
Hybrid Quinoline-Thiosemicarbazone Therapeutics as a New Treatment Opportunity for Alzheimer’s Disease‒Synthesis, In Vitro Cholinesterase Inhibitory Potential and Computational Modeling Analysis


 ,
,  and
and
Abstract
:
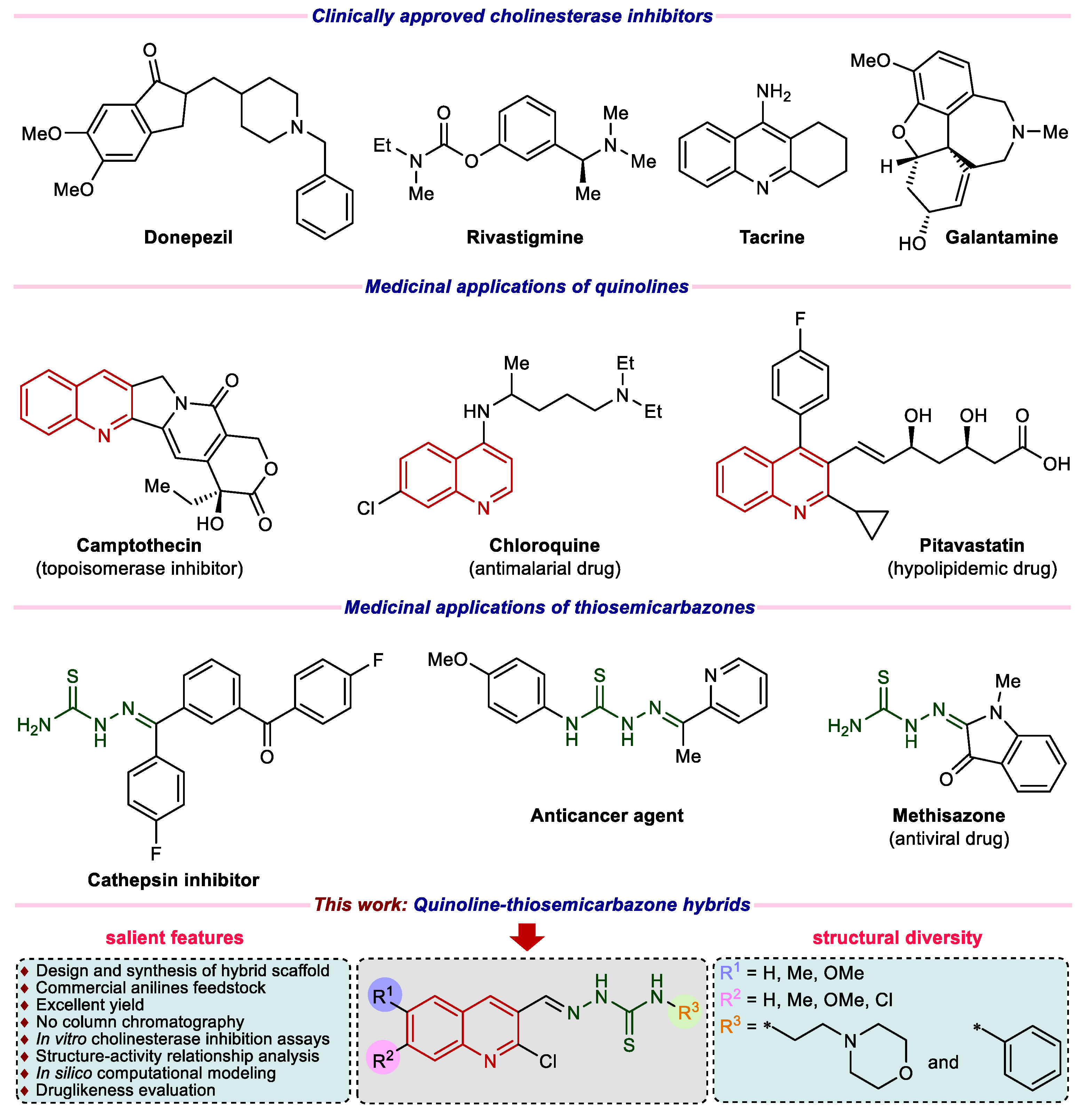
1. Introduction
2. Results and Discussion
