
Photochemistry of Thymine in Protic Polar Nanomeric Droplets Using Electrostatic Embeding TD-DFT/MM


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
2.1. Electrostatic Embedding Coupling in ESPF DFT/MM
2.2. Electrostatic Embedding Coupling in ESPF TDA-TDDFT/MM
2.3. Branching Update Method
3. Computational Details
4. Results and Discussion
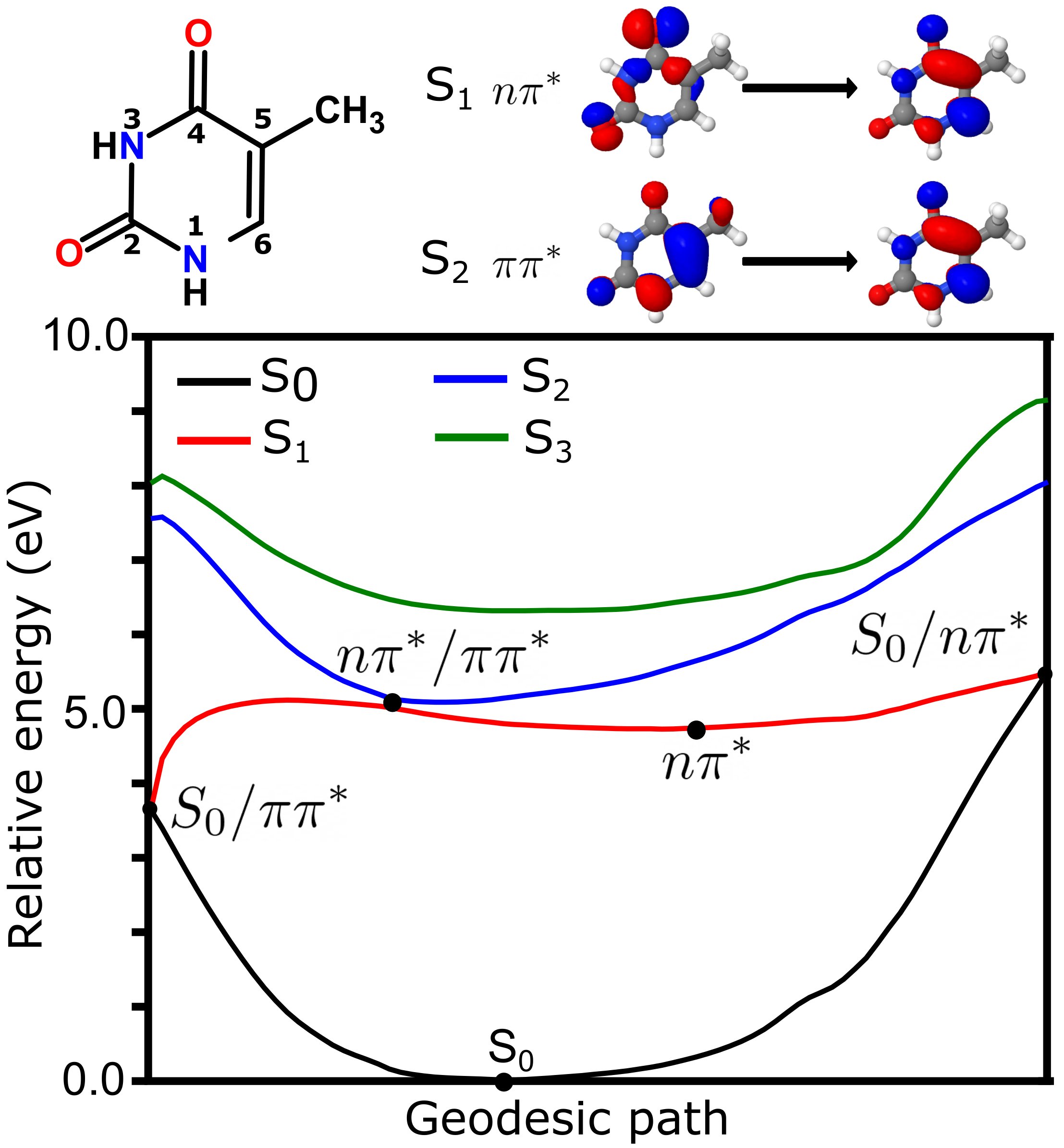
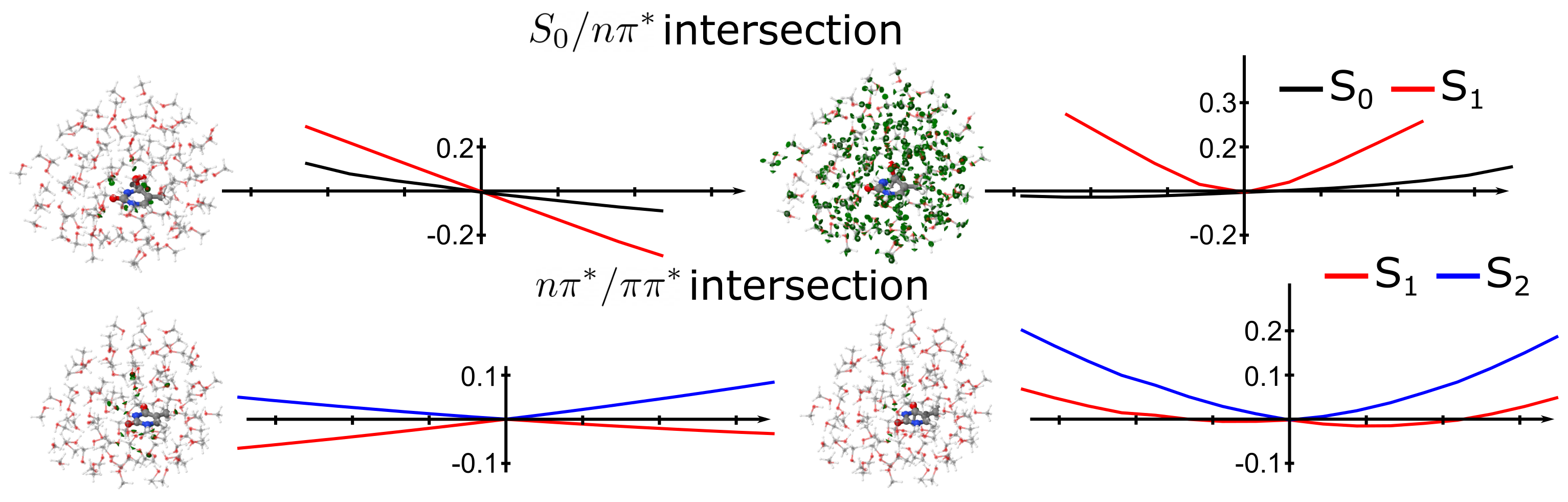
4.1. Photochemistry of Thymine in Gas Phase
4.2. Photochemistry of Thymine in Methanol Droplet
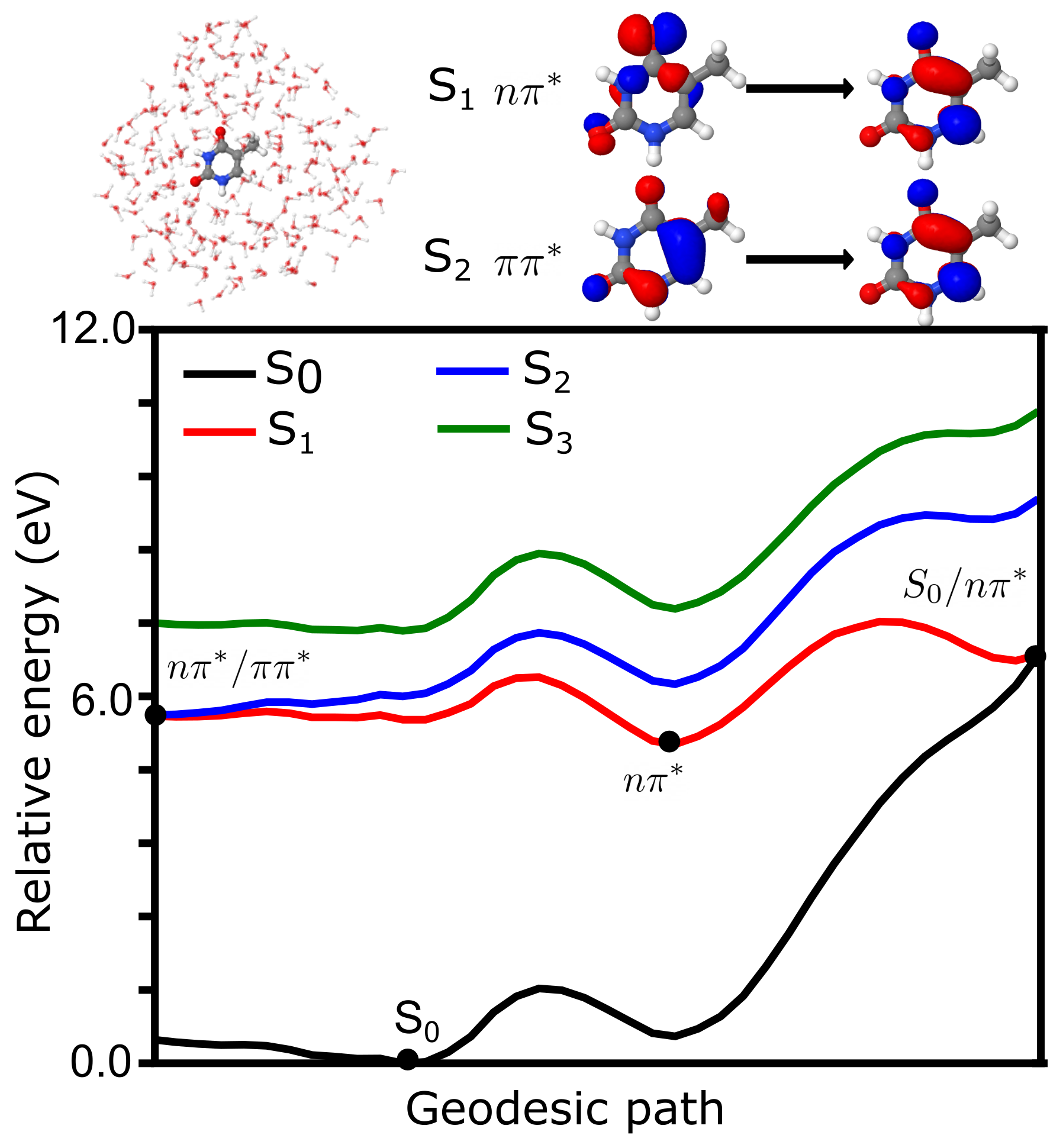
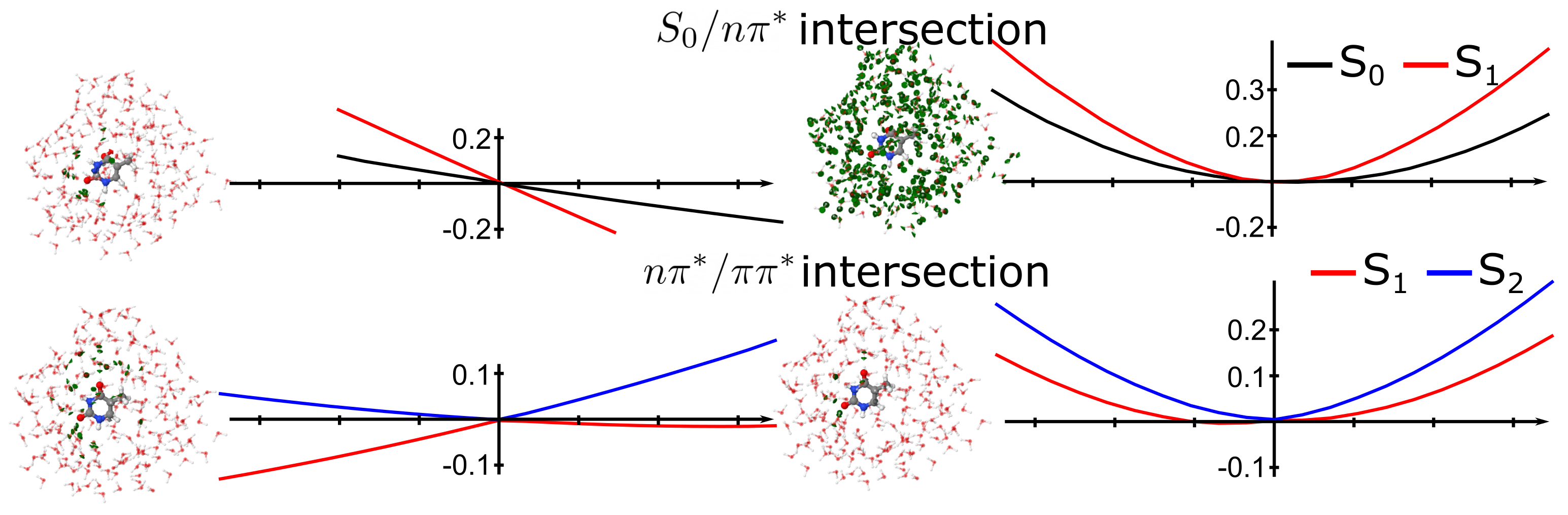
4.3. Photochemistry of Thymine in Water Droplet
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Gruijl, F.R. Skin cancer and solar UV radiation. Eur. J. Cancer 1999, 35, 2003–2009. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Nir, E.; Kleinermanns, K.; Grace, L.; de Vries, M.S. On the Photochemistry of Purine Nucleobases. J. Phys. Chem. A 2001, 105, 5106–5110. [Google Scholar] [CrossRef] [Green Version]
- Merchán, M.; González-Luque, R.; Climent, T.; Serrano-Andrés, L.; Rodríguez, E.; Reguero, M.; Peláez, D. Unified model for the ultrafast decay of pyrimidine nucleobases. J. Phys. Chem. B 2006, 110, 26471–26476. [Google Scholar] [CrossRef]
- Barbatti, M.; Aquino, A.J.A.; Szymczak, J.J.; Nachtigallová, D.; Hobza, P.; Lischka, H. Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. USA 2010, 107, 21453–21458. [Google Scholar] [CrossRef] [Green Version]
