Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) is an important threat as it causes serious hospital and community acquired infections with deathly outcomes oftentimes, therefore, development of new treatments against this bacterium is a priority. Shikimate kinase, an enzyme in the shikimate pathway, is considered a good target for developing antimicrobial drugs; this is given because of its pathway, which is essential in bacteria whereas it is absent in mammals. In this work, a computer-assisted drug design strategy was used to report the first potentials inhibitors for Shikimate kinase from methicillin-resistant Staphylococcus aureus (SaSK), employing approximately 5 million compounds from ZINC15 database. Diverse filtering criteria, related to druglike characteristics and virtual docking screening in the shikimate binding site, were performed to select structurally diverse potential inhibitors from SaSK. Molecular dynamics simulations were performed to elucidate the dynamic behavior of each SaSK–ligand complex. The potential inhibitors formed important interactions with residues that are crucial for enzyme catalysis, such as Asp37, Arg61, Gly82, and Arg138. Therefore, the compounds reported provide valuable information and can be seen as the first step toward developing SaSK inhibitors in the search of new drugs against MRSA.
1. Introduction
Today, antimicrobial resistance in bacteria has become a serious healthcare concern [1]; the World Health Organization (WHO) published a list of 12 bacteria in 2017 [2], their level of resistance to antibiotics has become so grave, that they represent an important threat. Therefore, these organisms have been declared priority pathogens to encourage antibiotic drug design by pharmaceutical companies [3]. From the Gram-positive bacteria shown on this list, drug-resistant Staphylococcus aureus has become one of the most problematic pathogens worldwide.
Methicillin-resistant Staphylococcus aureus (MRSA) has been widely known as a major cause of nosocomial and community-acquired infections that range from mild cases, such as skin and soft tissue infections, to more serious and deadlier, like bacteremia, osteomyelitis, and infective endocarditis [4]. In 2017, an estimated of 12,000 cases of MRSA infections and 20,000 deaths associated with it, occurred in the United States alone [5]. Over the years, MRSA has shown an alarming increase in antimicrobial resistance [6,7,8], developing resistance to vancomycin as well, the last resort treatment for MRSA infections [9]. Furthermore, the recent underdevelopment trend of new antimicrobial agents by pharmaceutical companies has added to the threat that this pathogen already presents [10,11]. Moreover, it has become apparent that research and discovery of new drugs, that possess unique mechanisms of action to prevent chances of developing drug resistance, are becoming urgently necessary.
Accordingly, pathways that are essential for bacterial survival, absent in humans, have become ideal targets to obtain new antimicrobial agents. The shikimate pathway (SP) enzymes are attractive targets for the development of antimicrobial drugs, given that this pathway is only present in bacteria, plants, fungi, apicomplexan parasites, and it is absent in mammals [12]. SP links carbohydrate metabolism to aromatic compounds biosynthesis by means of seven metabolic steps culminating in chorismate production, an important precursor for the synthesis of aromatic amino acids, folate, ubiquinone, among other essential molecules [13]. This route has been previously validated as an antimicrobial target [14], therefore, the design of inhibitors against enzymes of this route has gained significant attention over the years [15].
Shikimate kinase (SK, EC 2.7.1.71) is the fifth enzyme in SP, it catalyzes the conversion of shikimate to shikimate 3-phosphate by phosphorylation of the 3-hydroxyl group of shikimates using ATP as co-substrate; this enzyme has already been established as a promising target, it is essential in a variety of organisms like Mycobacterium tuberculosis, where the deletion of the aroK gene, which codes for SK, disrupts cell viability [16]. SK has been recognized as a member of the nucleoside monophosphate kinases (NMP) family; it possesses three domains, the CORE domain, which involves residues that belong to the conserved binding loop (P-loop) which forms the binding site of ATP and ADP, the LID domain that closes over the active site and contains important residues for ATP binding, and the NMP-binding domain, an important region that corresponds to the shikimate binding region [17]. In 2019, our group reported the biochemical, kinetic, and structural characterization of SK from MRSA (SaSK). In that work, it was found that SaSK shares common characteristics with other bacterial SKs, and by performing homology modeling and molecular dynamics studies important structural information was reported [18].
This study was initiated to work with a chemical library of around 5 million small molecules under a computer-assisted drug design strategy; it included virtual screening, ADME-Tox properties predictions, and molecular dynamics simulations; it was completed to report the first set of potential inhibitors from SaSK.
