
Molecular Dynamics Simulations Study of the Interactions between Human Dipeptidyl-Peptidase III and Two Substrates




Abstract
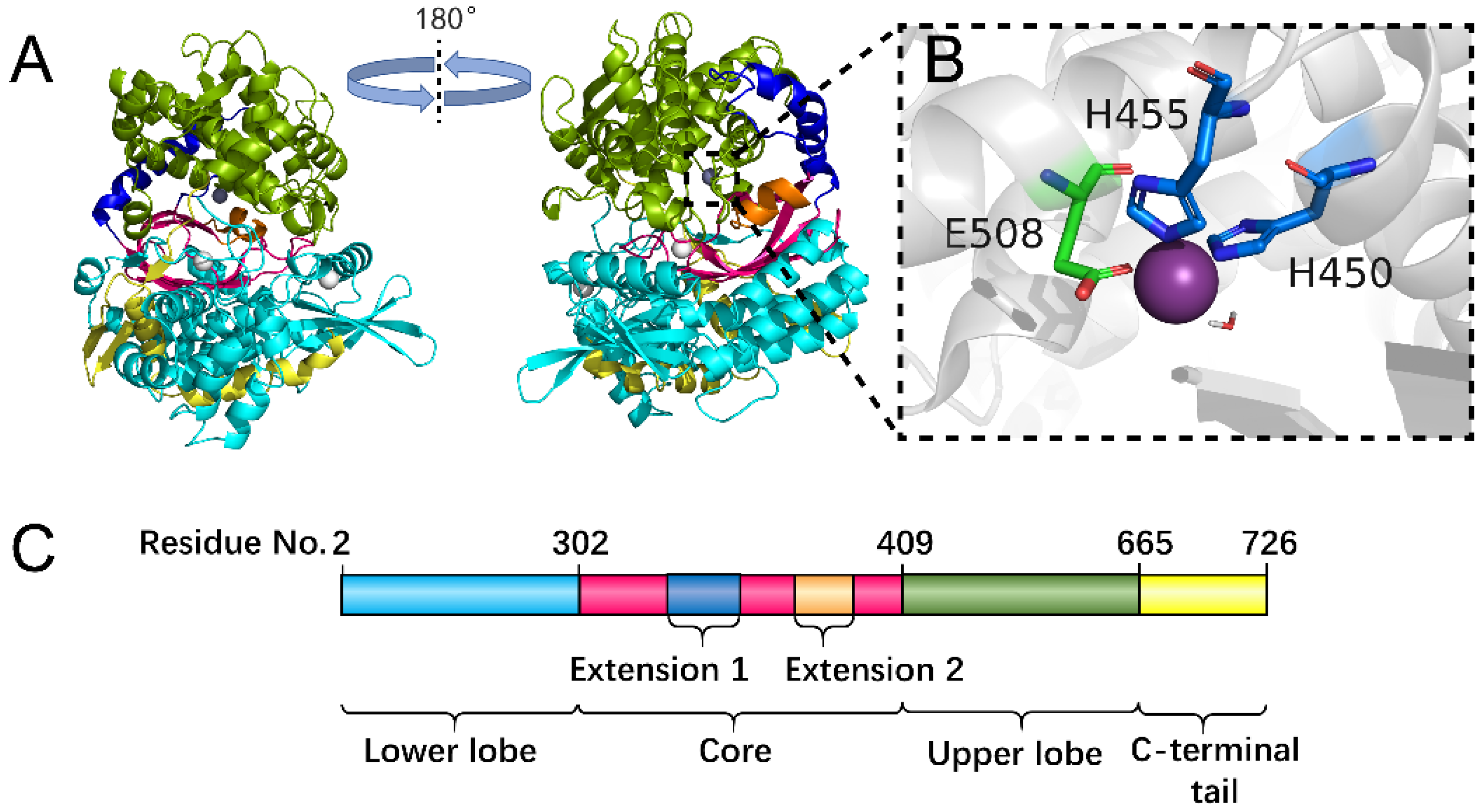
:1. Introduction
2. Results and Discussion
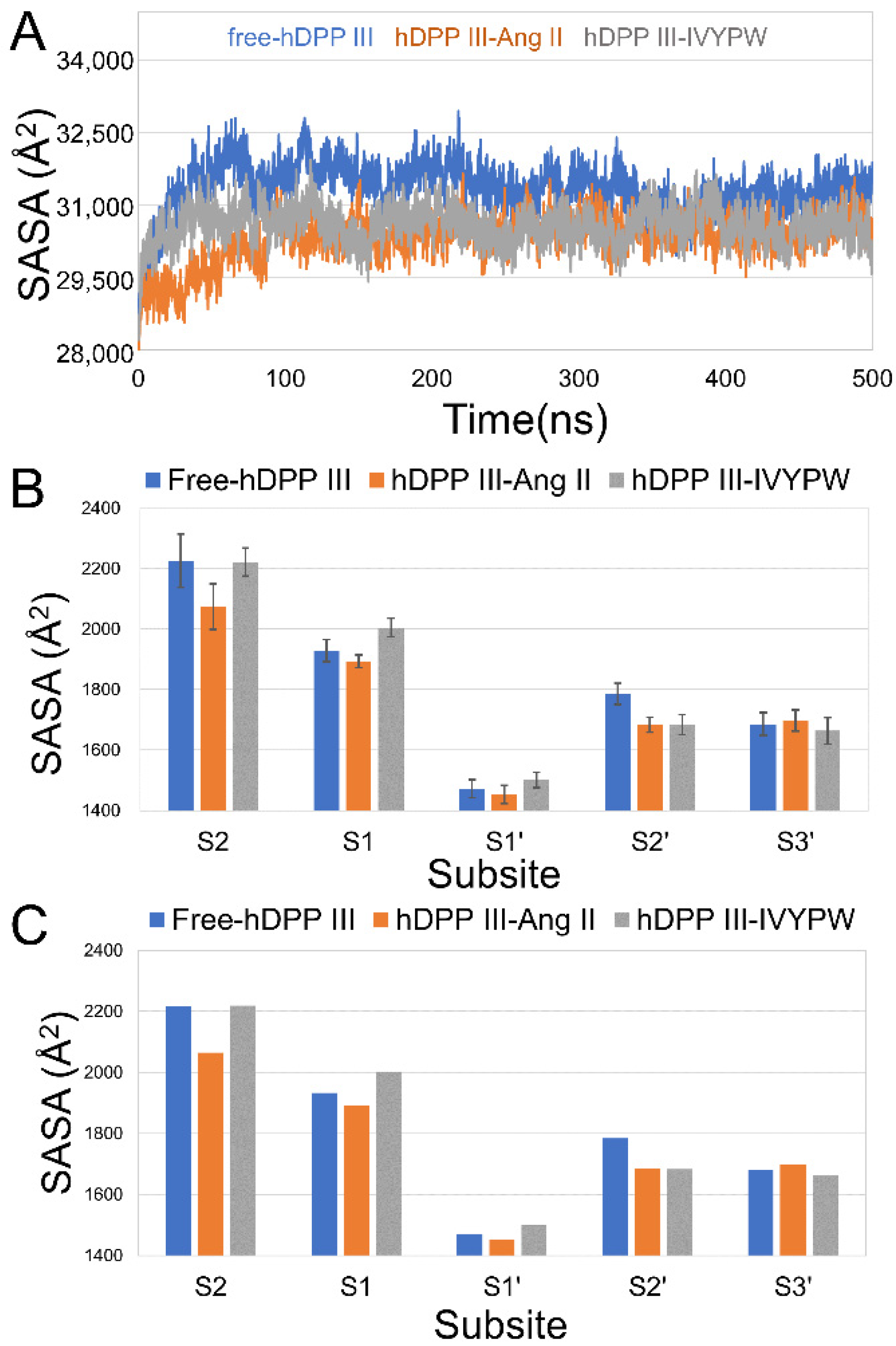
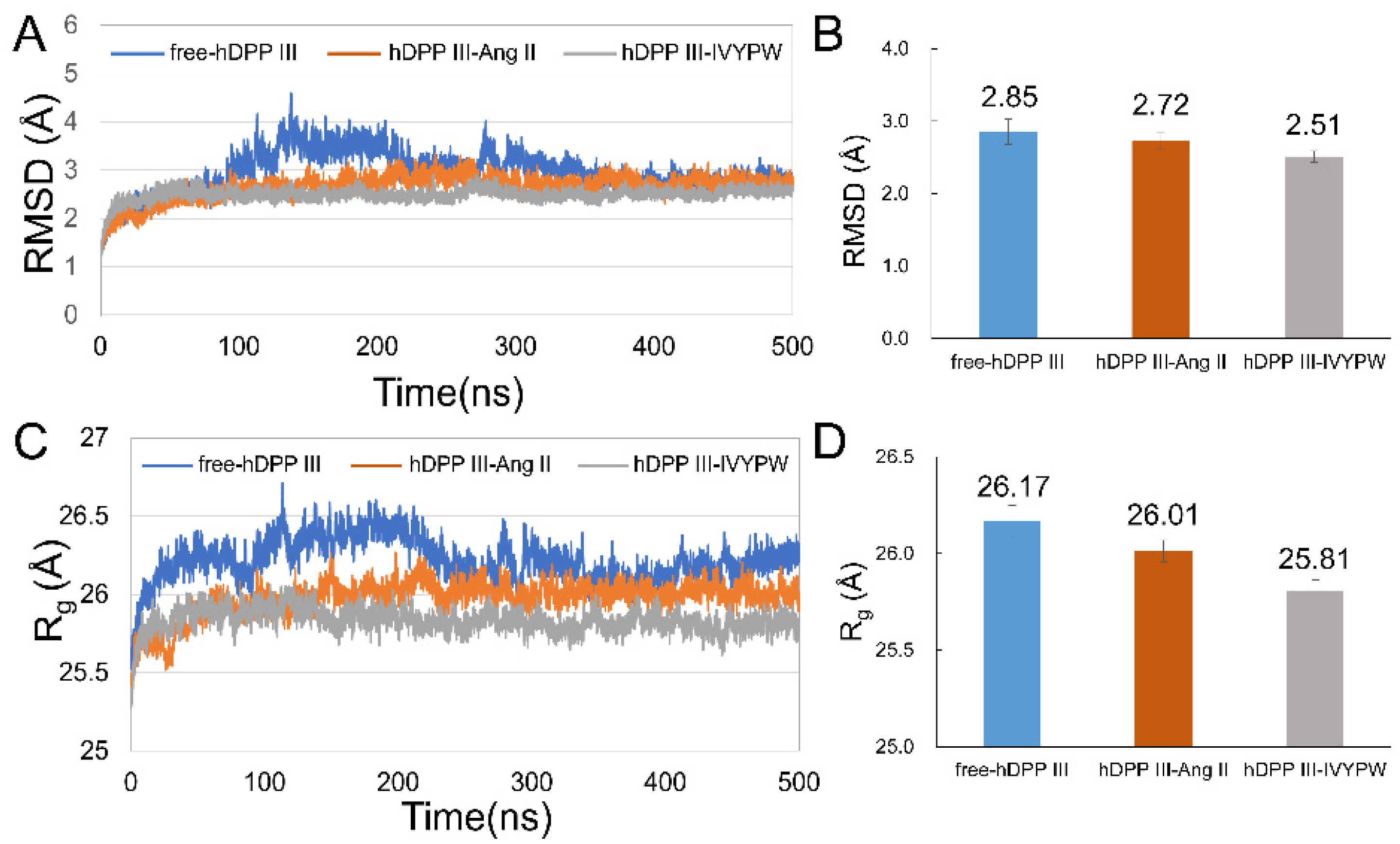
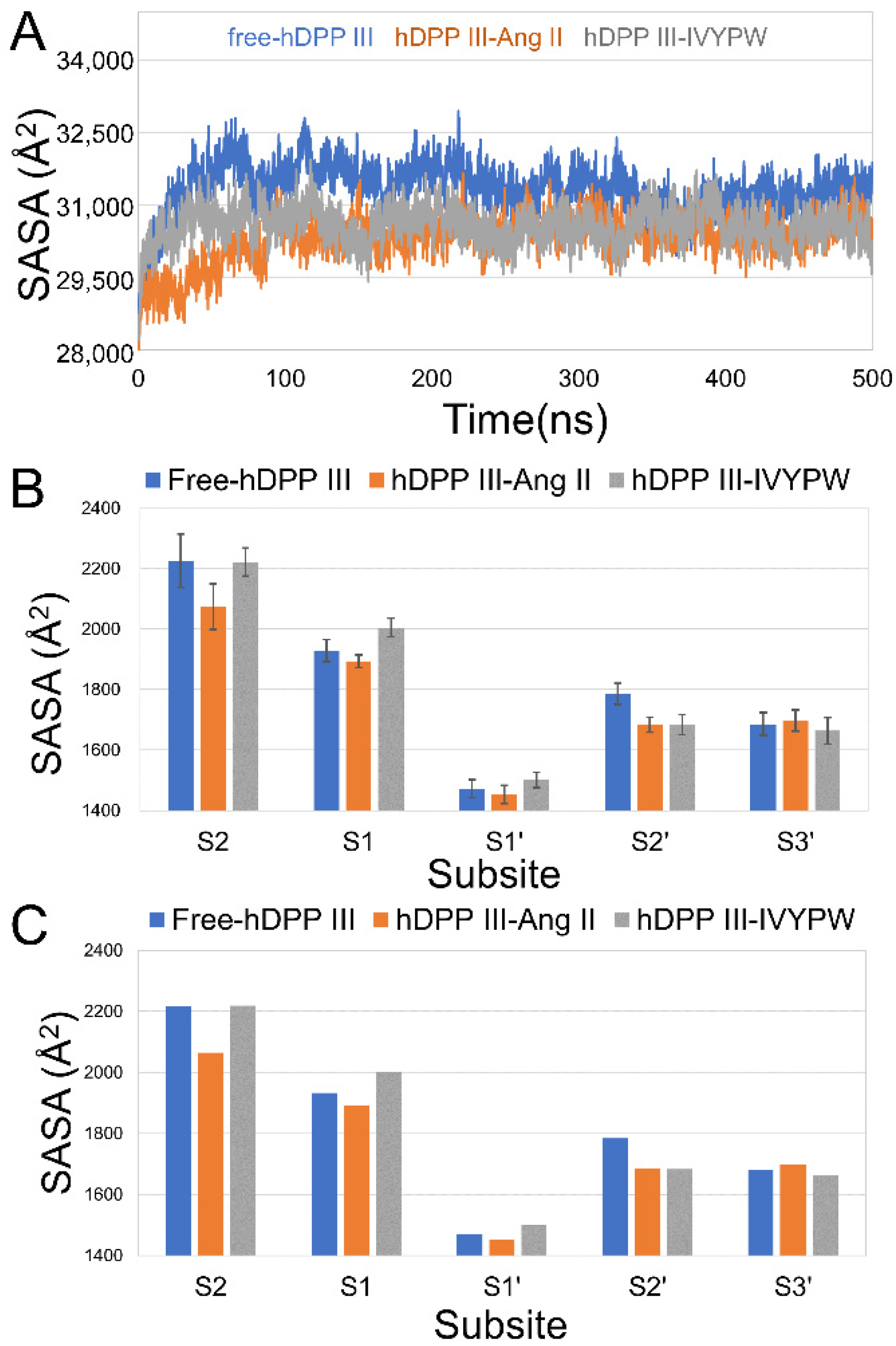
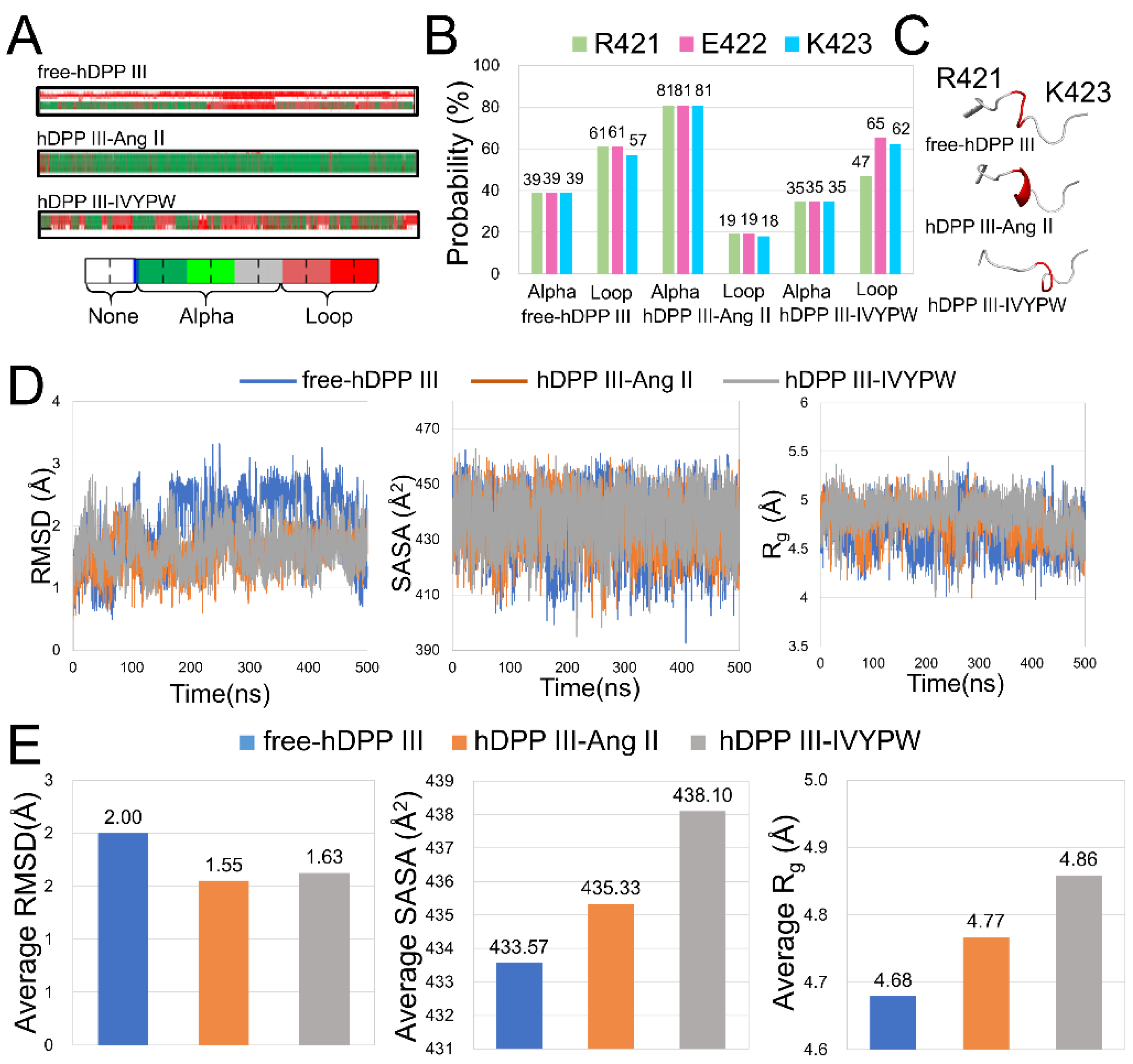
2.1. Stabilization and Hydrophilicity of the Systems
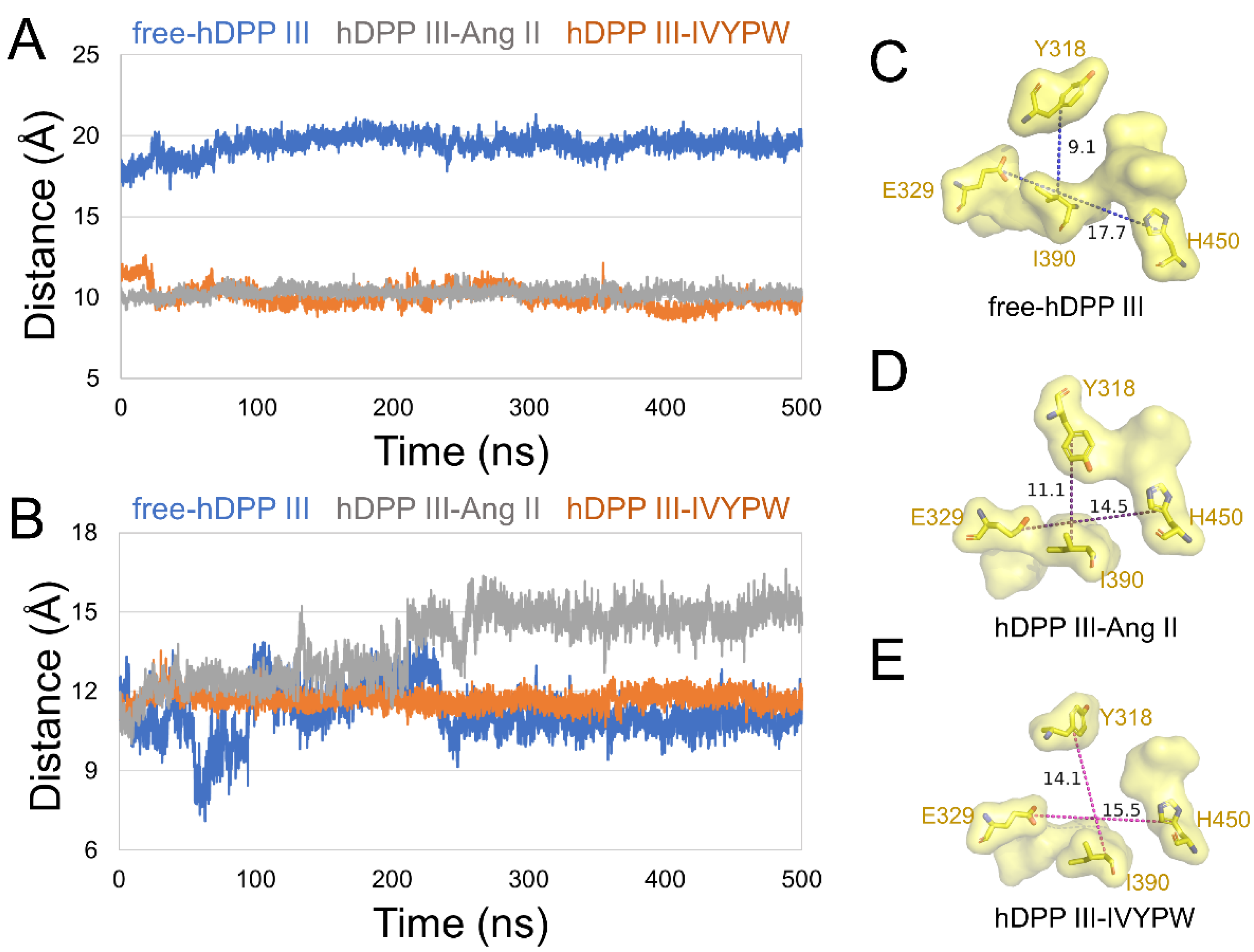
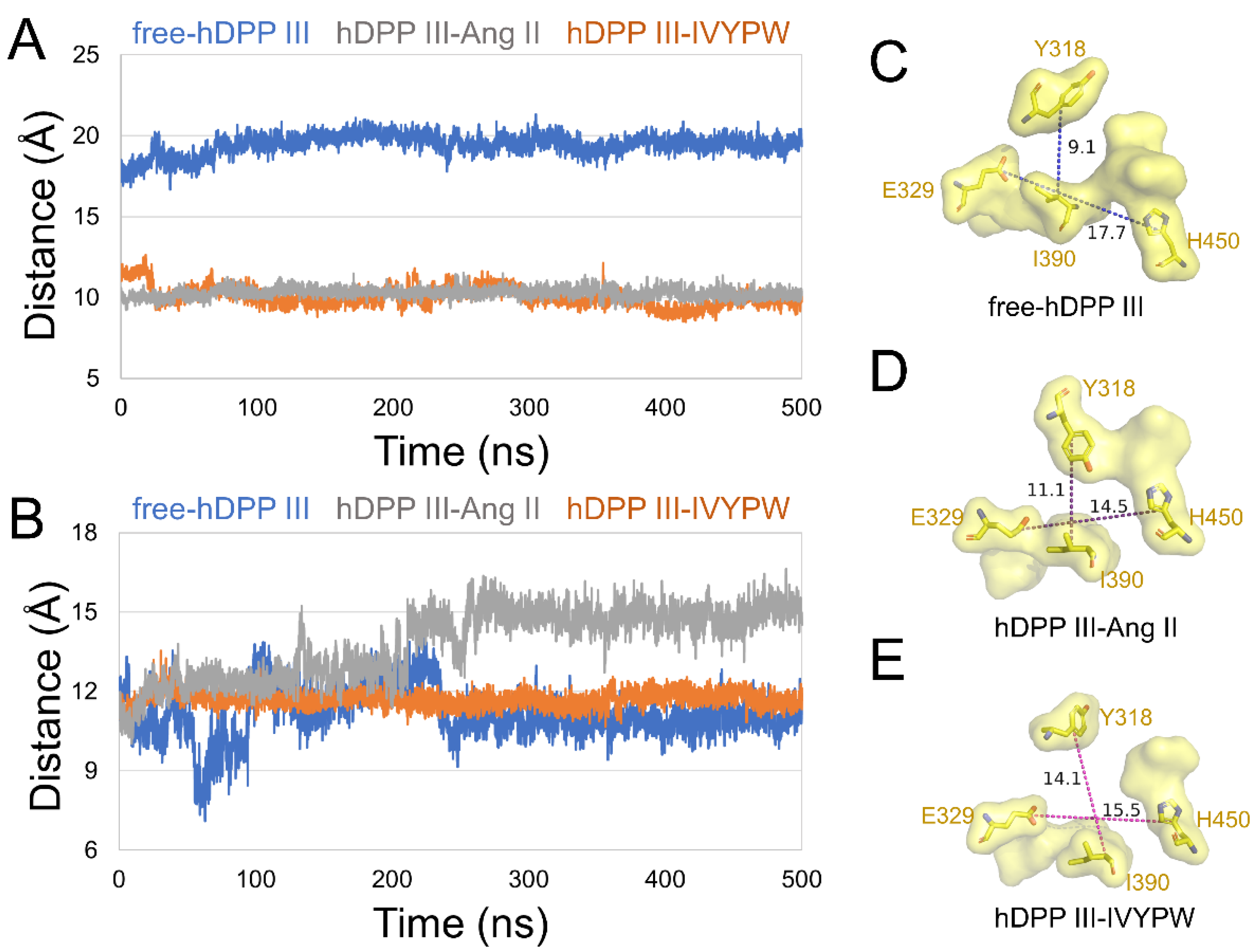
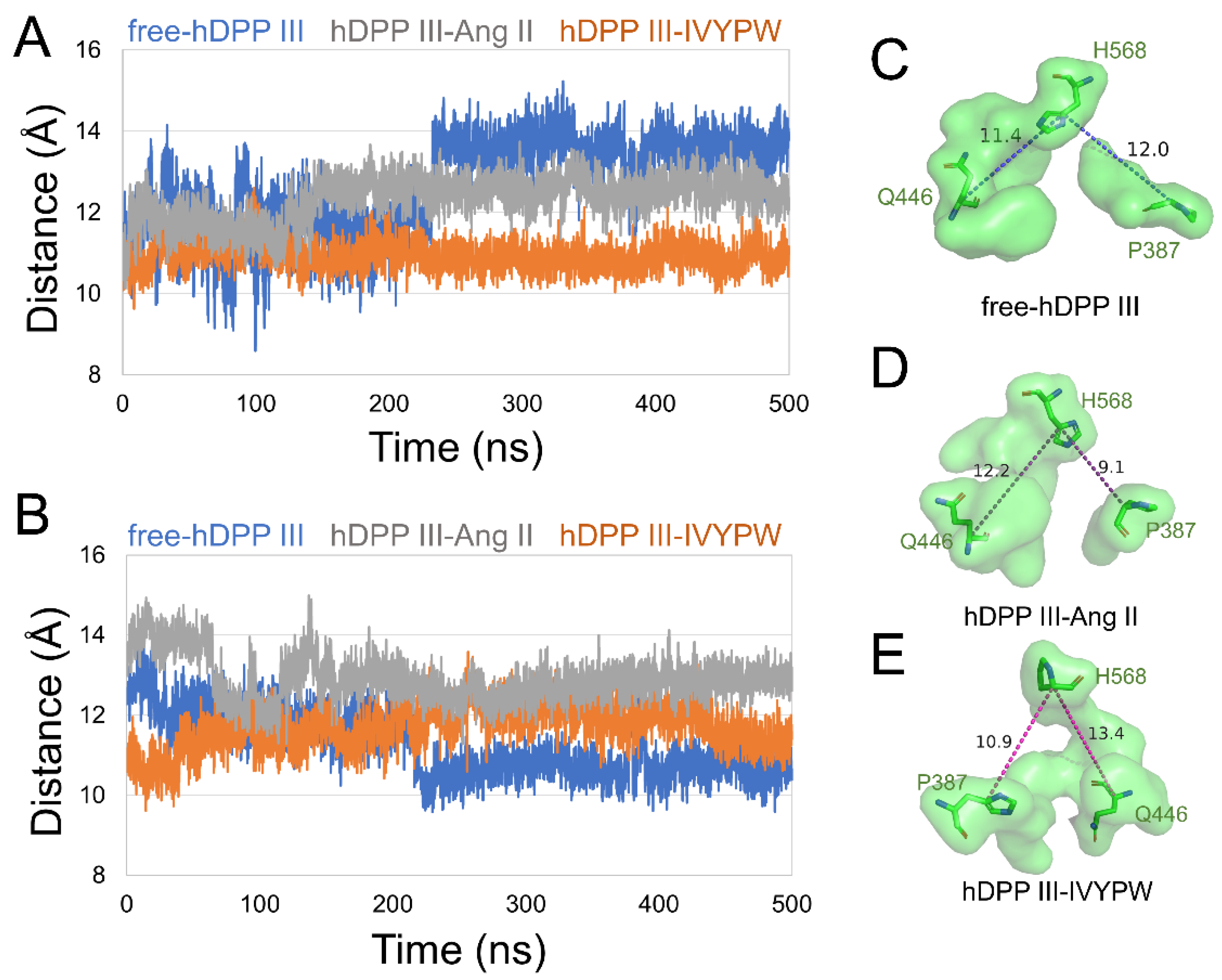
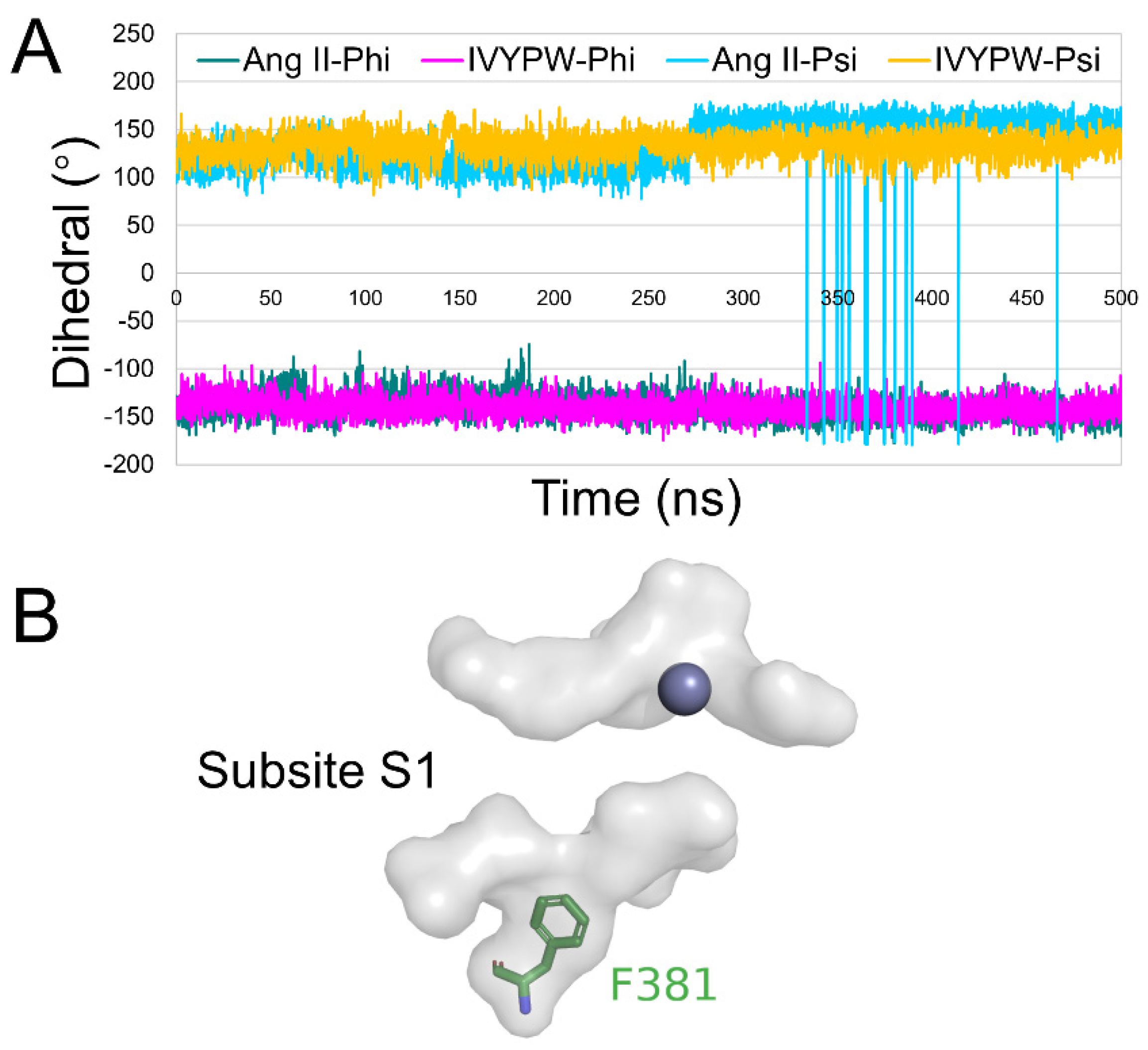
2.2. The Size of the Catalytic Subsites
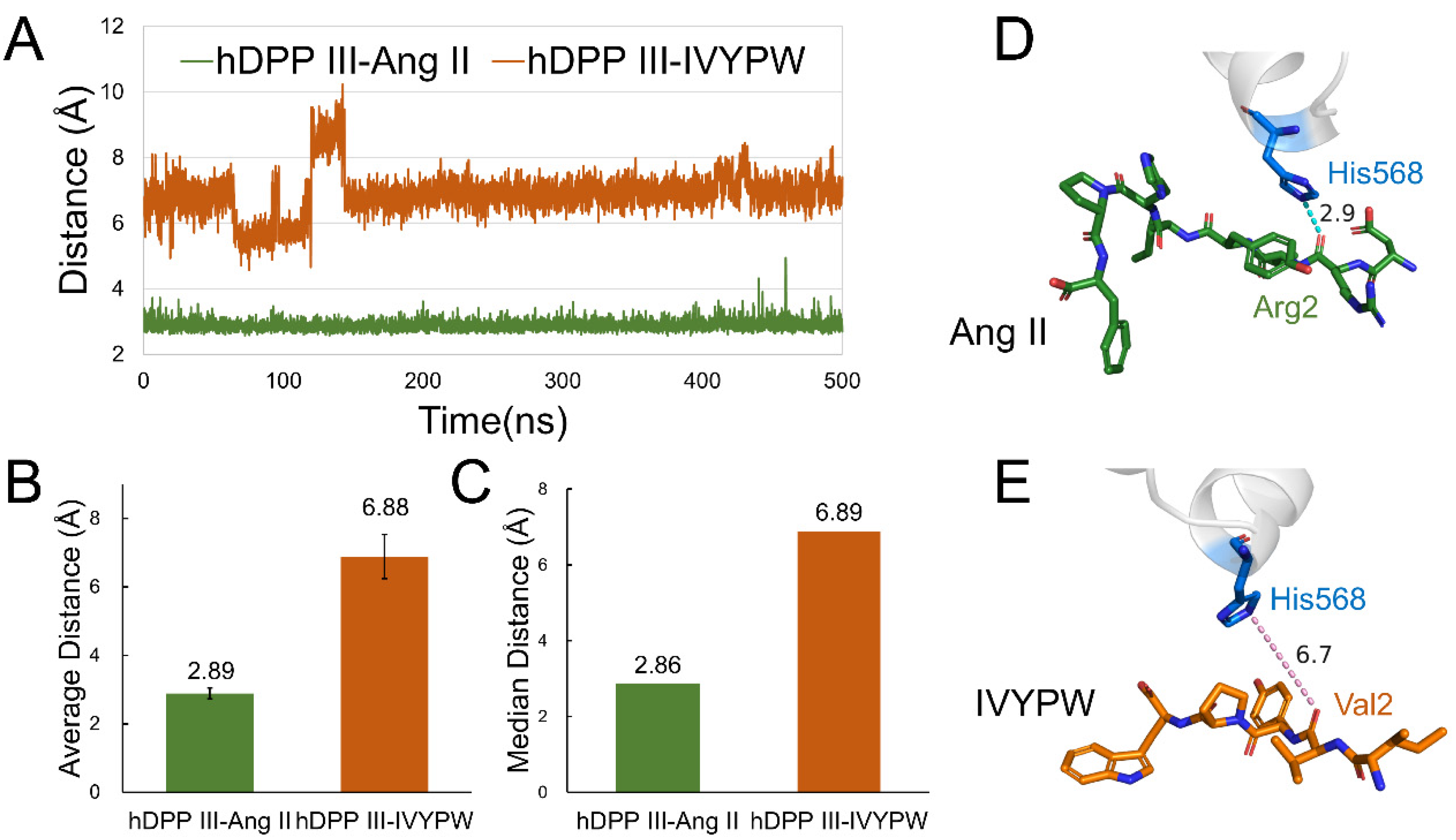
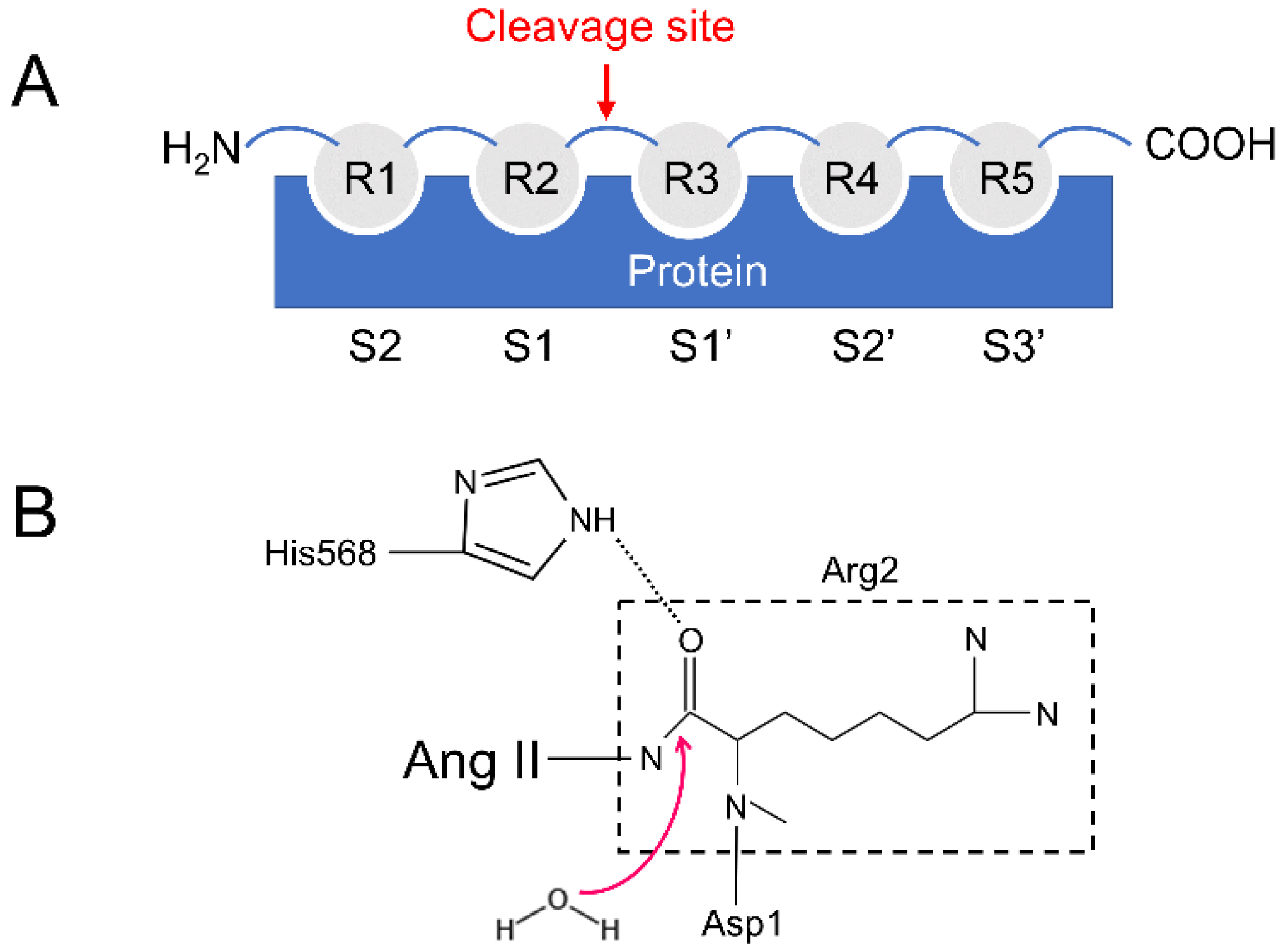
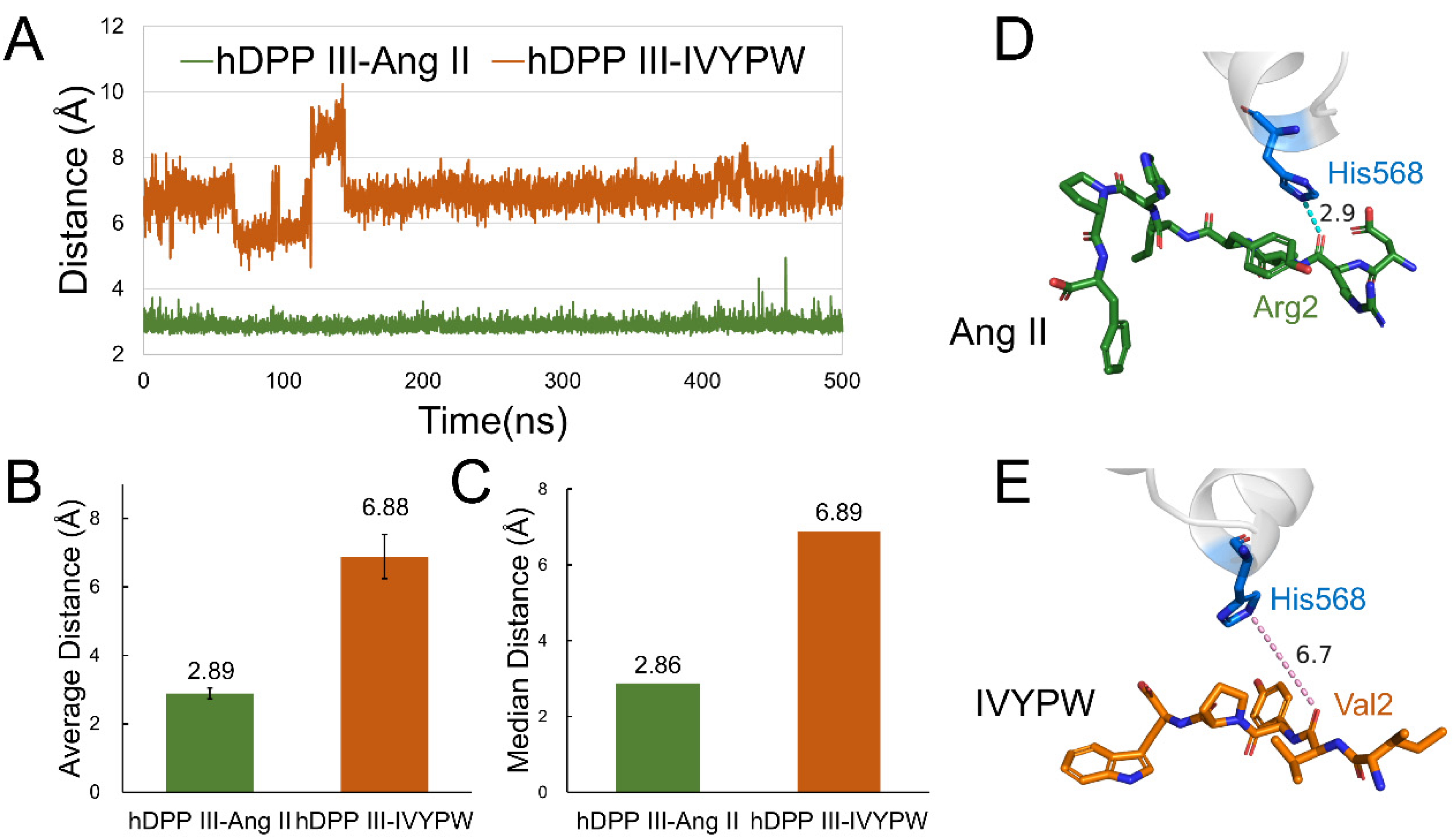
2.3. Analysis of Hydrogen Bonds
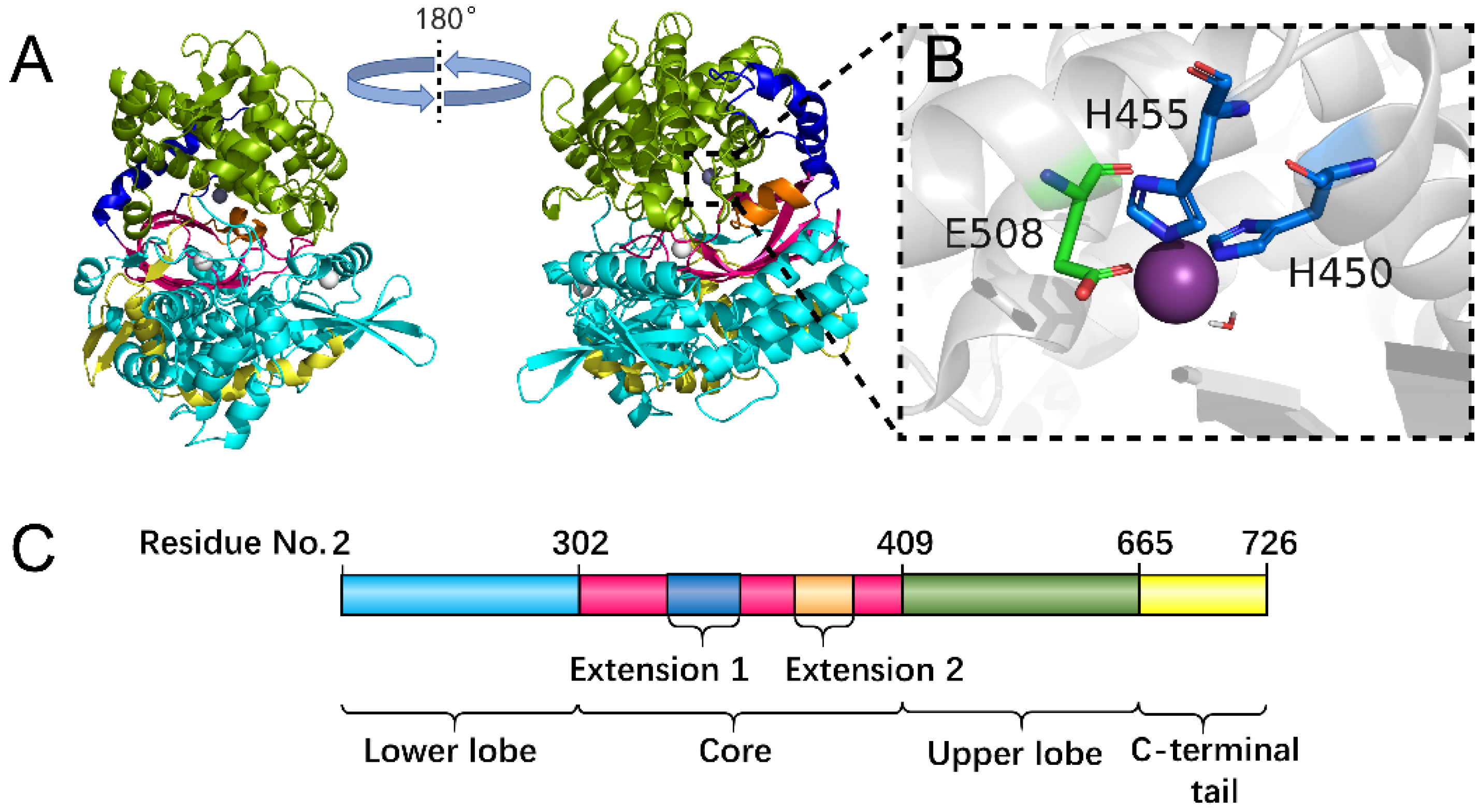
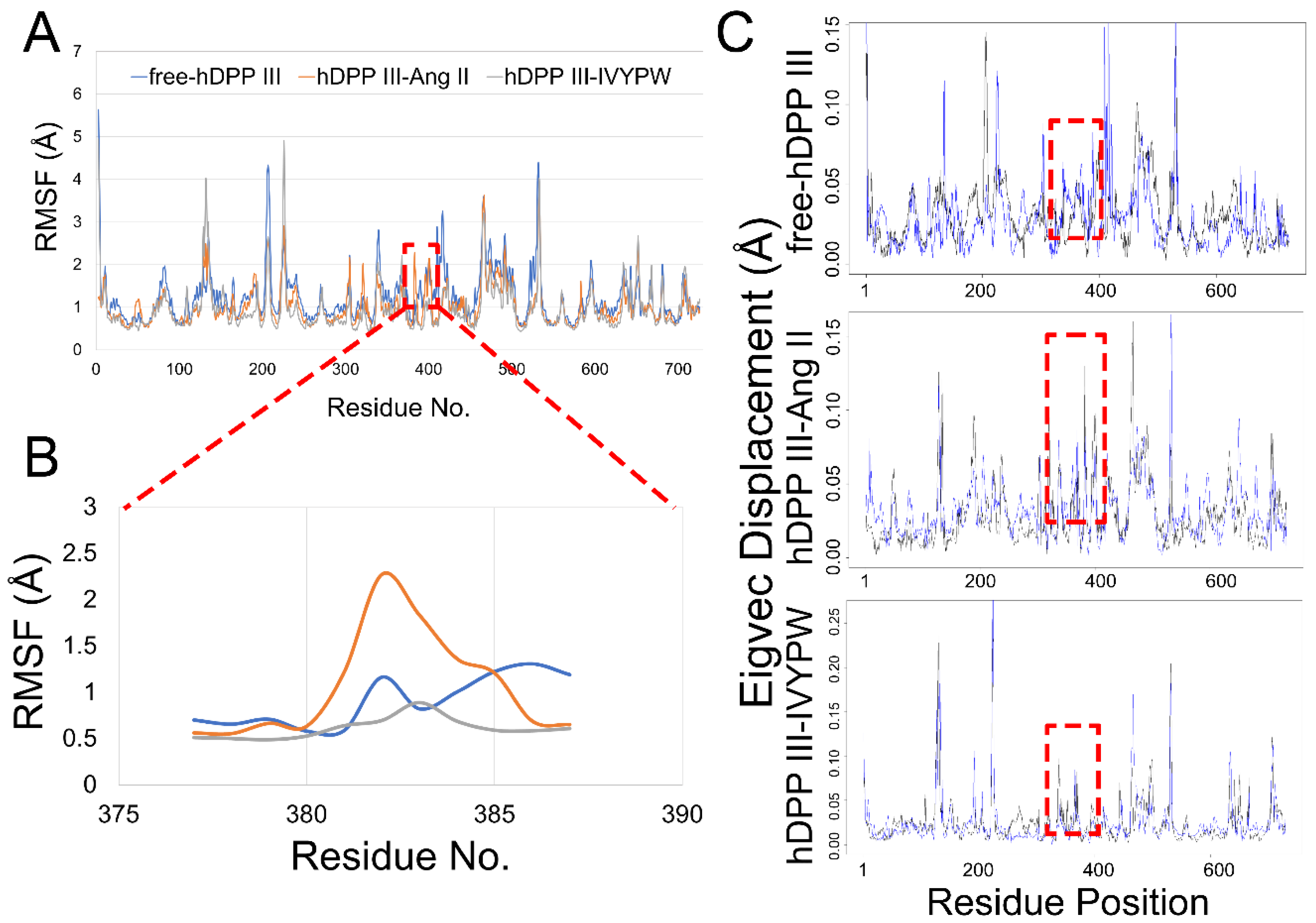
2.4. Flexibility Analysis of hDPP III
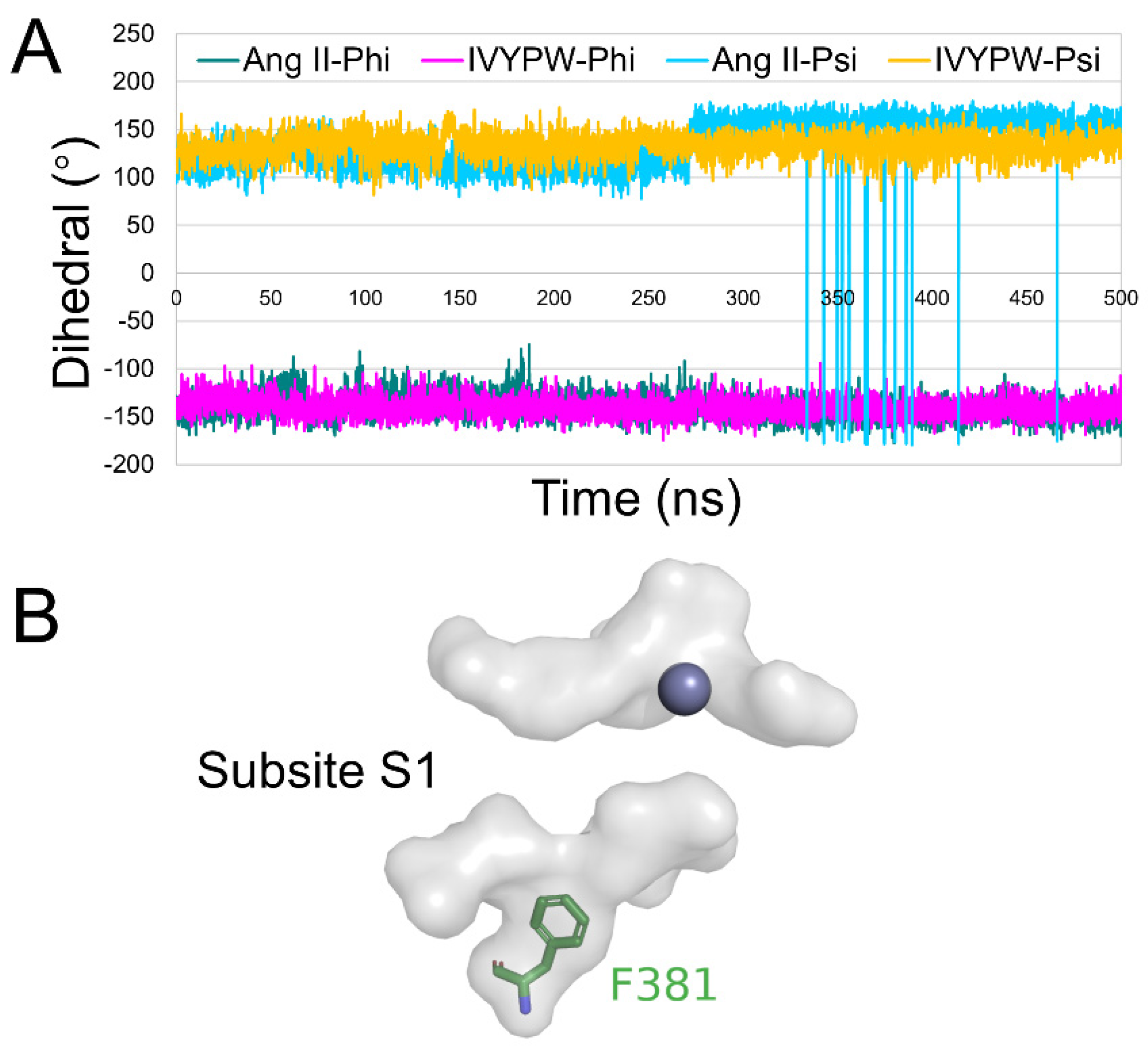
2.5. Conformational Changes for Inhibitor Binding
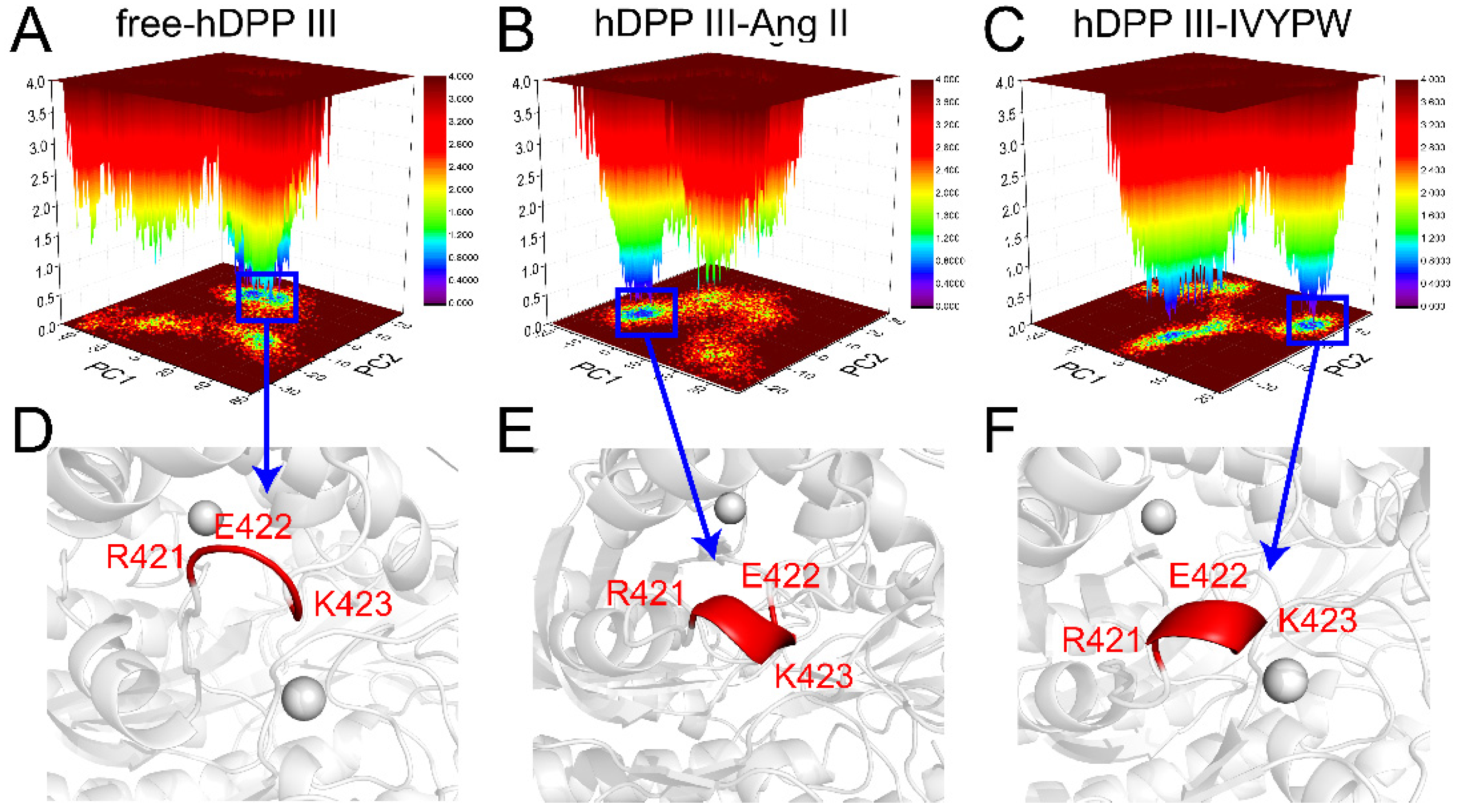
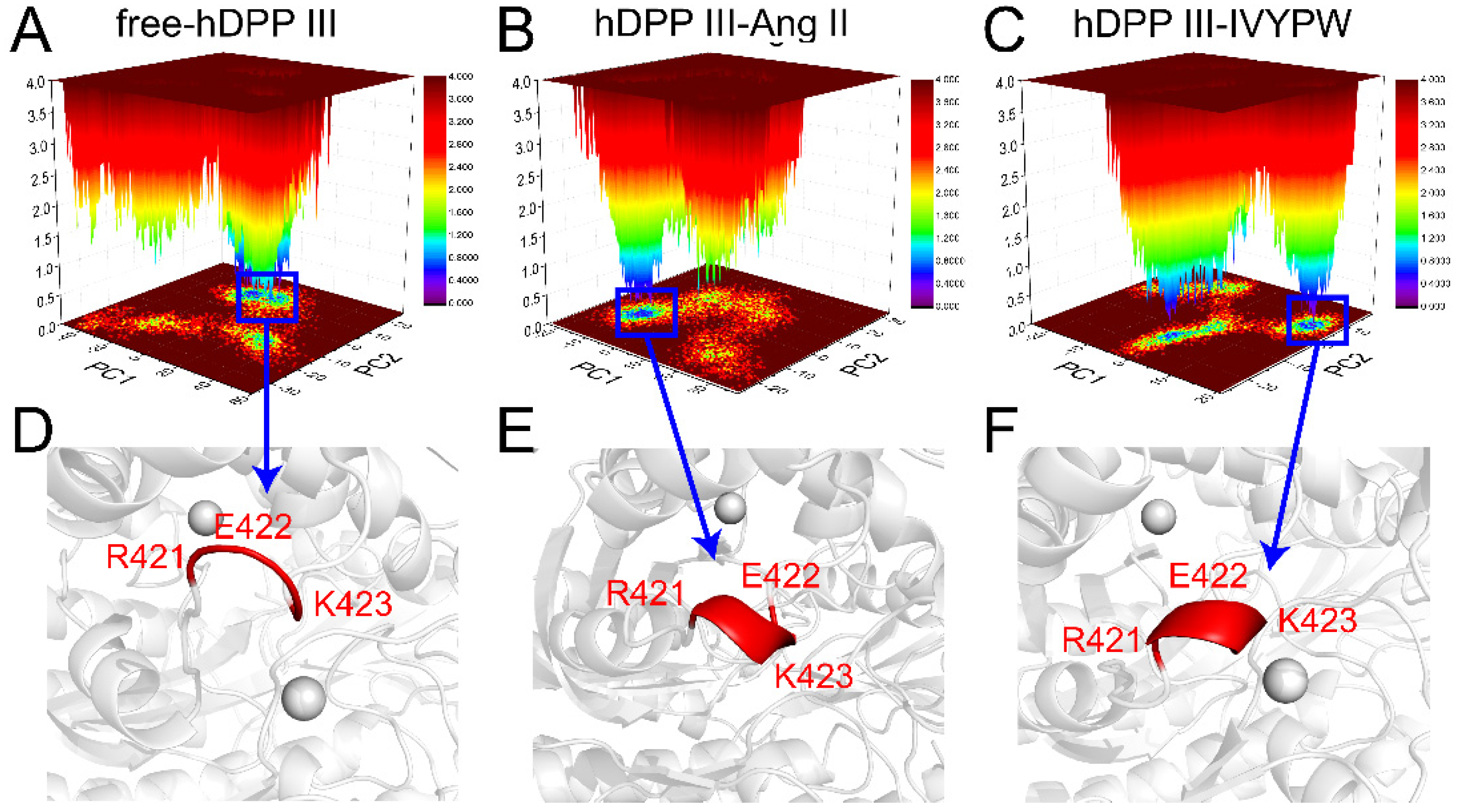
2.6. Principal Component Analysis (PCA) and Free Energy Landscape (FEL) Analysis
2.7. MM/PBSA and Interaction Energy Results
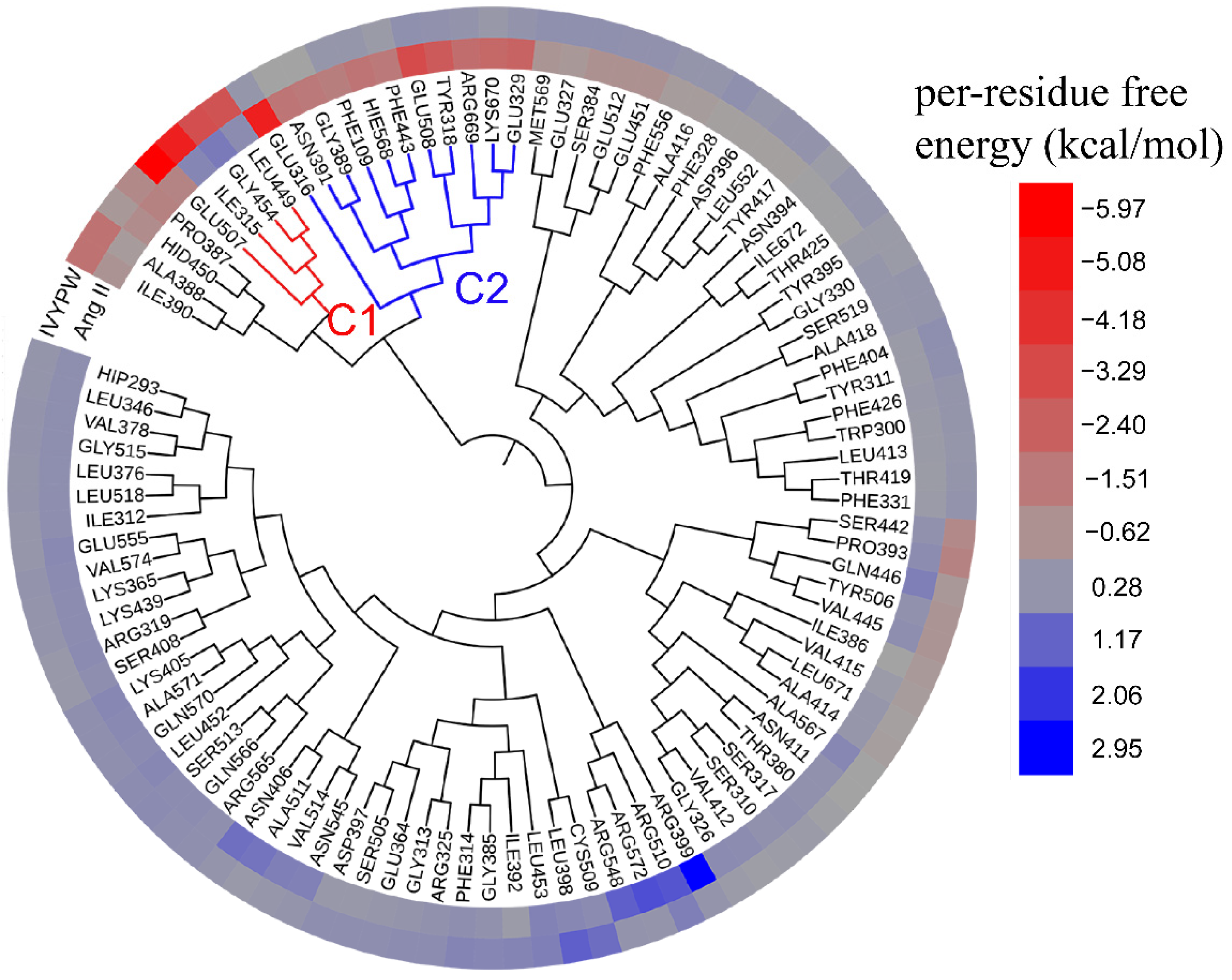
2.8. Hotspot Interaction Points Detected by Hierarchical Cluster (HC) Analysis
3. Discussion
4. Materials and Methods
4.1. MD Simulations
4.2. PCA and FEL Analysis
4.3. MM/PBSA Binding Free Energy Calculations, Interaction Entropy Calculations, and Ligand–Residue Energy Decomposition Analysis