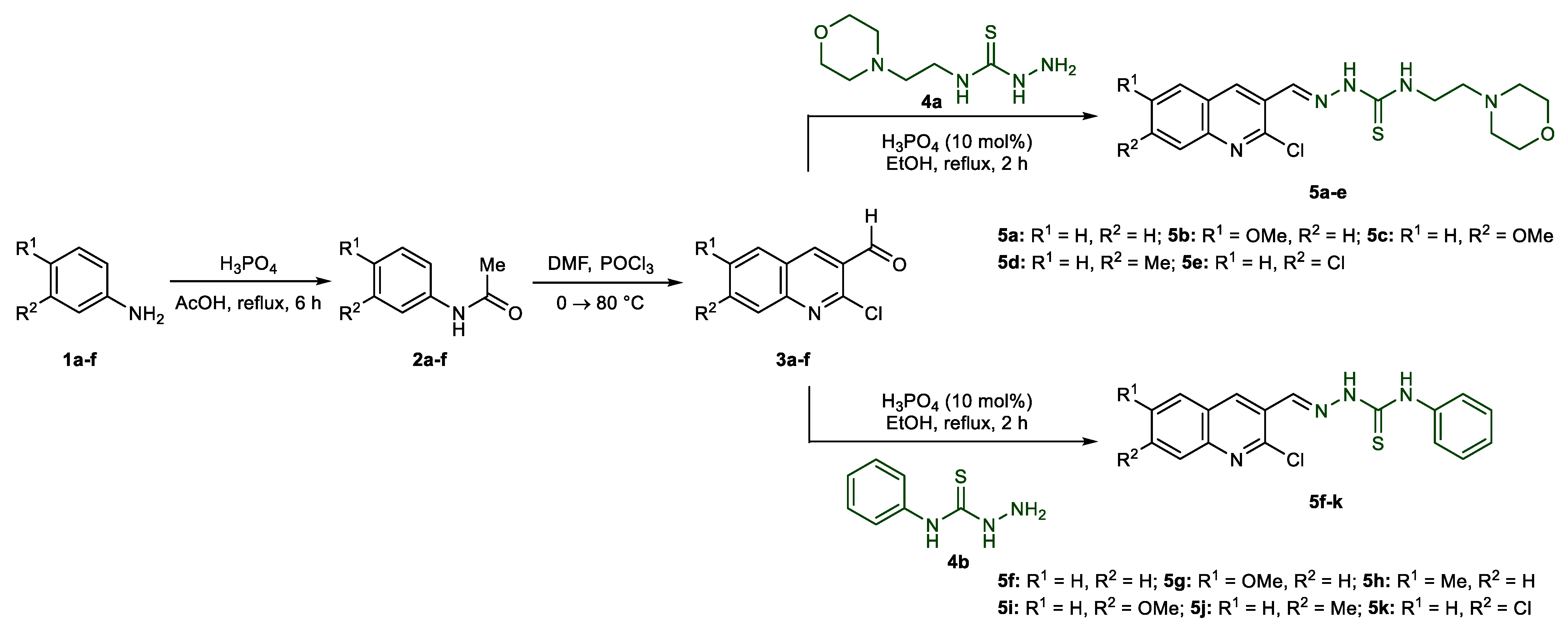
2.1. Synthetic Chemistry
2.2. Spectroscopic Characterization
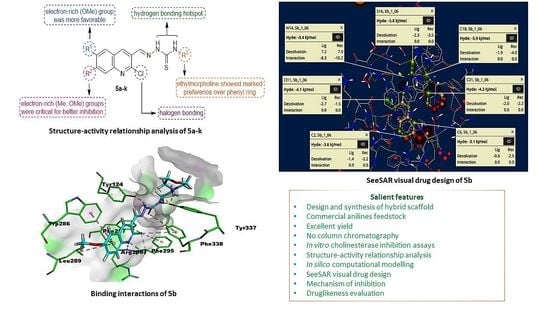