2. Results and Discussion
2.1. Compounds Filtering
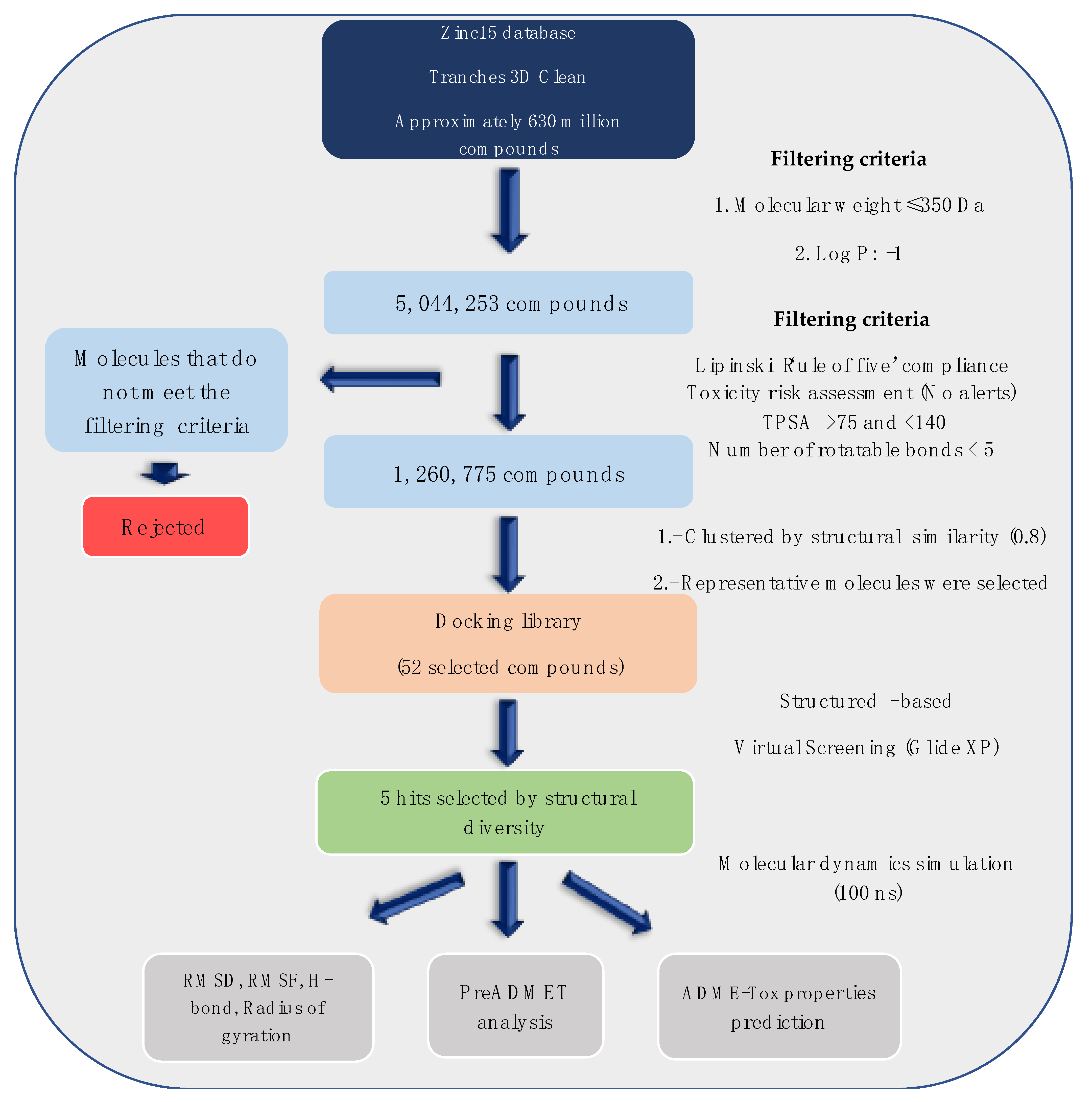
It is increasingly becoming apparent that toxic properties are crucial determinants for the successful development of new drugs. There are unfavorable characteristics that may lead to rejection of possible candidates in the later stages of the drug process [19]. In this study, a total of 5,044,253 compounds, based on SaSK substrate molecular weight and Log p value, were selected from ZINC15 Database (MW ≤ 350 Da and a Logp value of −1). Afterwards, a filtering strategy was applied considering different parameters, such as Lipinski “Rule of Five” compliance, risk of potential toxicity (Mutagenic, Tumorigenic, Reproductive, and Irritant effects), topological surface area (TPSA), number of rotatable bonds, and finally, a clustering by structure similarity, providing as a result a total of 52 molecules (Figure 1, Table S1).
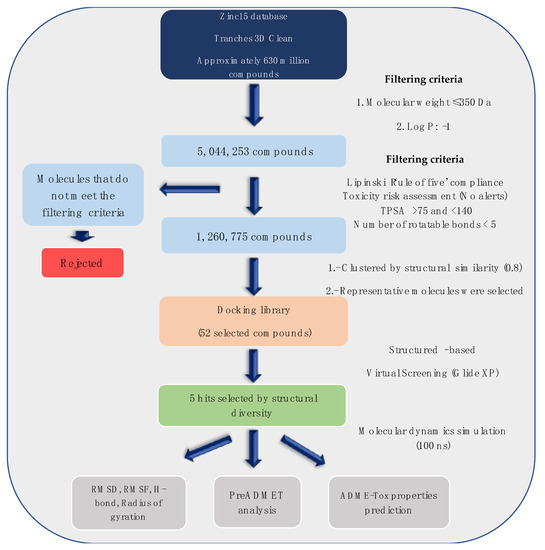
Figure 1.
Workflow to select the potential SaSK inhibitors by computer-assisted drug design.
2.2. Virtual Screening
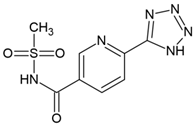
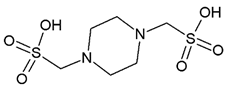
Currently, virtual screening has become essential for the drug design process, it permits an accurate prediction of the position and conformation of a ligand in the binding site of a target protein through an established scoring function [20]. After filtering, 52 compounds were docked into the SaSK active site using the protocol described in materials and methods; the five molecules with the highest docking score and structural diversity were selected in this study (Figure 1 and Table 1).

Table 1.
Glide XP docking results for the top five hits.
Docking results show that compounds interact with residues important for substrate binding or enzyme catalysis, such as Gly82 and Arg138, both are in charge of the stabilization and orientation of shikimate; moreover, Gly83 also participates in substrate stabilization [21]. Conversely, Arg120 is a residue present in the LID domain known for its function as a substrate stabilizing residue in the ATP-shikimate complex [22]. Finally, Asp37 participates in the interaction with the hydroxyl groups of shikimate molecule [23] while Asp35 is an important residue in the nucleotide binding domain that forms hydrogen bonds with other residues that facilitate the approximation of shikimate and ATP [21] (Table 1 and Figure 2).

Figure 2.
Interaction of top compounds. Two-dimensional representation of SaSK residues interacting with (a) C1, (b) C2, (c) C3, (d) C4, and (e) C5. Hydrogen bonds are depicted in pink, salt bridges are shown in red-blue, and Pi-cation interactions in red.