4.4. HC Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sabljić, I.; Meštrović, N.; Vukelić, B.; Macheroux, P.; Gruber, K.; Luić, M.; Abramić, M. Crystal structure of dipeptidyl peptidase III from the human gut symbiont Bacteroides thetaiotaomicron. PLoS ONE 2017, 12, e0187295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baral, P.K.; Jajcanin-Jozić, N.; Deller, S.; Macheroux, P.; Abramić, M.; Gruber, K. The first structure of dipeptidyl-peptidase III provides insight into the catalytic mechanism and mode of substrate binding. J. Biol. Chem. 2008, 283, 22316–22324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomić, A.; Brkić, H.; Matić, A.; Tomić, S. Unravelling the inhibitory zinc ion binding site and the metal exchange mechanism in human DPP III. Phys. Chem. Chem. Phys. 2021, 23, 13267–13275. [Google Scholar] [CrossRef] [PubMed]
- Agić, D.; Brkić, H.; Tomić, S.; Karačić, Z.; Špoljarević, M.; Lisjak, M.; Bešlo, D.; Abramić, M. Validation of flavonoids as potential dipeptidyl peptidase III inhibitors: Experimental and computational approach. Chem. Biol. Drug Des. 2017, 89, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.; Rawlings, N.; Woessner, J. Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Barsun, M.; Jajcanin, N.; Vukelić, B.; Spoljarić, J.; Abramić, M. Human dipeptidyl peptidase III acts as a post-proline-cleaving enzyme on endomorphins. Biol. Chem. 2007, 388, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Morton, F.R.; Kok, C.Y.; Kong, J.