- Wolf, T.J.A.; Gühr, M. Photochemical pathways in nucleobases measured with an X-ray FEL. Phil. Trans. R. Soc. A 2019, 377, 20170473. [Google Scholar] [CrossRef] [Green Version]
- Arslancan, S.; Martínez-Fernández, L.; Corral, I. Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules 2017, 22, 998. [Google Scholar] [CrossRef]
- Gustavsson, T.; Bányász, A.; Lazzarotto, E.; Markovitsi, D.; Scalmani, G.; Frisch, M.J.; Barone, V.; Improta, R. Singlet Excited-State Behavior of Uracil and Thymine in Aqueous Solution: A Combined Experimental and Computational Study of 11 Uracil Derivatives. J. Am. Chem. Soc. 2006, 128, 607–619. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V. The excited states of adenine and thymine nucleoside and nucleotide in aqueous solution: A comparative study by time-dependent DFT calculations. Theor. Chem. Acc. 2008, 120, 491–497. [Google Scholar] [CrossRef]
- Lischka, H.; Barbatti, M.; Siddique, F.; Das, A.; Aquino, A.J. The effect of hydrogen bonding on the nonadiabatic dynamics of a thymine-water cluster. Chem. Phys. 2018, 515, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Brown, A. Effects of hydrogen bonding with H2O on the resonance Raman spectra of uracil and thymine. Comput. Theor. Chem. 2017, 1100, 70–82. [Google Scholar] [CrossRef]
- Janicki, M.J.; Szabla, R.; §poner, J.; Góra, R.W. Solvation effects alter the photochemistry of 2-thiocytosine. Chem. Phys. 2018, 515, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Johns, H.; Rapaport, S.; Delbrück, M. Photochemistry of thymine dimers. J. Mol. Biol. 1962, 4, 104–114. [Google Scholar] [CrossRef]
