
RNA Activation—A Novel Approach to Therapeutically Upregulate Gene Transcription

 ,
, {kind=link}
{kind=link}
Abstract
1. RNA Activation Is a Novel Class of Therapeutics
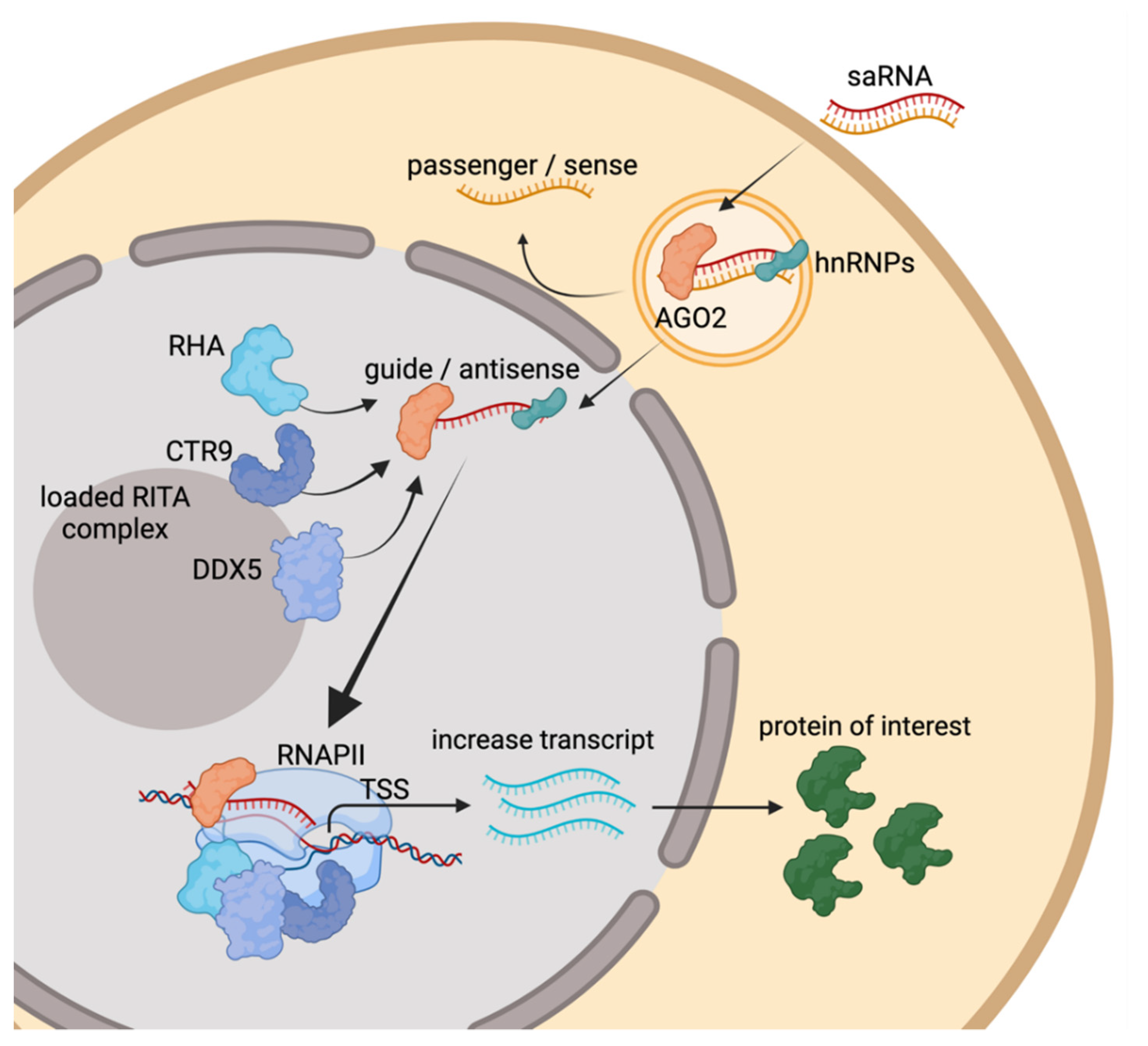
2. Molecular Mechanisms of RNAa
3. Applications of saRNA in Preclinical Disease Models
4. saRNA Targeting CEBPA Expression Demonstrates Anti-Tumor Activity
5. Formulated CEBPA-51 saRNA (MTL-CEBPA) Targets Immunosuppressive Myeloid Cells and Synergize with Other Anti-Cancer Agents
6. Promising Observations of MTL-CEBPA in Clinical Trials
7. Future Perspective for saRNA Therapeutics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef]
- Janowski, B.A.; Younger, S.T.; Hardy, D.B.; Ram, R.; Huffman, K.E.; Corey, D.R. Activating Gene Expression in Mammalian Cells with Promoter-Targeted Duplex RNAs. Nat. Chem. Biol. 2007, 3, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small DsRNAs Induce Transcriptional Activation in Human Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef]
- Britten, R.J.; Davidson, E.H. Gene Regulation for Higher Cells: A Theory. Science 1969, 165, 349–357. [Google Scholar] [CrossRef]
- Kuwabara, T.; Hsieh, J.; Nakashima, K.; Taira, K.; Gage, F.H. A Small Modulatory DsRNA Specifies the Fate of Adult Neural Stem Cells. Cell 2004, 116, 779–793. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Huang, V.; Qin, Y.; Wang, J.; Wang, X.; Place, R.F.; Lin, G.; Lue, T.F.; Li, L.-C. RNAa Is Conserved in Mammalian Cells. PLoS ONE 2010, 5, e8848. [Google Scholar] [CrossRef]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.P.; van der Brug, M.P.; Wahlestedt, C. Inhibition of Natural Antisense Transcripts in Vivo Results in Gene-Specific Transcriptional Upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Ahituv, N.; Moses, A.M.; Prabhakar, S.; Nobrega, M.A.; Shoukry, M.; Minovitsky, S.; Dubchak, I.; Holt, A.; Lewis, K.D.; et al. In Vivo Enhancer Analysis of Human Conserved Non-Coding Sequences. Nature 2006, 444, 499–502. [Google Scholar] [CrossRef]
- Visel, A.; Prabhakar, S.; Akiyama, J.A.; Shoukry, M.; Lewis, K.D.; Holt, A.; Plajzer-Frick, I.; Afzal, V.; Rubin, E.M.; Pennacchio, L.A. Ultraconservation Identifies a Small Subset of Extremely Constrained Developmental Enhancers. Nat. Genet. 2008, 40, 158–160. [Google Scholar] [CrossRef]
- Uesaka, M.; Agata, K.; Oishi, T.; Nakashima, K.; Imamura, T. Evolutionary Acquisition of Promoter-Associated Non-Coding RNA (PancRNA) Repertoires Diversifies Species-Dependent Gene Activation Mechanisms in Mammals. BMC Genom. 2017, 18, 285. [Google Scholar] [CrossRef]