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2008, 32, D160–D164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichna, J.; Janecka, A.; Costentin, J.; Do Rego, J.C. The endomorphin system and its evolving neurophysiological role. Pharmacol. Rev. 2007, 59, 88–123. [Google Scholar] [CrossRef]
- Bodnar, R.J. Endogenous opiates and behavior: 2013. Peptides 2014, 62, 67–136. [Google Scholar] [CrossRef]
- Jha, S.; Taschler, U.; Domenig, O.; Poglitsch, M.; Bourgeois, B.; Pollheimer, M.; Pusch, L.M.; Malovan, G.; Frank, S.; Madl, T.; et al. Dipeptidyl peptidase 3 modulates the renin-angiotensin system in mice. J. Biol. Chem. 2020, 295, 13711–13723. [Google Scholar] [CrossRef]
- Menale, C.; Robinson, L.J.; Palagano, E.; Rigoni, R.; Erreni, M.; Almarza, A.J.; Strina, D.; Mantero, S.; Lizier, M.; Forlino, A.; et al. Absence of Dipeptidyl Peptidase 3 Increases Oxidative Stress and Causes Bone Loss. J. Bone Miner. Res. 2019, 34, 2133–2148. [Google Scholar] [CrossRef]
- Ren, X.; Yu, J.; Guo, L.; Ma, H. Dipeptidyl-peptidase 3 protects oxygen-glucose deprivation/reoxygenation-injured hippocampal neurons by suppressing apoptosis, oxidative stress and inflammation via modulation of Keap1/Nrf2 signaling. Int. Immunopharmacol. 2021, 96, 107595. [Google Scholar] [CrossRef]
- Matj, S.; Kekez, I.; Tomin, M.; Bogár, F.; Šupljika, F.; Kazazj, S.; Hanj, M.; Jha, S.; Brkj, H.; Bourgeois, B.; et al. Binding of dipeptidyl peptidase III to the oxidative stress cell sensor Kelch-like ECH-associated protein 1 is a two-step process. J. Biomol. Struct. Dyn. 2020, 39, 6870–6881. [Google Scholar]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.; Major, M.B. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agj, D.; Brki, H.; Kazazi, S.; Tomic, A.; Abramj, M. Aprotinin interacts with substrate-binding site of human dipeptidyl peptidase III. J. Biomol. Struct. Dyn. 2018, 37, 3596–3606. [Google Scholar]
- Agić, D.; Karnaš, M.; Šubarić, D.; Lončarić, M.; Tomić, S.; Karačić, Z.; Bešlo, D.; Rastija, V.; Molnar, M.; Popović, B.M.; et al. Coumarin Derivatives Act as Novel Inhibitors of Human Dipeptidyl Peptidase III: Combined In Vitro and In Silico Study. Pharmaceuticals 2021, 14, 540. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, G.; Dobrovetsky, E.; Viertlmayr, R.; Dong, A.; Binter, A.; Abramj, M.; Macheroux, P.; Dhe-Paganon, S.; Gruber, K. Entropy-driven binding of opioid peptides induces a large domain motion in human dipeptidyl peptidase III. Proc. Natl. Acad. Sci. USA 2012, 109, 6525–6530. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Reithofer, V.; Reisinger, M.; Wallner, S.; Pavkov-Keller, T.; Macheroux, P.; Gruber, K. Substrate complexes of human dipeptidyl peptidase III reveal the mechanism of enzyme inhibition. Sci. Rep. 2016, 6, 23787. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, S.C.; Chauhan, S.S. Dipeptidyl peptidase III: A multifaceted oligopeptide N-end cutter. FEBS J. 2011, 278, 3256–3276. [Google Scholar] [CrossRef]