- LeClerc, J.E.; Borden, A.; Lawrence, C.W. The thymine-thymine pyrimidine-pyrimidone(6-4) ultraviolet light photoproduct is highly mutagenic and specifically induces 3’ thymine-to-cytosine transitions in Escherichia coli. Proc. Natl. Acad. Sci. USA 1991, 88, 9685–9689. [Google Scholar] [CrossRef] [Green Version]
- Otoshi, E.; Yagi, T.; Mori, T.; Matsunaga, T.; Nikaido, O.; Kim, S.T.; Hitomi, K.; Ikenaga, M.; Todo, T. Respective Roles of Cyclobutane Pyrimidine Dimers, (6–4)Photoproducts, and Minor Photoproducts in Ultraviolet Mutagenesis of Repair-deficient Xeroderma Pigmentosum A Cells. Cancer Res. 2000, 60, 1729–1735. [Google Scholar]
- Marguet, S.; Markovitsi, D. Time-Resolved Study of Thymine Dimer Formation. J. Am. Chem. Soc. 2005, 127, 5780–5781. [Google Scholar] [CrossRef] [Green Version]
- Rauer, C.; Nogueira, J.J.; Marquetand, P.; González, L. Cyclobutane Thymine Photodimerization Mechanism Revealed by Nonadiabatic Molecular Dynamics. J. Am. Chem. Soc. 2016, 138, 15911–15916. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Zuehlsdorff, T.J.; Payne, M.C.; Hine, N.D.M. Photophysics and Photochemistry of DNA Molecules: Electronic Excited States Leading to Thymine Dimerization. J. Phys. Chem. C 2018, 122, 11633–11640. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Lee, S.; Huix-Rotllant, M.; Filatov, M.; Choi, C.H. Impact of the Dynamic Electron Correlation on the Unusually Long Excited-State Lifetime of Thymine. J. Phys. Chem. Lett. 2021, 12, 4339–4346. [Google Scholar] [CrossRef]