- Meng, X.; Jiang, Q.; Chang, N.; Wang, X.; Liu, C.; Xiong, J.; Cao, H.; Liang, Z. Small Activating RNA Binds to the Genomic Target Site in a Seed-Region-Dependent Manner. Nucleic Acids Res. 2016, 44, 2274–2282. [Google Scholar] [CrossRef]
- Voutila, J.; Reebye, V.; Roberts, T.C.; Protopapa, P.; Andrikakou, P.; Blakey, D.C.; Habib, R.; Huber, H.; Saetrom, P.; Rossi, J.J.; et al. Development and Mechanism of Small Activating RNA Targeting CEBPA, a Novel Therapeutic in Clinical Trials for Liver Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2705–2714. [Google Scholar] [CrossRef]
- Place, R.F.; Noonan, E.J.; Földes-Papp, Z.; Li, L.-C. Defining Features and Exploring Chemical Modifications to Manipulate RNAa Activity. Curr. Pharm. Biotechnol. 2010, 11, 518–526. [Google Scholar] [CrossRef]
- Chu, Y.; Yue, X.; Younger, S.T.; Janowski, B.A.; Corey, D.R. Involvement of Argonaute Proteins in Gene Silencing and Activation by RNAs Complementary to a Non-Coding Transcript at the Progesterone Receptor Promoter. Nucleic Acids Res. 2010, 38, 7736–7748. [Google Scholar] [CrossRef]
- Zhao, X.; Reebye, V.; Hitchen, P.; Fan, J.; Jiang, H.; Sætrom, P.; Rossi, J.; Habib, N.A.; Huang, K.W. Mechanisms Involved in the Activation of C/EBPα by Small Activating RNA in Hepatocellular Carcinoma. Oncogene 2019, 38, 3446–3457. [Google Scholar] [CrossRef]
- Fimiani, C.; Goina, E.; Su, Q.; Gao, G.; Mallamaci, A. RNA Activation of Haploinsufficient Foxg1 Gene in Murine Neocortex. Sci. Rep. 2016, 6, 39311. [Google Scholar] [CrossRef] [PubMed]
- Huang, V.; Place, R.F.; Portnoy, V.; Wang, J.; Qi, Z.; Jia, Z.; Yu, A.; Shuman, M.; Yu, J.; Li, L.-C. Upregulation of Cyclin B1 by MiRNA and Its Implications in Cancer. Nucleic Acids Res. 2012, 40, 1695–1707. [Google Scholar] [CrossRef]
- Wu, H.-L.; Li, S.-M.; Hu, J.; Yu, X.; Xu, H.; Chen, Z.; Ye, Z.-Q. Demystifying the Mechanistic and Functional Aspects of P21 Gene Activation with Double-Stranded RNAs in Human Cancer Cells. J. Exp. Clin. Cancer Res. CR 2016, 35, 145. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Z.; Xia, D.; Wu, J.; Xu, H.; Ye, Z.-Q. Promoter-Associated Small Double-Stranded RNA Interacts with Heterogeneous Nuclear Ribonucleoprotein A2/B1 to Induce Transcriptional Activation. Biochem. J. 2012, 447, 407–416. [Google Scholar] [CrossRef]
- Portnoy, V.; Huang, V.; Place, R.F.; Li, L.-C. Small RNA and Transcriptional Upregulation. Wiley Interdiscip. Rev. RNA 2011, 2, 748–760. [Google Scholar] [CrossRef]