- Soisson, S.M.; Patel, S.B.; Abeywickrema, P.D.; Byrne, N.J.; Diehl, R.E.; Hall, D.L.; Ford, R.E.; Reid, J.C.; Rickert, K.W.; Shipman, J.M.; et al. Structural definition and substrate specificity of the S28 protease family: The crystal structure of human prolylcarboxypeptidase. BMC Struct. Biol. 2010, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Blumberger, J.; Lamoureux, G.; Klein, M. Peptide Hydrolysis in ThermolysinZ Ab Initio QM/MM Investigation of the Glu143-Assisted Water Addition Mechanism. J. Chem. Theory Comput. 2007, 3, 1837–1850. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hashimoto, J.; Shimamura, M.; Yamaguchi, T.; Hazato, T. Characterization of tynorphin, a potent endogenous inhibitor of dipeptidyl peptidaseIII. Peptides 2000, 21, 503–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Bogunia, M.; Makowski, M. Influence of Ionic Strength on Hydrophobic Interactions in Water: Dependence on Solute Size and Shape. J. Phys. Chem. B 2020, 124, 10326–10336. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016, University of California, San Francisco. 2016. Available online: http://ambermd.org/index.php (accessed on 13 April 2016).
- Essmann, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.-C.; Pedersen, L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Rosenberg, R.O.; Boughaleb, Y.; Nitzan, A.; Ratner, M.A. Effective potentials from Langevin dynamic simulations of framework solid electrolytes. Solid State Ion. 1986, 18–19, 127–135. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grant, B.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.; Caves, L. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Lindström, A.; Edvinsson, L.; Johansson, A.; Andersson, C.D.; Andersson, I.; Raubacher, F.; Linusson, A. Postprocessing of Docked Protein-Ligand Complexes Using Implicit Solvation Models. J. Chem. Inf. Model. 2011, 51, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Liu, X.; Zhang, J.Z. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wang, P.; Tu, G.; Yang, F.; Zheng, G.; Li, X.; Li, X.; Chen, Y.; Yao, X.; Zhu, F. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Phys. Chem. Chem. Phys. PCCP 2018, 20, 6606–6616. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, X.; Fu, T.; Li, S.; Li, B.; Xue, W.; Yao, X.; Chen, Y.; Zhu, F. Differentiating Physicochemical Properties between Addictive and Nonaddictive ADHD Drugs Revealed by Molecular Dynamics Simulation Studies. ACS Chem. Neurosci. 2017, 8, 1416–1428. [Google Scholar] [CrossRef]
- Zhang, Y.; Ying, J.B.; Hong, J.J.; Li, F.C.; Fu, T.T.; Yang, F.Y.; Zheng, G.X.; Yao, X.J.; Lou, Y.; Qiu, Y.; et al. How Does Chirality Determine the Selective Inhibition of Histone Deacetylase 6? A Lesson from Trichostatin A Enantiomers Based on Molecular Dynamics. ACS Chem. Neurosci. 2019, 10, 2467–2480. [Google Scholar] [CrossRef]
- Xue, W.; Yang, F.; Wang, P.; Zheng, G.; Chen, Y.; Yao, X.; Zhu, F. What Contributes to Serotonin-Norepinephrine Reuptake Inhibitors’ Dual-Targeting Mechanism? The Key Role of Transmembrane Domain 6 in Human Serotonin and Norepinephrine Transporters Revealed by Molecular Dynamics Simulation. ACS Chem. Neurosci. 2018, 9, 1128–1140. [Google Scholar] [CrossRef]