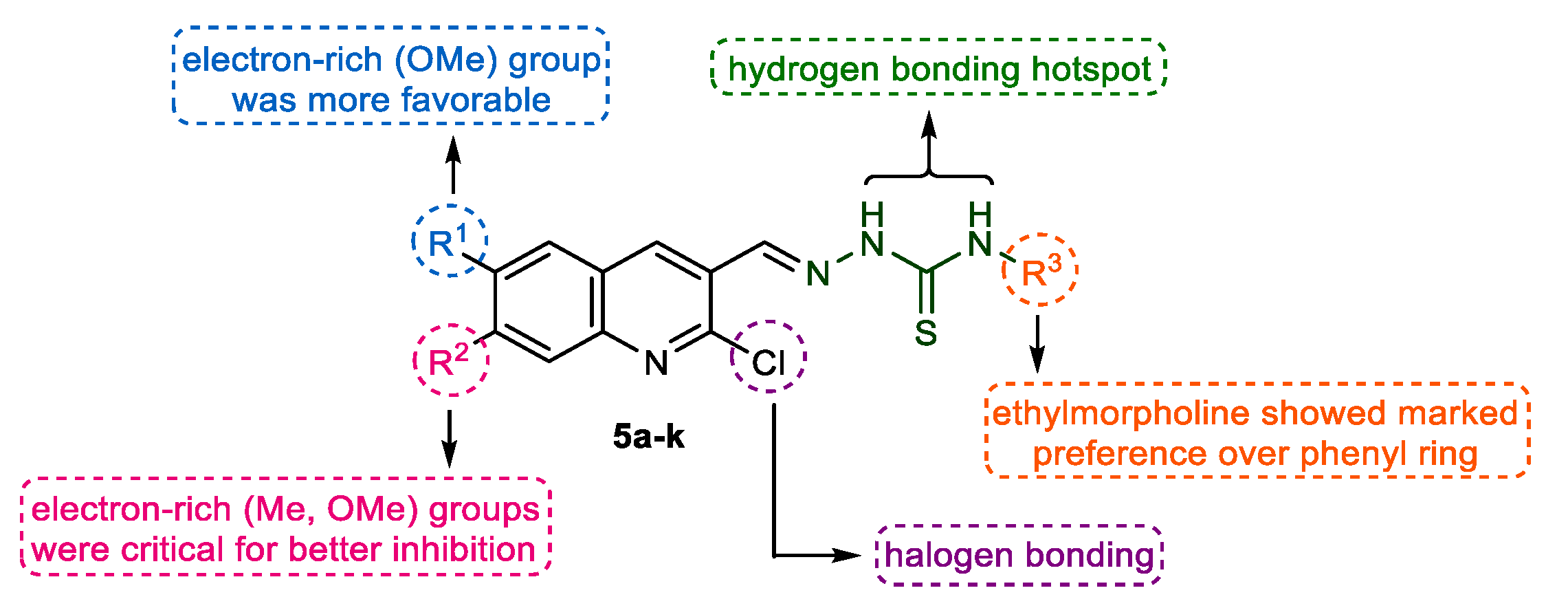
2.3. In Vitro Cholinesterase Inhibition and Structure–Activity Relationship Analyses
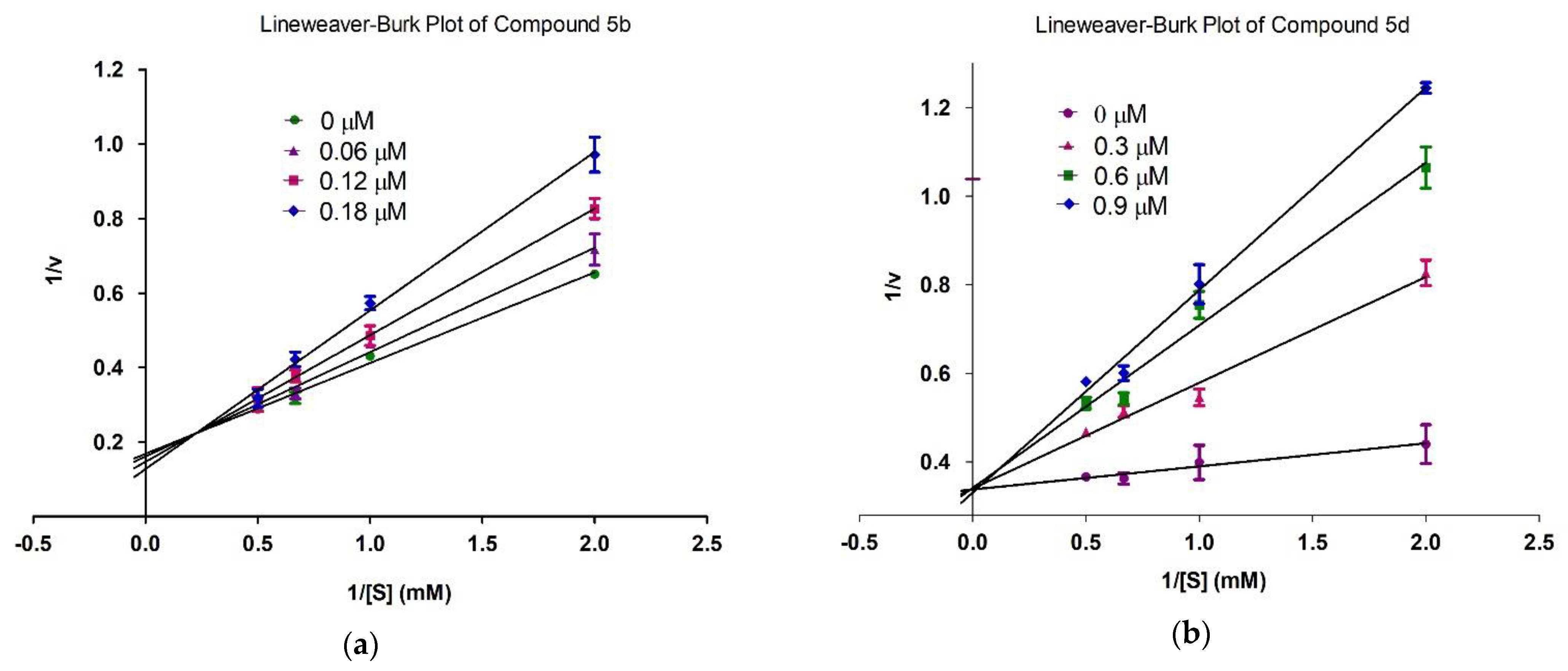