- Wang, B.; Sun, J.; Shi, J.; Guo, Q.; Tong, X.; Zhang, J.; Hu, N.; Hu, Y. Small-Activating RNA Can Change Nucleosome Positioning in Human Fibroblasts. J. Biomol. Screen. 2016, 21, 634–642. [Google Scholar] [CrossRef]
- Portnoy, V.; Lin, S.H.S.; Li, K.H.; Burlingame, A.; Hu, Z.-H.; Li, H.; Li, L.-C. SaRNA-Guided Ago2 Targets the RITA Complex to Promoters to Stimulate Transcription. Cell Res. 2016, 26, 320–335. [Google Scholar] [CrossRef]
- Yang, K.; Shen, J.; Xie, Y.-Q.; Lin, Y.-W.; Qin, J.; Mao, Q.-Q.; Zheng, X.-Y.; Xie, L.-P. Promoter-Targeted Double-Stranded Small RNAs Activate PAWR Gene Expression in Human Cancer Cells. Int. J. Biochem. Cell Biol. 2013, 45, 1338–1346. [Google Scholar] [CrossRef]
- Dar, S.A.; Kumar, M. SaRNAdb: Resource of Small Activating RNAs for Up-Regulating the Gene Expression. J. Mol. Biol. 2018, 430, 2212–2218. [Google Scholar] [CrossRef]
- Kang, M.R.; Li, G.; Pan, T.; Xing, J.C.; Li, L.C. Development of therapeutic dsP21-322 for cancer treatment. Adv. Exp. Med. Biol. 2017, 983, 217–229. [Google Scholar]
- Place, R.F.; Wang, J.; Noonan, E.J.; Meyers, R.; Manoharan, M.; Charisse, K.; Duncan, R.; Huang, V.; Wang, X.; Li, L.-C. Formulation of Small Activating RNA Into Lipidoid Nanoparticles Inhibits Xenograft Prostate Tumor Growth by Inducing P21 Expression. Mol. Ther. Nucleic Acids 2012, 1, e15. [Google Scholar] [CrossRef]
- Wei, J.; Zhao, J.; Long, M.; Han, Y.; Wang, X.; Lin, F.; Ren, J.; He, T.; Zhang, H. P21WAF1/CIP1 Gene Transcriptional Activation Exerts Cell Growth Inhibition and Enhances Chemosensitivity to Cisplatin in Lung Carcinoma Cell. BMC Cancer 2010, 10, 632. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Liu, X.; Wang, J.; Li, F.; Li, C.; Shan, B. Up-Regulation of P21WAF1/CIP1 by Small Activating RNA Inhibits the in Vitro and in Vivo Growth of Pancreatic Cancer Cells. Tumori 2012, 98, 804–811. [Google Scholar] [CrossRef]
- Kang, M.R.; Yang, G.; Place, R.F.; Charisse, K.; Epstein-Barash, H.; Manoharan, M.; Li, L.C. Intravesical Delivery of Small Activating RNA Formulated into Lipid Nanoparticles Inhibits Orthotopic Bladder Tumor Growth. Cancer Res. 2012, 72, 5069–5079. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Feng, C.L.; Zheng, W.S.; Huang, S.; Zhang, W.X.; Wu, H.N.; Zhan, Y.; Han, Y.X.; Wu, S.; Jiang, J.D. Tumor-Selective Lipopolyplex Encapsulated Small Active RNA Hampers Colorectal Cancer Growth in Vitro and in Orthotopic Murine. Biomaterials 2017, 141, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Meng, L.; Xu, F.; Lian, J.; Xu, Y.; Xie, X.; Wang, X.; He, H.; Wang, C.; Zhu, Y. Enhanced Wild-Type P53 Expression by Small Activating RNA DsP53-285 Induces Cell Cycle Arrest and Apoptosis in Pheochromocytoma Cell Line PC12. Oncol. Rep. 2017, 38, 3160–3166. [Google Scholar] [CrossRef][Green Version]
