for Wildlife Conservation and Development (NCWCD) Publishing: Riyadh, Saudi Arab, 1999. [Google Scholar]
- Tackholm, V. Students’ Flora of Egypt; Cairo University Publishing: Cairo, Egypt, 1974. [Google Scholar]
- Ragab, E.A. Highly methoxylated flavonoids and the anti-eczematous activity of Rhamnus disperma roots. J. Pharmacogn. Phytochem. 2015, 4, 01–08. [Google Scholar]
- Marzouk, M.S.; El-Toumy, S.A.A.; Merfort, I.; Nawwar, M.A.M. Polyphenolic metabolites of Rhamnus disperma. Phytochemistry 1999, 52, 943–946. [Google Scholar] [CrossRef]
- Ammar, R.B.; Bhouri, W.; Sghaier, M.B.; Boubaker, J.; Skandrani, I.; Neffati, A.; Bouhlel, I.; Kilani, S.; Mariotte, A.M.; Chekir-Ghedira, L.; et al. Antioxidant and free radical-scavenging properties of three flavonoids isolated from the leaves of Rhamnus alaternus L. (Rhamnaceae): A structure-activity relationship study. Food Chem. 2009, 116, 258–264. [Google Scholar] [CrossRef]
- Benamar, H.; Rarivoson, E.; Tomassini, L.; Frezza, C.; Marouf, A.; Bennaceur, M.; Nicoletti, M. Phytochemical profiles, antioxidant and anti-acetylcholinesterasic activities of the leaf extracts of Rhamnus lycioides subsp. oleoides (L.) Jahand. & Maire in different solvents. Nat. Prod. Res. 2019, 33, 1456–1462. [Google Scholar]
- Chen, G.; Li, X.; Saleri, F.; Guo, M. Analysis of flavonoids in Rhamnus davurica and its antiproliferative activities. Molecules 2016, 21, 1275. [Google Scholar] [CrossRef]
- Chen, G.; Wu, J.; Li, N.; Guo, M. Screening for anti-proliferative and anti-inflammatory components from Rhamnus davurica Pall. using bio-affinity ultrafiltration with multiple drug targets. Anal. Bioanal. Chem. 2018, 410, 3587–3595. [Google Scholar] [CrossRef]
- Hamed, M.M.; Refahy, L.A.; Abdel-Aziz, M.S. Evaluation of antimicrobial activity of some compounds isolated from Rhamnus cathartica L. Orient J. Chem. 2015, 31, 1133–1140. [Google Scholar] [CrossRef]
- Ammar, R.B.; Kilani, S.; Bouhlel, I.; Skandrani, I.; Naffeti, A.; Boubaker, J.; Sghaier, M.B.; Bhouri, W.; Mahmoud, A.; Chekir-Ghedira, L.; et al. Antibacterial and cytotoxic activities of extracts from (Tunisian) Rhamnus alaternus (Rhamnaceae). Ann. Microbiol. 2007, 57, 453–460. [Google Scholar] [CrossRef]
- Mohammed, H.A. Antioxidant Flavonol Rhamnosides from Rhamnus disperma Roots. Int. J. Pharm. Phytopharm. Res. 2015, 5, 2249–6084. [Google Scholar]
- Harborne, J.B. The Flavonoids–Advances in Research since 1986; Chapman and Hall/CRC Publishing: London, UK, 1994. [Google Scholar]
- Gorin, P.A.J.; Mazurek, M. Further studies on the assignment of signals in 13C magnetic resonance spectra of aldoses and derived methyl glycosides. Can. J. Chem. 1975, 53, 1212–1223. [Google Scholar] [CrossRef]
- Markham, K.R.; Ternai, B.; Stanley, R.; Geiger, H.; Mabry, T.J. Carbon-13 NMR studies of flavonoids—III: Naturally occurring flavonoid glycosides and their acylated derivatives. Tetrahedron 1978, 34, 1389–1397. [Google Scholar] [CrossRef]
- Wang, D.-M.; Pu, W.-J.; Wang, Y.-H.; Zhang, Y.-J.; Wang, S.-S. A new isorhamnetin glycoside and other phenolic compounds from Callianthemum taipaicum. Molecules 2012, 17, 4595–4603. [Google Scholar] [CrossRef] [Green Version]
- Wagner, H.; Chari, V.M.; Sonnenbichler, J. 13C-NMR-spektren natürlich vorkommender flavonoide. Tetrahedron Lett. 1976, 17, 1799–1802. [Google Scholar] [CrossRef]
- Wen, P.; Han, H.; Wang, R.; Wang, N.; Yao, X. C-glycosylfavones and aromatic glycosides from Campylotropis hirtella (Franch.) Schindl. Asian J. Tradit. Med. 2007, 2, 149–153. [Google Scholar]
- Mastelić, J.; Jerković, I.; Vinković, M.; Džolić, Z.; Vikić-Topić, D. Synthesis of selected naturally occurring glucosides of volatile compounds. Their chromatographic and spectroscopic properties. Croat. Chem. Acta 2004, 77, 491–500. [Google Scholar]