Although, there are no reports in literature of inhibitors for SaSK, other shikimate kinases, in particular for M. tuberculosis and H. pylori, have been shown [22,24]. These molecules comprise several scaffolds, such as pyrazolone derivatives, manzamines, 2-aminobenzothiazoles, and substrate analogues, that particularly target the shikimate binding site [25,26,27].
Docking results in this study were able to select compounds that possess a particular orientation that allows them to perform important interactions with residues that are vital for enzyme catalysis, such as Arg61, Arg138, and Gly82, which are reported to be critical for ligand stabilization and orientation in studies performed in M. tuberculosis shikimate kinase [21,22,23,28].
2.3. Molecular Dynamics Studies
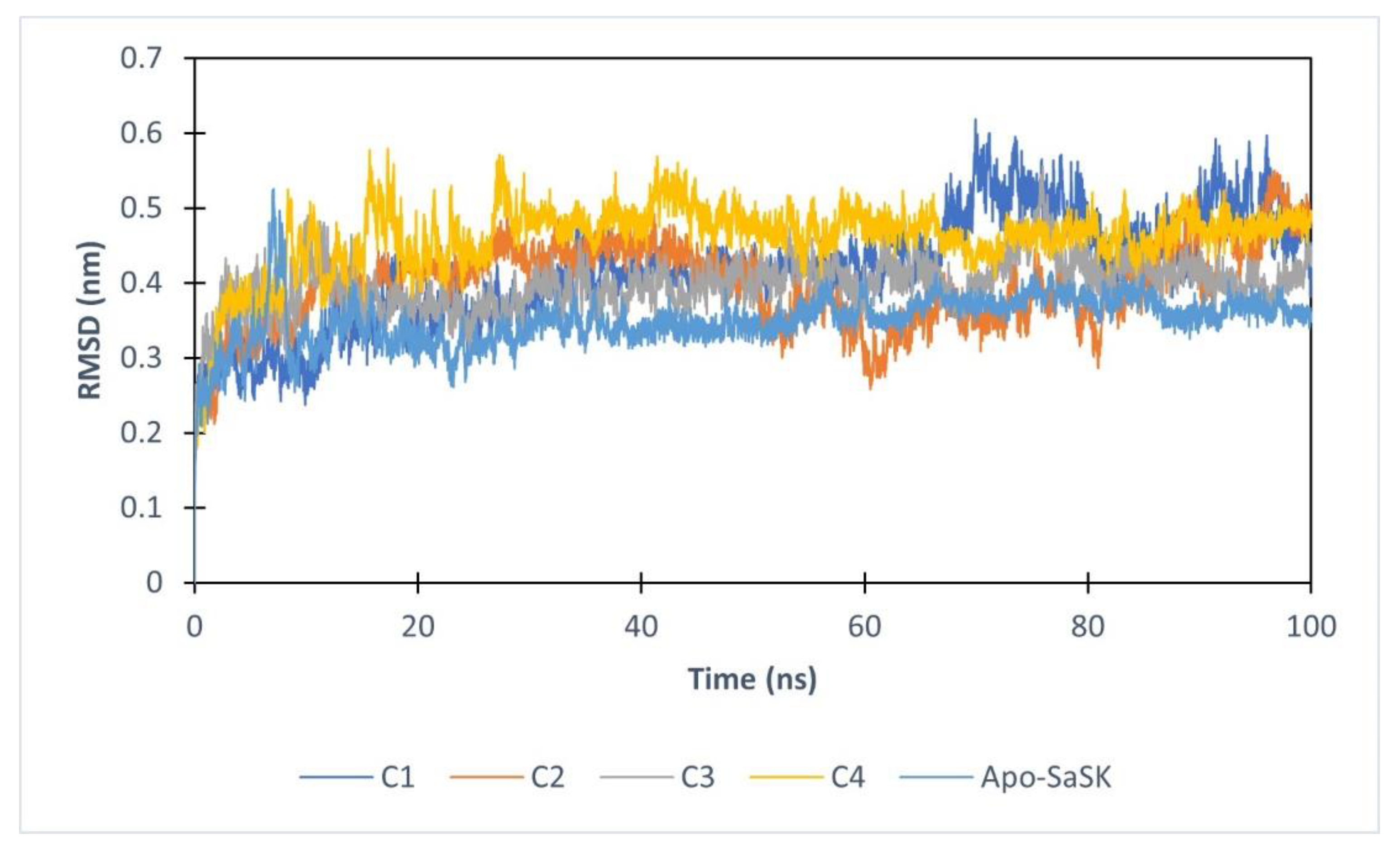
For purposes of gathering added information about SaSK-potential inhibitor complex, molecular dynamics simulations of 100 ns were performed. First, protein–ligand complex stability was evaluated analyzing the root mean square deviation (RMSD) of the Cα protein atoms. The data show that after 20 ns, four of the complexes reach stability, however, C5 leaves the binding site after 6 ns of simulation, therefore, no analysis was performed for this compound. Average RMSD values obtained are 0.416, 0.397, 0.397, and 0.459 nm for SaSK-C1, SaSK-C2, SaSK-C3, and SaSK-C4 complexes, respectively, indicating that all remain stable during simulation time (Figure 3).
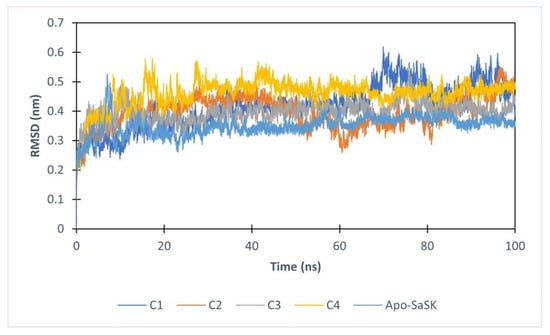
Figure 3.
Root mean square deviation analysis for free enzyme and the different protein–ligand complexes.