- Nowell, C.S.; Radtke, F. Notch as a Tumour Suppressor. Nat. Rev. Cancer 2017, 17, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jiang, K.; Jiang, P.; He, H.; Chen, K.; Shao, J.; Deng, G. Mechanism of Notch1-SaRNA-1480 Reversing Androgen Sensitivity in Human Metastatic Castration-Resistant Prostate Cancer. Int. J. Mol. Med. 2020, 46, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gao, P.; Han, Y.; Zhang, L.; Ren, J.; Lin, F.; Min, L.; Wang, X.; He, T.; Dong, K.; et al. Double Strand RNA-Guided Endogeneous E-Cadherin up-Regulation Induces the Apoptosis and Inhibits Proliferation of Breast Carcinoma Cells in Vitro and in Vivo. Cancer Sci. 2010, 101, 1790–1796. [Google Scholar] [CrossRef]
- Dai, J.; He, H.; Lin, D.; Wang, C.; Zhu, Y.; Xu, D. Up-Regulation of E-Cadherin by SaRNA Inhibits the Migration and Invasion of Renal Carcinoma Cells. Int. J. Clin. Exp. Pathol. 2018, 11, 5792–5800. [Google Scholar]
- Li, F.; Li, J.; Yu, J.; Pan, T.; Yu, B.; Sang, Q.; Dai, W.; Hou, J.; Yan, C.; Zang, M.; et al. Identification of ARGLU1 as a Potential Therapeutic Target for Gastric Cancer Based on Genome-Wide Functional Screening Data. EBioMedicine 2021, 69, 103436. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Stechman, M.J.; Loh, N.Y.; Thakker, R.V. Genetic Causes of Hypercalciuric Nephrolithiasis. Pediatric Nephrol. 2009, 24, 2321–2332. [Google Scholar] [CrossRef]
- Zeng, T.; Duan, X.; Zhu, W.; Liu, Y.; Wu, W.; Zeng, G. SaRNA-Mediated Activation of TRPV5 Reduces Renal Calcium Oxalate Deposition in Rat via Decreasing Urinary Calcium Excretion. Urolithiasis 2018, 46, 271–278. [Google Scholar] [CrossRef]
- Kimura, Y.; Iwanaga, E.; Iwanaga, K.; Endo, S.; Inoue, Y.; Tokunaga, K.; Nagahata, Y.; Masuda, K.; Kawamoto, H.; Matsuoka, M. A Regulatory Element in the 3′-untranslated Region of CEBPAis Associated with Myeloid/NK/T-cell Leukemia. Eur. J. Haematol. 2020, 96, 613–617. [Google Scholar]
- Lourenço, A.R.; Coffer, P.J. A Tumor Suppressor Role for C/EBPα in Solid Tumors: More than Fat and Blood; Nature Publishing Group: Berlin, Germany, 2017; Volume 36, pp. 5221–5230. [Google Scholar]