- Tippmann, S. Programming tools: Adventures with R. Nature 2015, 517, 109–110. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Residue No. | Receptor Length | Ligand | Ligand Length | Ligand Residues |
|---|---|---|---|---|---|
| 5E33 | 2-726 | 725 | - | - | - |
| 5E2Q | 2-726 | 725 | Ang II | 8 | DRVYIHPF |
| 5E3C | 3-726 | 724 | IVYPW | 5 | IVYPW |
| Substrate | Subsite | Acceptor | Donor H | Donor | Probability (%) |
|---|---|---|---|---|---|
| Ang II | S2 | Asp1@O | Asn391@H | Asn391@N | 98.97 |
| S1′ | Val3@O | Ala388@H | Ala388@N | 97.5 | |
| S1, S1′ | Gly389@O | Val3@H | Val3@N | 90.39 | |
| S1 | Tyr318@O | Arg2@H | Arg2@N | 88.05 | |
| S1 | Arg2@O | His568@H | His568@N | 74.65 | |
| S1, S1′ | Val2@O | Gly389@H | Gly389@N | 67.18 | |
| S2′ | Tyr4@O | Arg572@H | Arg572@N | 65.58 | |
| S1 | Glu329@O | Arg2@H | Arg2@N1 | 63.39 | |
| S2′ | Tyr4@O | Arg572@H | Arg572@N | 61.65 | |
| S1 | Glu329@O1 | Arg2@H | Arg2@N2 | 60.54 | |
| S1 | Glu329@O2 | Arg2@H | Arg2@N3 | 56.13 | |
| IVYPW | S2 | Ile1@O | Asn391@H | Asn391@N | 98.87 |
| S1′, S2′ | Tyr3@O | Ala388@H | Ala388@N | 94.92 | |
| S1, S1′ | Gly389@O | Tyr3@H | Tyr3@N | 90.09 | |
| S1, S1′ | TYR_727@O | Gly389@H | Gly389@N | 79.04 | |
| S2 | Asn391@O | Ile1@H1 | Ile1@N | 31.29 | |
| S2 | Asn391@O | Ile1@H2 | Ile1@N | 31.11 | |
| S2 | Asn391@O | Ile1@H3 | Ile1@N | 30.92 |
| PC1 (%) | PC2 (%) | |
|---|---|---|
| Free-hDPP III | 28.33 | 15.35 |