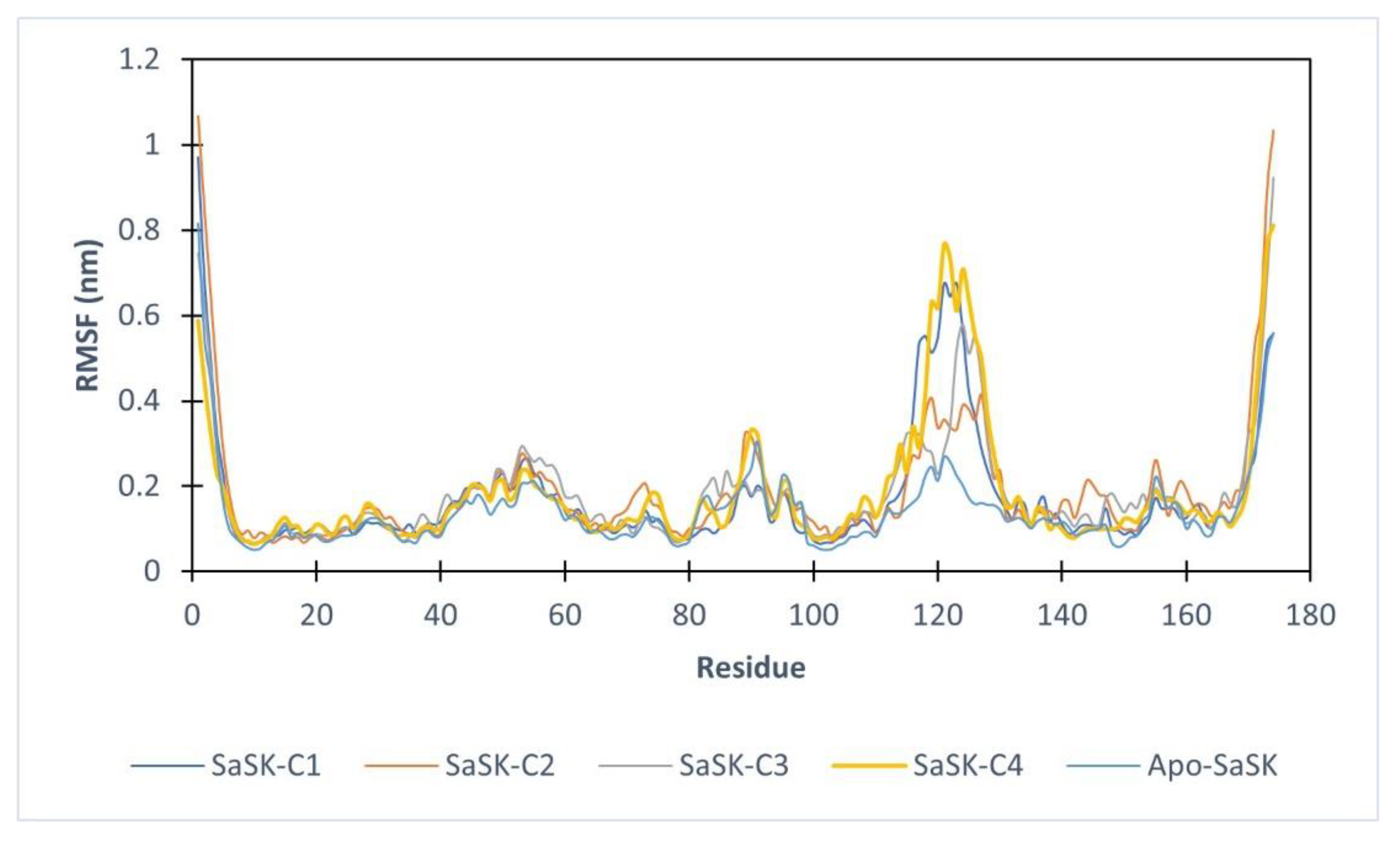
Furthermore, root mean square fluctuations analysis (RMSF) was performed, the results show that in all four complexes, the highest fluctuation observed corresponds to the LID domain (residues 120–135 in SaSK), a region that it is known for its great flexibility [29], in this case, this movement can be attributed to an essential movement to accommodate the compound within the binding site. These fluctuations are more notorious in SaSK-C1, SaSK-C3, and SaSK-C4 complexes (Figure 4). Average RMSF values obtained are 0.172, 0.191, 0.184, and 0.186 nm for SaSK-C1, SaSK-C2, SaSK-C3, and SaSK-C4, respectively.
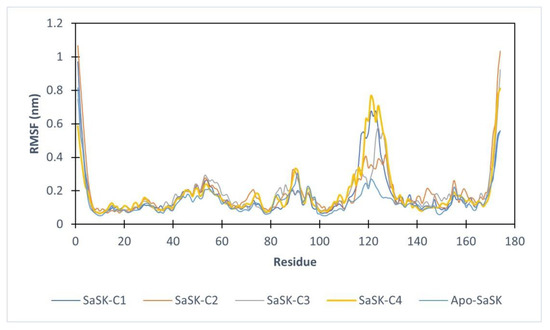
Figure 4.
Root mean square fluctuation analysis for free enzyme and the different protein–ligand complexes.