- Ly, T.N.; Yamauchi, R.; Shimoyamada, M.; Kato, K. Isolation and structural elucidation of some glycosides from the rhizomes of smaller galanga (Alpinia officinarum Hance). J. Agric. Food Chem. 2002, 50, 4919–4924. [Google Scholar] [CrossRef]
- Sak, K. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn. Rev. 2014, 8, 122–146. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.-H.; Chen, W.-J.; Lin-Shiau, S.-Y.; Ho, C.-T.; Lin, J.-K. Tangeretin induces cell-cycle G1 arrest through inhibiting cyclin-dependent kinases 2 and 4 activities as well as elevating Cdk inhibitors p21 and p27 in human colorectal carcinoma cells. Carcinogenesis 2002, 23, 1677–1684. [Google Scholar] [CrossRef] [Green Version]
- Benavente-Garcia, O.; Castillo, J. Update on uses and properties of citrus flavonoids: New findings in anticancer, cardiovascular, and anti-inflammatory activity. J. Agric. Food Chem. 2008, 56, 6185–6205. [Google Scholar] [CrossRef]
- Walle, T. Methoxylated flavones, a superior cancer chemopreventive flavonoid subclass? Semin. Cancer Biol. 2007, 17, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Neves, A.R.; Correia-da-Silva, M.; Silva, P.; Ribeiro, D.; Sousa, E.; Bousbaa, H.; Pinto, M. Synthesis of new glycosylated flavonoids with inhibitory activity on cell growth. Molecules 2018, 23, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, H.A.; Al-Omar, M.S.; El-Readi, M.Z.; Alhowail, A.H.; Aldubayan, M.A.; Abdellatif, A.A.H. Formulation of Ethyl Cellulose Microparticles Incorporated Pheophytin A Isolated from Suaeda vermiculata for Antioxidant and Cytotoxic Activities. Molecules 2018, 24, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, H.A.; Khan, R.A.; Abdel-Hafez, A.A.; Abdel-Aziz, M.; Ahmed, E.; Enany, S.; Mahgoub, S.; Al-Rugaie, O.; Alsharidah, M.; Aly, M.S.A.; et al. Phytochemical Profiling, In Vitro and In Silico Anti-Microbial and Anti-Cancer Activity Evaluations and Staph GyraseB and h-TOP-IIβ Receptor-Docking Studies of Major Constituents of Zygophyllum coccineum L. Aqueous-Ethanolic Extract and Its Subsequent Fra. Molecules 2021, 26, 577. [Google Scholar] [CrossRef]
- Shaheen, U.; Ragab, E.A.; Abdalla, A.N.; Bader, A. Triterpenoidal saponins from the fruits of Gleditsia caspica with proapoptotic properties. Phytochemistry 2018, 145, 168–178. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MCF 7 | A2780 | HT 29 | MRC5 |
|---|---|---|---|---|
| 1 | 2.17 ± 0.26 | 0.53 ± 0.45 | 2.16 ± 0.02 | 4.40 ± 1.45 |
| 2 | 6.81 ± 0.04 | 8.97 ± 1.72 | 9.07 ± 0.22 | 5.46 ± 1.57 |
| 3 | 2.76 ± 0.16 | 3.73 ± 1.75 | 2.71 ± 1.25 | 11.73 ± 1.58 |
| 9 | 2.190 ± 0.64 | - | 3.18 ± 0.64 | 19.04 ± 2.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, H.A.; Abd El-Wahab, M.F.; Shaheen, U.; Mohammed, A.E.-S.I.; Abdalla, A.N.; Ragab, E.A. Isolation, Characterization, Complete Structural Assignment, and Anticancer Activities of the Methoxylated Flavonoids from Rhamnus disperma Roots. Molecules 2021, 26, 5827. https://doi.org/10.3390/molecules26195827
Mohammed HA, Abd El-Wahab MF, Shaheen U, Mohammed AE-SI, Abdalla AN, Ragab EA. Isolation, Characterization, Complete Structural Assignment, and Anticancer Activities of the Methoxylated Flavonoids from Rhamnus disperma Roots. Molecules. 2021; 26(19):5827. https://doi.org/10.3390/molecules26195827
Chicago/Turabian StyleMohammed, Hamdoon A., Mohammed F. Abd El-Wahab, Usama Shaheen, Abd El-Salam I. Mohammed, Ashraf N. Abdalla, and Ehab A. Ragab. 2021. "Isolation, Characterization, Complete Structural Assignment, and Anticancer Activities of the Methoxylated Flavonoids from Rhamnus disperma Roots" Molecules 26, no. 19: 5827. https://doi.org/10.3390/molecules26195827
APA StyleMohammed, H. A., Abd El-Wahab, M. F., Shaheen, U., Mohammed, A. E.-S. I., Abdalla, A. N., & Ragab, E. A. (2021). Isolation, Characterization, Complete Structural Assignment, and Anticancer Activities of the Methoxylated Flavonoids from Rhamnus disperma Roots. Molecules, 26(19), 5827. https://doi.org/10.3390/molecules26195827