- Loomis, K.D.; Zhu, S.; Yoon, K.; Johnson, P.F.; Smart, R.C. Genetic Ablation of CCAAT/Enhancer Binding Protein α in Epidermis Reveals Its Role in Suppression of Epithelial Tumorigenesis. Cancer Res. 2007, 67, 6768–6776. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bennett, K.L.; Hackanson, B.; Smith, L.T.; Morrison, C.D.; Lang, J.C.; Schuller, D.E.; Weber, F.; Eng, C.; Plass, C. Tumor Suppressor Activity of CCAAT/Enhancer Binding Protein a Is Epigenetically down-Regulated in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2007, 67, 4657–4664. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Yamada, N.; Ogawa, Y.; Ikegami, M. CCAAT/Enhancer-Binding Protein-α Suppresses Lung Tumor Development in Mice through the P38α MAP Kinase Pathway. PLoS ONE 2013, 8, e57013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shim, M.; Powers, K.L.; Ewing, S.J.; Zhu, S.; Smart, R.C. Diminished Expression of C/EBPa in Skin Carcinomas Is Linked to Oncogenic Ras and Reexpression of C/EBPa in Carcinoma Cells Inhibits Proliferation. Cancer Res. 2005, 65, 861–867. [Google Scholar]
- Lu, J.; Du, C.; Yao, J.; Wu, B.; Duan, Y.; Zhou, L.; Xu, D.; Zhou, F.; Gu, L.; Zhou, H.; et al. C/EBPα Suppresses Lung Adenocarcinoma Cell Invasion and Migration by Inhibiting β-Catenin. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 1779–1788. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Li, M.; Tian, C.-C.; Wang, T.-T.; Mi, S.-F. CCAAT Enhancer Binding Protein α Suppresses Proliferation, Metastasis, and Epithelial-Mesenchymal Transition of Ovarian Cancer Cells via Suppressing the Wnt/β-Catenin Signaling. Neoplasma 2021, 68, 602–612. [Google Scholar] [CrossRef]
- Reebye, V.; Saetrom, P.; Mintz, P.J.; Huang, K.-W.; Swiderski, P.; Peng, L.; Liu, C.; Liu, X.; Lindkaer-Jensen, S.; Zacharoulis, D.; et al. Novel RNA Oligonucleotide Improves Liver Function and Inhibits Liver Carcinogenesis in Vivo. Hepatology 2014, 59, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.W.; Reebye, V.; Mintz, P.; Tien, Y.W.; Lai, H.S.; Sætrom, P.; Reccia, I.; Swiderski, P.; Armstrong, B.; et al. Targeted Delivery of C/EBPα -SaRNA by Pancreatic Ductal Adenocarcinoma-Specific RNA Aptamers Inhibits Tumor Growth in Vivo. Mol. Ther. 2016, 24, 1106–1116. [Google Scholar] [CrossRef]
- Yoon, S.; Huang, K.-W.; Andrikakou, P.; Vasconcelos, D.; Swiderski, P.; Reebye, V.; Sodergren, M.; Habib, N.; Rossi, J.J. Targeted Delivery of C/EBPα-SaRNA by RNA Aptamers Shows Anti-Tumor Effects in a Mouse Model of Advanced PDAC. Mol. Ther. Nucleic Acids 2019, 18, 142–154. [Google Scholar] [CrossRef]
- Bégay, V.; Smink, J.J.; Loddenkemper, C.; Zimmermann, K.; Rudolph, C.; Scheller, M.; Steinemann, D.; Leser, U.; Schlegelberger, B.; Stein, H.; et al. Deregulation of the Endogenous C/EBPβ LIP Isoform Predisposes to Tumorigenesis. J. Mol. Med. 2015, 93, 39–49. [Google Scholar] [CrossRef]