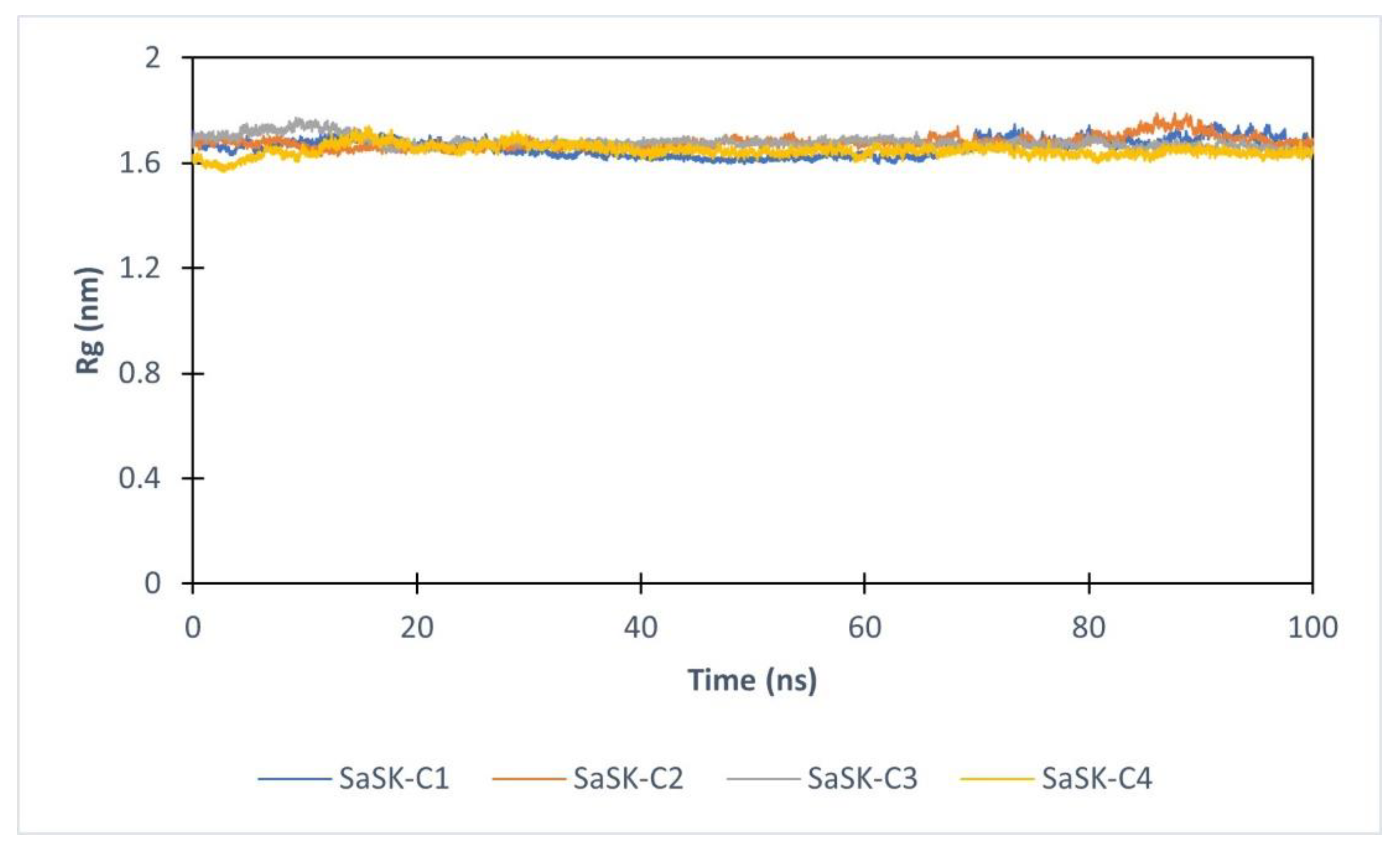
Furthermore, to assess the effect of compound binding in protein tertiary structure, the radius of gyration (Rg) analysis was realized. As it can be seen, the value of Rg in each complex keeps constant during entire simulation time, suggesting that none of the potential inhibitors alters the structure of the protein (Figure 5).
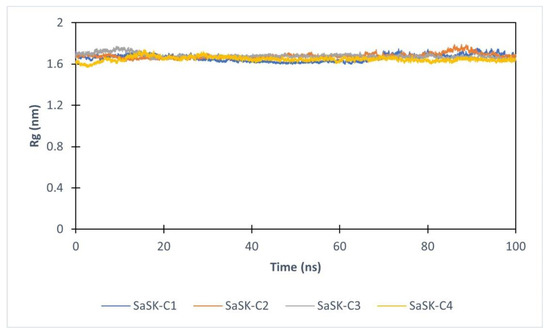
Figure 5.
Radius of gyration for SaSK in complex with C1(Blue), C2 (Orange), C3 (Gray), C4 (Yellow).
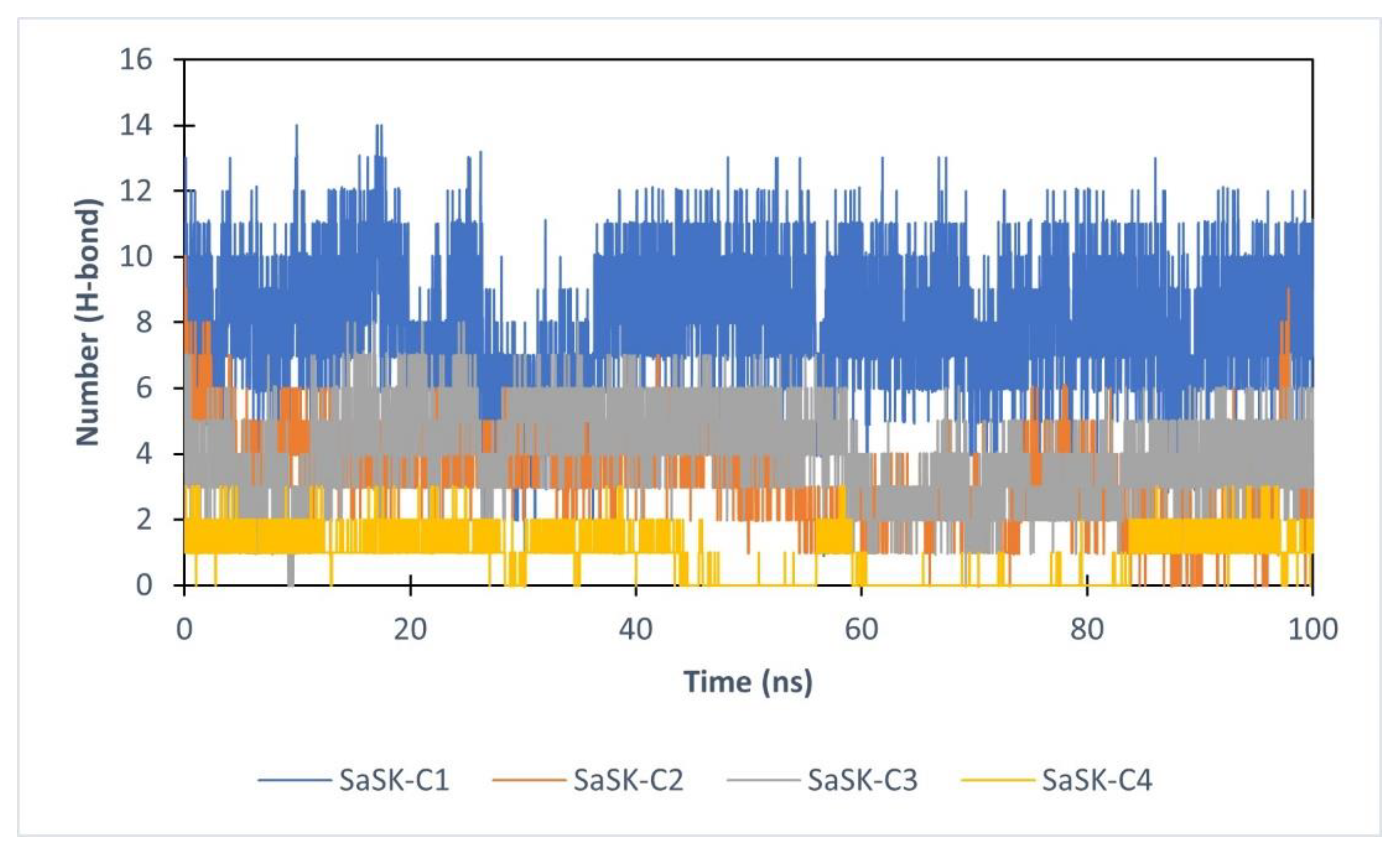
Finally, hydrogen bond analysis was performed. The data show that the number of H-bonds in the complexes vary during simulation time with an average of 7, 4, 3, and 1 for SaSK-C1, SaSK-C2, SaSK-C3, and SaSK-C4, respectively, suggesting a better binding mode for compound C1 (Figure 6 and Table 2).
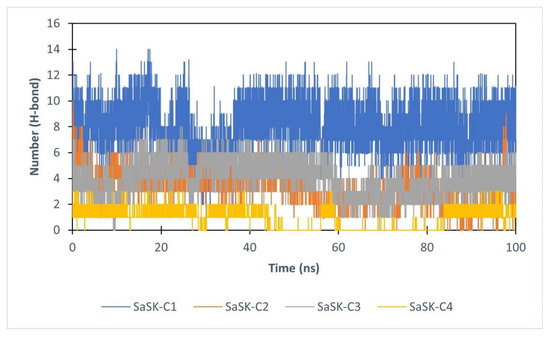
Figure 6.
H-Bond analysis for each SaSK–ligand complex.
Table 2.
Interacting residues through the 100 ns simulation time at selected time frames.
2.4. Linear Interaction Energy
The linear interaction energy (LIE) [30] approach is an extensively described method to compute binding affinities. It permits to combine explicit conformational sampling (of the protein-bound and unbound-ligand states) with efficiency to be able to calculate quantitative values for the protein–ligand binding free energy ΔGbind.
In this study, the LIE method was employed to calculate the binding affinities of the four complexes evaluated by molecular dynamics. Results show that C1 and C4 obtained a negative value for ΔGbind because of a combination of Van der Waals and electrostatic interaction energies, while C2 and C3 obtained a positive one, indicating that the former make a more stable complex with SaSK than the latter (Table 3).
Table 3.
Binding free energies calculated by the LIE method for each complex in MD simulation.
2.5. ADME-Tox Evaluation