- McFarland, B.K.; Farrell, J.P.; Miyabe, S.; Tarantelli, F.; Aguilar, A.; Berrah, N.; Bostedt, C.; Bozek, J.D.; Bucksbaum, P.H.; Castagna, J.C.; et al. Ultrafast X-ray Auger probing of photoexcited molecular dynamics. Nat. Commun. 2014, 5, 4235. [Google Scholar] [CrossRef] [Green Version]
- Hudock, H.R.; Levine, B.G.; Thompson, A.L.; Satzger, H.; Townsend, D.; Gador, N.; Ullrich, S.; Stolow, A.; Martínez, T.J. Ab Initio Molecular Dynamics and Time-Resolved Photoelectron Spectroscopy of Electronically Excited Uracil and Thymine. J. Phys. Chem. A 2007, 111, 8500–8508. [Google Scholar] [CrossRef] [Green Version]
- Picconi, D.; Barone, V.; Lami, A.; Santoro, F.; Improta, R. The Interplay between ππ*/nπ* Excited States in Gas-Phase Thymine: A Quantum Dynamical Study. ChemPhysChem 2011, 12, 1957–1968. [Google Scholar] [CrossRef]
- Casida, M.; Huix-Rotllant, M. Progress in Time-Dependent Density-Functional Theory. Ann. Rev. Phys. Chem. 2012, 63, 287–323. [Google Scholar] [CrossRef] [Green Version]
- Ferré, N.; Ángyán, J.G. Approximate electrostatic interaction operator for QM/MM calculations. Chem. Phys. Lett. 2002, 356, 331–339. [Google Scholar] [CrossRef]
- Schwinn, K.; Ferré, N.; Huix-Rotllant, M. Analytic QM/MM atomic charge derivatives avoiding the scaling of coupled perturbed equations with the MM subsystem size. J. Chem. Phys. 2019, 151, 041102. [Google Scholar] [CrossRef]