- Reebye, V.; Huang, K.W.; Lin, V.; Jarvis, S.; Cutilas, P.; Dorman, S.; Ciriello, S.; Andrikakou, P.; Voutila, J.; Saetrom, P.; et al. Gene Activation of CEBPA Using SaRNA: Preclinical Studies of the First in Human SaRNA Drug Candidate for Liver Cancer. Oncogene 2018, 37, 3216–3228. [Google Scholar] [CrossRef]
- Avellino, R.; Delwel, R. Expression and Regulation of C/EBPα in Normal Myelopoiesis and in Malignant Transformation. Blood 2017, 129, 2083–2091. [Google Scholar] [CrossRef]
- Mackert, J.R.; Qu, P.; Min, Y.; Johnson, P.F.; Yang, L.; Lin, P.C. Dual Negative Roles of C/EBPα in the Expansion and pro-Tumor Functions of MDSCs. Sci. Rep. 2017, 7, 14048. [Google Scholar] [CrossRef]
- Torroella-Kouri, M.; Ma, X.; Perry, G.; Ivanova, M.; Cejas, P.J.; Owen, J.L.; Iragavarapu-Charyulu, V.; Lopez, D.M. Diminished Expression of Transcription Factors Nuclear Factor KappaB and CCAAT/Enhancer Binding Protein Underlies a Novel Tumor Evasion Mechanism Affecting Macrophages of Mammary Tumor-Bearing Mice. Cancer Res. 2005, 65, 10578–10584. [Google Scholar] [CrossRef]
- Hashimoto, A.; Sarker, D.; Reebye, V.; Jarvis, S.; Sodergren, M.H.; Kossenkov, A.; Sanseviero, E.; Raulf, N.; Vasara, J.; Andrikakou, P.; et al. Up-Regulation of C/EBPα Inhibits Suppressive Activity of Myeloid Cells and Potentiates Antitumor Response in Mice and Cancer Patients. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Huang, K.-W.; Tan, C.P.; Reebye, V.; Chee, C.E.; Zacharoulis, D.; Habib, R.; Blakey, D.C.; Rossi, J.J.; Habib, N.; Sodergren, M.H. MTL-CEBPA Combined with Immunotherapy or RFA Enhances Immunological Anti-Tumor Response in Preclinical Models. Int. J. Mol. Sci. 2021, 22, 9168. [Google Scholar] [CrossRef]
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy with Immunotherapy in Cancer. Front. Oncol. 2021, 11, 637504. [Google Scholar] [CrossRef]
- Williams, C.S.; Tsujii, M.; Reese, J.; Dey, S.K.; DuBois, R.N. Host Cyclooxygenase-2 Modulates Carcinoma Growth. J. Clin. Investig. 2000, 105, 1589–1594. [Google Scholar] [CrossRef]
- Qi, X.; Zhao, Y.; Li, H.; Guo, X.; Han, G. Management of Hepatocellular Carcinoma: An Overview of Major Findings from Meta-Analyses. Oncotarget 2016, 7, 34703–34751. [Google Scholar] [CrossRef]