In the final analysis to complete the characterization of these potential inhibitors, an important issue during first steps of drug design process is the prediction of the ADME-Tox properties of the molecules. In this context, a detailed study was performed for the four compounds using the SwissADME online tool [31] and PreADMET server [32]. The data show that, in general, all compounds obtained evaluations in the permitted range of each characteristic, which indicates the drugability potential of these compounds (Table 4 and Table 5). It is important to note that this type of characterization has not been reported for inhibitors against other SKs from bacteria [22,24].
Table 4.
Physicochemical properties of potential SaSK inhibitors.
Table 5.
PreADMET results characteristics for potential SaSK inhibitors.
3. Materials and Methods
3.1. Small Molecules Chemical Library
In this study, A 3D small molecule database was retrieved from ZINC15 database Tranches (http://zinc15.docking.org/, accessed on 9 January 2021) [33]. First, based on substrate structure, a Log p value of −1 and a molecular weight ≤ 350 Da, were used as selection criteria. By the time this work was finished, the number of compounds was neighboring 5 million.
Prior to virtual screening by docking, compounds were filtered according to Lipinski’s Rule of Five’ [34] to select those that possess physicochemical properties present in potential drug candidates only. Additionally, given that unfavorable structural alerts that can produce toxicity may lead to a compound being rejected in further studies [19,35], an in silico toxicity risk assessment for Mutagenicity, Tumorigenic, Irritant, and Reproductive effects was performed using Osiris Data Warrior software [36]. The presence of a single toxic parameter was enough to eliminate a given compound. Furthermore, the topological surface area (TPSA) was also calculated where a value ranging from >75 to <140 was necessary to be included, along with a number of rotatable bonds of less than 5. Finally, compounds were clustered according to their structure and similarity using as criteria that the highest similarity value fell below 0.8 according to Data Warrior Software.