- Schwinn, K.; Ferré, N.; Huix-Rotllant, M. Efficient Analytic Second Derivative of Electrostatic Embedding QM/MM Energy: Normal Mode Analysis of Plant Cryptochrome. J. Chem. Theor. Comput. 2020, 16, 3816–3824. [Google Scholar] [CrossRef]
- Huix-Rotllant, M.; Ferré, N. Analytic Energy, Gradient, and Hessian of Electrostatic Embedding QM/MM Based on Electrostatic Potential-Fitted Atomic Charges Scaling Linearly with the MM Subsystem Size. J. Chem. Theor. Comput. 2021, 17, 538–548. [Google Scholar] [CrossRef]
- Schwinn, K.; Ferré, N.; Huix-Rotllant, M. UV-visible absorption spectrum of FAD and its reduced forms embedded in a cryptochrome protein. Phys. Chem. Chem. Phys. 2020, 22, 12447–12455. [Google Scholar] [CrossRef]
- Huix-Rotllant, M.; Schwinn, K.; Ferré, N. Infrared spectroscopy from electrostatic embedding QM/MM: Local normal mode analysis of infrared spectra of arabidopsis thaliana plant cryptochrome. Phys. Chem. Chem. Phys. 2021, 23, 1666–1674. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K.; Morokuma, K. Updated Branching Plane for Finding Conical Intersections without Coupling Derivative Vectors. J. Chem. Theory Comput. 2010, 6, 1538–1545. [Google Scholar] [CrossRef]
- Fetter, A. Quantum Theory of Many-Particle Systems; Dover Publications: Mineola, NY, USA, 2003. [Google Scholar]
- Casida, M.E. Time-Dependent Density Functional Response Theory for Molecules. In Recent Advances in Density Functional Methods; World Scientific: Singapore, 1995; Volume 1, pp. 155–192. [Google Scholar]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Handy, N.C.; Schaefer, H.F. On the evaluation of analytic energy derivatives for correlated wave functions. J. Chem. Phys. 1984, 81, 5031–5033. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Ponder, J.W. TINKER: Software Tools for Molecular Design; Version 6.3.3; Washington University School of Medicine: Saint Louis, MO, USA, 2014. [Google Scholar]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Zhu, X.; Thompson, K.C.; Martínez, T.J. Geodesic interpolation for reaction pathways. J. Chem. Phys. 2019, 150, 164103. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.G.; Ko, C.; Quenneville, J.; MartÍnez, T.J. Conical intersections and double excitations in time-dependent density functional theory. Mol. Phys. 2006, 104, 1039–1051. [Google Scholar] [CrossRef]
- Burghardt, I.; Cederbaum, L.S.; Hynes, J.T. Environmental effects on a conical intersection: A model study. Faraday Discuss. 2004, 127, 395–411. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huix-Rotllant, M. Photochemistry of Thymine in Protic Polar Nanomeric Droplets Using Electrostatic Embeding TD-DFT/MM. Molecules 2021, 26, 6021. https://doi.org/10.3390/molecules26196021
Huix-Rotllant M. Photochemistry of Thymine in Protic Polar Nanomeric Droplets Using Electrostatic Embeding TD-DFT/MM. Molecules. 2021; 26(19):6021. https://doi.org/10.3390/molecules26196021
Chicago/Turabian StyleHuix-Rotllant, Miquel. 2021. "Photochemistry of Thymine in Protic Polar Nanomeric Droplets Using Electrostatic Embeding TD-DFT/MM" Molecules 26, no. 19: 6021. https://doi.org/10.3390/molecules26196021