| hDPP III-Ang II | 17.44 | 14.13 |
| hDPP III-IVYPW | 13.74 | 11.5 |
| hDPP III-Ang II | hDPP III-IVYPW | |
|---|---|---|
| EvdW | −88.68 ± 0.34 | −67.55 ± 0.22 |
| Eelec | −553.42 ± 1.43 | −261.78 ± 1.67 |
| EPB | 512.63 ± 1.24 | 284.67 ± 1.65 |
| Enpolar | −83.09 ± 0.10 | −52.75 ± 0.10 |
| Edisper | 150.05 ± 0.10 | 95.39 ± 0.10 |
| ΔGgas | −642.01 ± 1.39 | −329.33 ± 1.66 |
| ΔGsolv | 579.58 ± 1.25 | 327.31 ± 1.65 |
| ΔTotal | −62.52 ± 0.78 | −2.03 ± 0.54 |
| IE | 65.99 ± 0.05 | 37.59 ± 1.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Lv, S.; Fu, X.; Han, L.; Han, W.; Li, W. Molecular Dynamics Simulations Study of the Interactions between Human Dipeptidyl-Peptidase III and Two Substrates. Molecules 2021, 26, 6492. https://doi.org/10.3390/molecules26216492
Zhang S, Lv S, Fu X, Han L, Han W, Li W. Molecular Dynamics Simulations Study of the Interactions between Human Dipeptidyl-Peptidase III and Two Substrates. Molecules. 2021; 26(21):6492. https://doi.org/10.3390/molecules26216492
Chicago/Turabian StyleZhang, Shitao, Shuai Lv, Xueqi Fu, Lu Han, Weiwei Han, and Wannan Li. 2021. "Molecular Dynamics Simulations Study of the Interactions between Human Dipeptidyl-Peptidase III and Two Substrates" Molecules 26, no. 21: 6492. https://doi.org/10.3390/molecules26216492
APA StyleZhang, S., Lv, S., Fu, X., Han, L., Han, W., & Li, W. (2021). Molecular Dynamics Simulations Study of the Interactions between Human Dipeptidyl-Peptidase III and Two Substrates. Molecules, 26(21), 6492. https://doi.org/10.3390/molecules26216492