3.2. Docking Studies
The previously reported SaSK 3D homology model [18] was employed for docking studies, the protein structure was prepared with the Protein Preparation Wizard in Maestro (Schrödinger Suite Release 2019-4) [37]. Bonds order was assigned, hydrogen atoms were added, and formal charges were treated, protein minimization was applied with the OPLS3e forcefield. The grid box was generated with default settings, with a 10Å × 10Å × 10Å size, using the center between amino acids Met14, Asp37, Ile48, Phe60, Arg61, Gly81, Gly82, Gly83 Pro118, and Arg138, which correspond to amino acids forming the shikimate binding site of the enzyme [26,29]. After filtering criteria (Figure 1), the 3D ligand structures of compounds selected were prepared using Ligprep, at a selected pH range of 7 ± 2, where ionization states were generated; the energy was minimized using the OPLS3e force field. Docking studies were carried out with Glide [38] implemented in the Maestro software, using the extra precision (XP) mode [39] that provides further elimination of false positives by applying extensive sampling and a more stringent scoring function, the best five poses of each compound were retained as output.
3.3. Molecular Dynamics Studies
Molecular dynamics simulations were performed using GROMACS version 2019.3 [40] and CHARMM 36 forcefield [41]. Before MD simulation, compounds were parametrized using SwissParam Server (http://swissparam.ch/, accessed 17 May 2021) [42]. SaSK coordinates and topology were constructed using GROMACS, then each ligand was merged into a complex with SaSK and the system was immersed into the center of a dodecahedral box, the solute box distance was set at 1.0 nm. The system was solvated by the addition of TIP3P waters [43] and counterions were added to reach a salt concentration of 0.15 M.
MD simulations began with an energy minimization (EM) simulation as the first step, which was performed during 100 ps to reach a local minimum employing the steepest descent algorithm. Afterwards, the system was submitted to temperature and pressure equilibration by performing two 100 ps equilibration steps namely, an isothermal-isochoric (NVT) ensemble followed by an isothermal-isobaric (NPT) ensemble under no position restraint, thus bringing the system to a 310 K temperature and 1 bar pressure. Temperature and pressure were maintained by employing the velocity-rescale thermostat [44] and the Parrinello-Rahman pressure coupling methods [45]. Finally, a 100 ns timescale MD was carried out, employing a 1.2 nm for short-range interactions and the leap-frog integrator algorithm. MD simulations were then analyzed using Visual Molecular Dynamics (VMD) software [46]. Furthermore, to explore the structural and dynamic behavior of the protein-ligand complex, analysis of the MD data involved root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and hydrogen bond analysis (H-bonding).
3.4. Linear Interaction Energy Calculations
Binding free energies were obtained for each of the complexes based on the linear interaction energy (LIE) method calculated by the Equation (1):
where (VLJ)bound indicates the average Lennard–Jones energy for ligand–protein interaction; (VLJ)free is the average Lennard–Jones energy for ligand–solvent interaction; (VCL)bound is the average electrostatic energy for ligand–protein interaction; (VCL)free is the average electrostatic energy for ligand–solvent interaction; the LIE coefficients are given by α, β, and γ which for small drug-like ligands correspond to α = 0.18, β = 0.50, and γ = 0.00 [30,47,48].
∆Gbind = α[(VLJ)bound − (VLJ)free] + β[(VCL)bound − (VCL)free] + γ
3.5. ADME Properties Prediction
Absorption, distribution, metabolism, and excretion properties of each potential inhibitor were predicted using SwissADME web tool [31] and the online PreADMET server (http://preadmet.bmdrc.org, accessed 23 August 2021) [32].
4. Conclusions
The study of an around 5 million small molecules database through a computer-assisted drug design strategy, permitted to find the first set of potential inhibitors of SaSK. According to the structural analysis, these compounds formed interactions with residues important for enzyme catalysis. Furthermore, they demonstrated good ADME-Tox and druglike characteristics, which make these molecules an attractive starting point for the development of new drugs against MRSA.
Supplementary Materials
The following is available online. Table S1: Structures of the compounds selected for virtual screening.
Author Contributions
Conceptualization, A.T.-V. and C.A.-D.; methodology, L.R.-S., E.S.-C., A.T.-V. and C.A.-D.; formal analysis, L.R.-S., M.V.-S., J.C.-M., M.G.P.-G. and A.C.-V.; writing—original draft preparation, L.R.-S.; writing—review and editing, M.G.P.-G., A.T.-V. and C.A.-D.; funding acquisition, A.T.-V. and C.A.-D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT), grants 257848 (A.T.-V.) and 258694 (C.A.-D.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
L.R.-S. acknowledge Consejo Nacional de Ciencia y Tecnología (CONACyT) for the Ph. D. fellowship granted. (No. 597625).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, N.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. WHO Pathogens Priority List Working Group. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: http://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 15 August 2019).