- Habib, N.A.; Huang, K.-W.; Reebye, V.; Sodergren, M.; Rossi, J. Abstract 3856: MTLCEBPA, a Drug Candidate for Hepatocellular-Carcinoma Enhances Efficacy of Sorafenib. Cancer Res. 2019, 79, 3856. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; de Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Plummer, E.R.; Basu, B.; Meyer, T.; Huang, K.-W.; Evans, T.R.J.; Spalding, D.; Ma, Y.T.; Palmer, D.H.; Chee, C.E.; et al. Preliminary Results of a First-in-Human, First-in-Class Phase I Study of MTL-CEBPA, a Small Activating RNA (SaRNA) Targeting the Transcription Factor C/EBP-α in Patients with Advanced Liver Cancer. J. Clin. Oncol. 2018, 36, 2509. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, Y.H.; Chiu, C.J.; Lu, L.C.; Liao, C.C.; Liang, C.W.; Hsu, C.H.; Cheng, A.L. Targeting Tumor-Infiltrating Ly6G+ Myeloid Cells Improves Sorafenib Efficacy in Mouse Orthotopic Hepatocellular Carcinoma. Int. J. Cancer 2018, 142, 1878–1889. [Google Scholar] [CrossRef]
- Kingwell, K. Small Activating RNAs Lead the Charge to Turn up Gene Expression. Nat. Rev. Drug Discov. 2021, 20, 573–574. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.; Basu, B.; Chee, C.E.; Huang, K.-W.; Palmer, D.H.; Ma, Y.T.; Evans, T.R.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic up-Regulating C/EBP-α, in Patients with Advanced Liver Cancer: A First-in-Human, Multi-Centre, Open-Label, Phase I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef]
- Kwok, A.; Raulf, N.; Habib, N. Developing Small Activating RNA as a Therapeutic: Current Challenges and Promises. Ther. Deliv. 2019, 10, 151–164. [Google Scholar] [CrossRef]
- Debacker, A.J.; Voutila, J.; Catley, M.; Blakey, D.; Habib, N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol. Ther. 2020, 28, 1759–1771. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.P.; Sinigaglia, L.; Gomez, V.; Nicholls, J.; Habib, N.A. RNA Activation—A Novel Approach to Therapeutically Upregulate Gene Transcription. Molecules 2021, 26, 6530. https://doi.org/10.3390/molecules26216530
Tan CP, Sinigaglia L, Gomez V, Nicholls J, Habib NA. RNA Activation—A Novel Approach to Therapeutically Upregulate Gene Transcription. Molecules. 2021; 26(21):6530. https://doi.org/10.3390/molecules26216530
Chicago/Turabian StyleTan, Choon Ping, Laura Sinigaglia, Valentí Gomez, Joanna Nicholls, and Nagy A. Habib. 2021. "RNA Activation—A Novel Approach to Therapeutically Upregulate Gene Transcription" Molecules 26, no. 21: 6530. https://doi.org/10.3390/molecules26216530
APA StyleTan, C. P., Sinigaglia, L., Gomez, V., Nicholls, J., & Habib, N. A. (2021). RNA Activation—A Novel Approach to Therapeutically Upregulate Gene Transcription. Molecules, 26(21), 6530. https://doi.org/10.3390/molecules26216530