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections—United States. MMWR Morb. Mortal Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, R.H. Emerging options for treatment of invasive, multidrug-resistant Staphylococcus aureus infections. Pharmacotherapy 2007, 27, 227–249. [Google Scholar] [CrossRef]
- McCallum, N.; Berger-Bächi, B.; Senn, M.M. Regulation of antibiotic resistance in Staphylococcus aureus. Int. J. Med. Microbiol. 2007, 300, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M. Multidrug-Resistant Staphylococcus aureus, India, 2013–2015. Emerg. Infect. Dis. 2016, 22, 1666–1667. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar]
- Duval, R.E.; Grare, M.; Demoré, B. Fight Against Antimicrobial Resistance: We Always Need New Antibacterials but for Right Bacteria. Molecules 2019, 24, 3152. [Google Scholar] [CrossRef] [Green Version]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [Green Version]
- Bentley, R. The shikimate pathway—A metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 307–384. [Google Scholar] [CrossRef] [PubMed]
- Haslam, E.H. (Ed.) 1—The Shikimate Pathway: Biosynthesis of the Aromatic Amino Acids. In Biosynthesis of Natural Products Series, The Shikimate Pathway; Butterworth-Heinemann: Oxford, UK, 1974; pp. 3–48. ISBN 9780408705691. [Google Scholar]
- Coggins, J.R.; Abell, C.; Evans, L.B.; Frederickson, M.; Robinson, D.A.; Roszak, A.W.; Lapthorn, A.P. Experiences with the shikimate-pathway enzymes as targets for rational drug design. Biochem. Soc. Trans. 2003, 31 Pt 3, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Simithy, J.; Goodwin, D.C.; Calderón, A.I. Selective Mycobacterium tuberculosis Shikimate Kinase Inhibitors as Potential Antibacterials. Perspect. Med. Chem. 2015, 7, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parish, T.; Stoker, N.G. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 2002, 148 Pt 10, 3069–3077. [Google Scholar] [CrossRef] [Green Version]
- Gan, J.; Gu, Y.; Li, Y.; Yan, H.; Ji, X. Crystal structure of Mycobacterium tuberculosis shikimate kinase in complex with shikimic acid and an ATP analogue. Biochemistry 2006, 45, 8539–8545. [Google Scholar] [CrossRef]
- Favela-Candia, A.; Téllez-Valencia, A.; Campos-Almazán, M.; Sierra-Campos, E.; Valdez-Solana, M.; Oria-Hernández, J.; Castillo-Villanueva, A.; Nájera, H.; Avitia-Domínguez, C. Biochemical, Kinetic, and Computational Structural Characterization of Shikimate Kinase from Methicillin-Resistant Staphylococcus aureus. Mol. Biotechnol. 2019, 61, 274–285. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.D.; Bourenkov, G.P.; Oberschall, A.; Strizhov, N.; Bartunik, H.D. Mechanism of phosphoryl transfer catalyzed by shikimate kinase from Mycobacterium tuberculosis. J. Mol. Biol. 2006, 364, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Prado, V.; Lence, E.; Otero, J.M.; Garcia-Doval, C.; van Raaij, M.J.; Llamas-Saiz, A.L.; Lamb, H.; Hawkins, A.R.; González-Bello, C. Mycobacterium tuberculosis Shikimate Kinase Inhibitors: Design and Simulation Studies of the Catalytic Turnover. J. Am. Chem. Soc. 2013, 135, 12366–12376. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.H.; de Oliveira, J.S.; Canduri, F.; Dias, M.V.; Palma, M.S.; Basso, L.A.; Santos, D.S.; de Azevedo, W.F., Jr. Structure of shikimate kinase from Mycobacterium tuberculosis reveals the binding of shikimic acid. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, D60, 2310–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Zhang, J.; Chen, L.; Chen, K.; Shen, X.; Jiang, H. Discovery of Helicobacter pylori shikimate kinase inhibitors: Bioassay and molecular modeling. Bioorganic Med. Chem. 2007, 15, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Rajput, V.S.; Gupta, M.; Chib, R.; Kumar, A.; Wazir, P.; Khan, I.A.; Nargotra, A. Benzothiazole Derivative as a Novel Mycobacterium tuberculosis Shikimate Kinase Inhibitor: Identification and Elucidation of Its Allosteric Mode of Inhibition. J. Chem. Inf. Model. 2016, 56, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Rajput, V.S.; Mehra, R.; Kumar, S.; Nargotra, A.; Singh, P.P.; Khan, I.A. Screening of antitubercular compound library identifies novel shikimate kinase inhibitors of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2016, 100, 5415–5426. [Google Scholar] [CrossRef] [PubMed]
- Simithy, J.; Fuanta, N.R.; Alturki, M.; Hobrath, J.V.; Wahba, A.E.; Pina, I.; Rath, J.; Hamann, M.T.; DeRuiter, J.; Goodwin, D.C.; et al. Slow-Binding Inhibition of Mycobacterium tuberculosis Shikimate Kinase by Manzamine Alkaloids. Biochemistry 2018, 57, 4923–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.C.; Chang, Y.N.; Wang, W.C. Structural basis for shikimate-binding specificity of Helicobacter pylori shikimate kinase. J. Bacteriol. 2005, 187, 8156–8163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Reshetnikova, L.; Li, Y.; Wu, Y.; Yan, H.; Singh, S.; Ji, X. Crystal structure of shikimate kinase from Mycobacterium tuberculosis reveals the dynamic role of the LID domain in catalysis. J. Mol. Biol. 2002, 319, 779–789. [Google Scholar] [CrossRef]
- Åqvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. Des. Sel. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Seul, South Corea: Bioinformatics and Molecular Design Research Center; PreADMET Program. 2004. Available online: http://preadmet.bmdrc.org (accessed on 23 August 2021).
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Limban, C.; Nuţă, D.C.; Chiriţă, C.; Negreș, S.; Arsene, A.L.; Goumenou, M.; Karakitsios, S.P.; Tsatsakis, A.M.; Sarigiannis, D.A. The use of structural alerts to avoid the toxicity of pharmaceuticals. Toxicol. Rep. 2018, 5, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Schrödinger Suite Release 2019-4; Schrödinger, LLC: New York, NY, USA, 2019.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; MacKerell, A.D. CHARMM36 All-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Boonstra, S.; Onck, P.R.; van der Giessen, E. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Punkvang, A.; Saparpakorn, P.; Hannongbua, S.; Wolschann, P.; Beyer, A.; Pungpo, P. Investigating the structural basis of arylamides to improve potency against M. tuberculosis strain through molecular dynamics simulations. Eur. J. Med. Chem. 2010, 45, 5585–5593. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Méndez-Lucio, O.; Medina-Franco, J.L.; Castillo, R.; Yépez-Mulia, L.; Hernández-Luis, F.; Hernández-Campos, A. Towards the identification of the binding site of benzimidazoles to β-tubulin of Trichinella spiralis: Insights from computational and experimental data. J. Mol. Graph. Model. 2013, 41, 12–19. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).