2.4. Mechanism of Inhibition
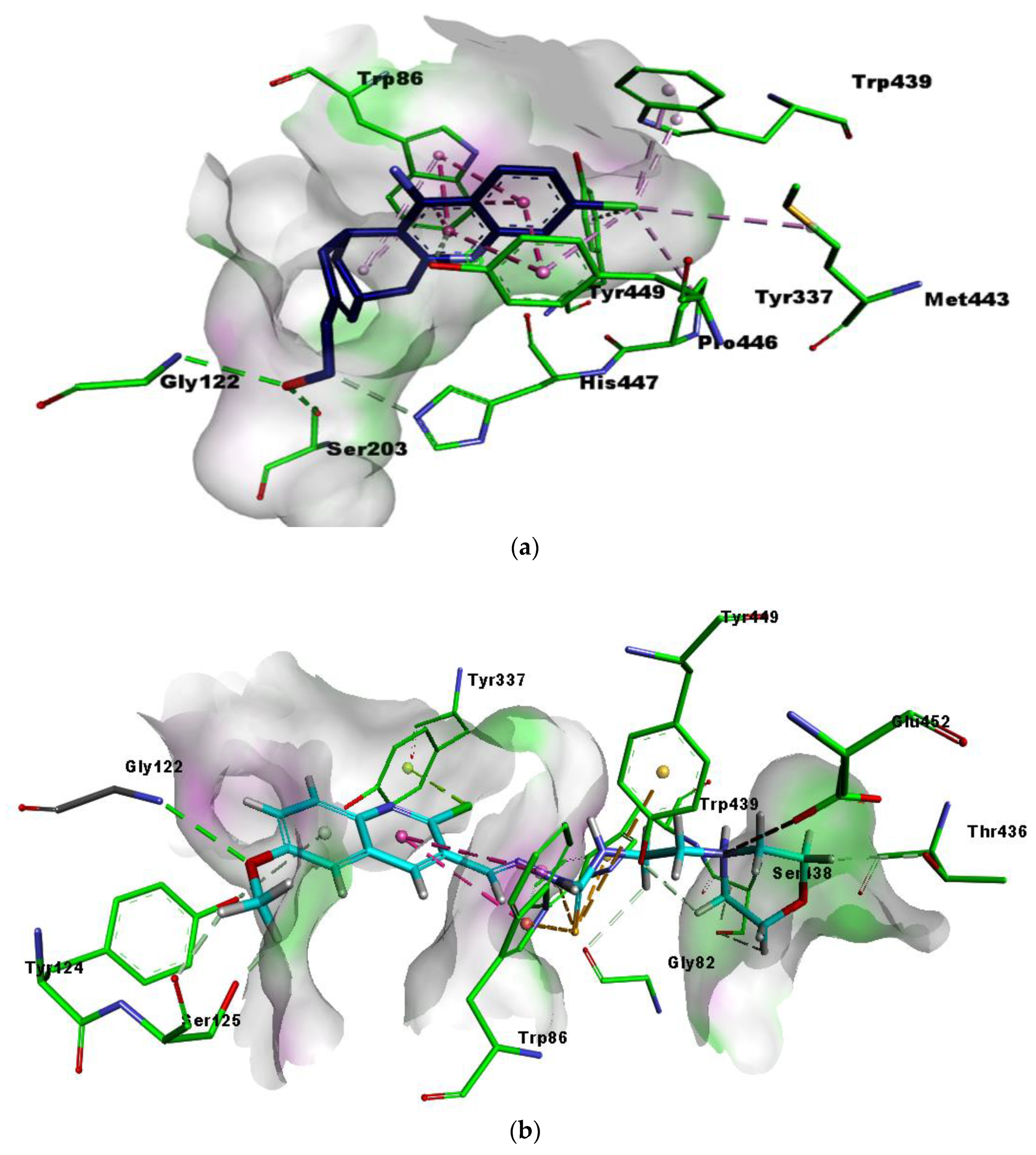
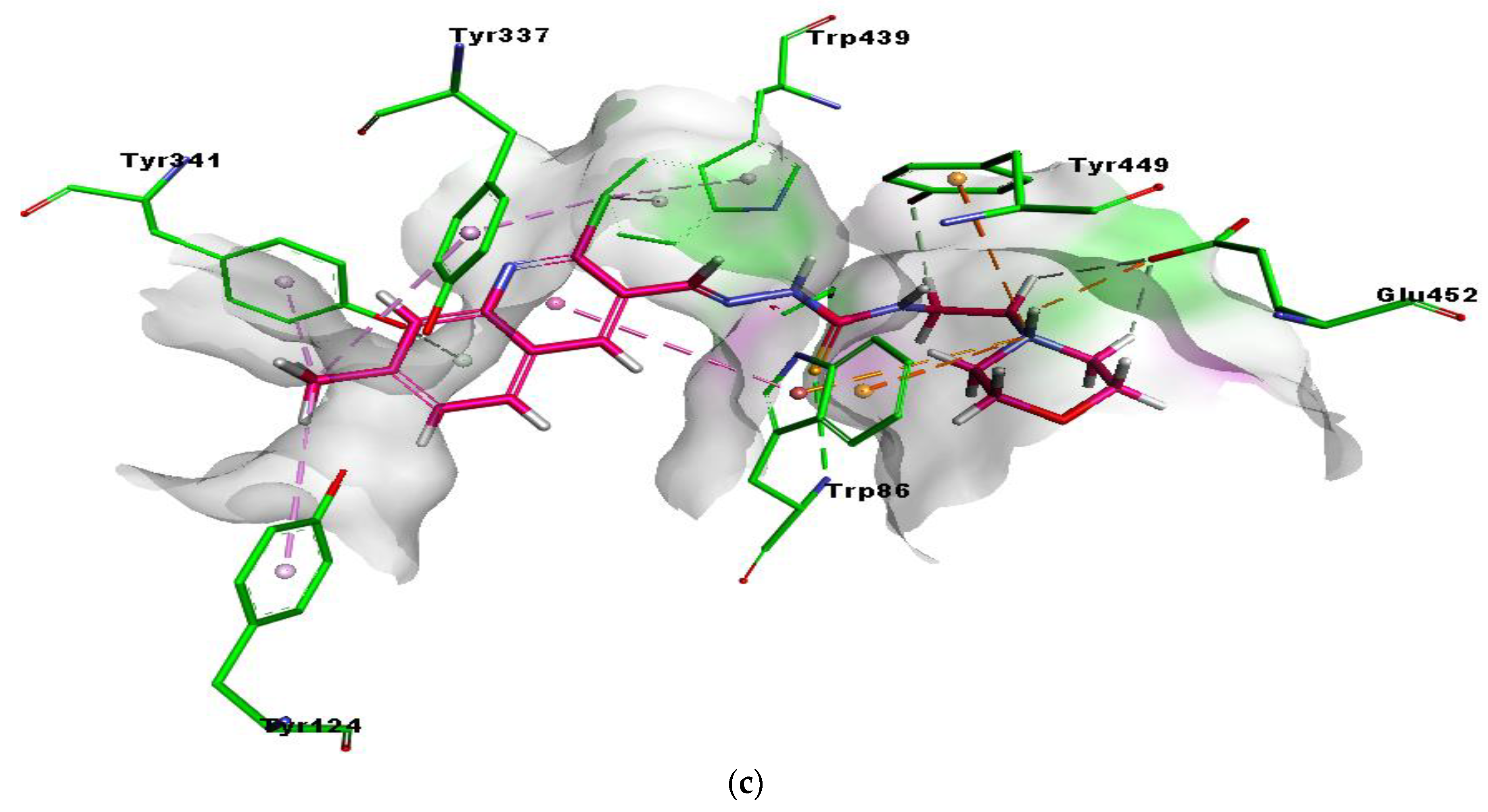
2.5. Molecular Docking Studies
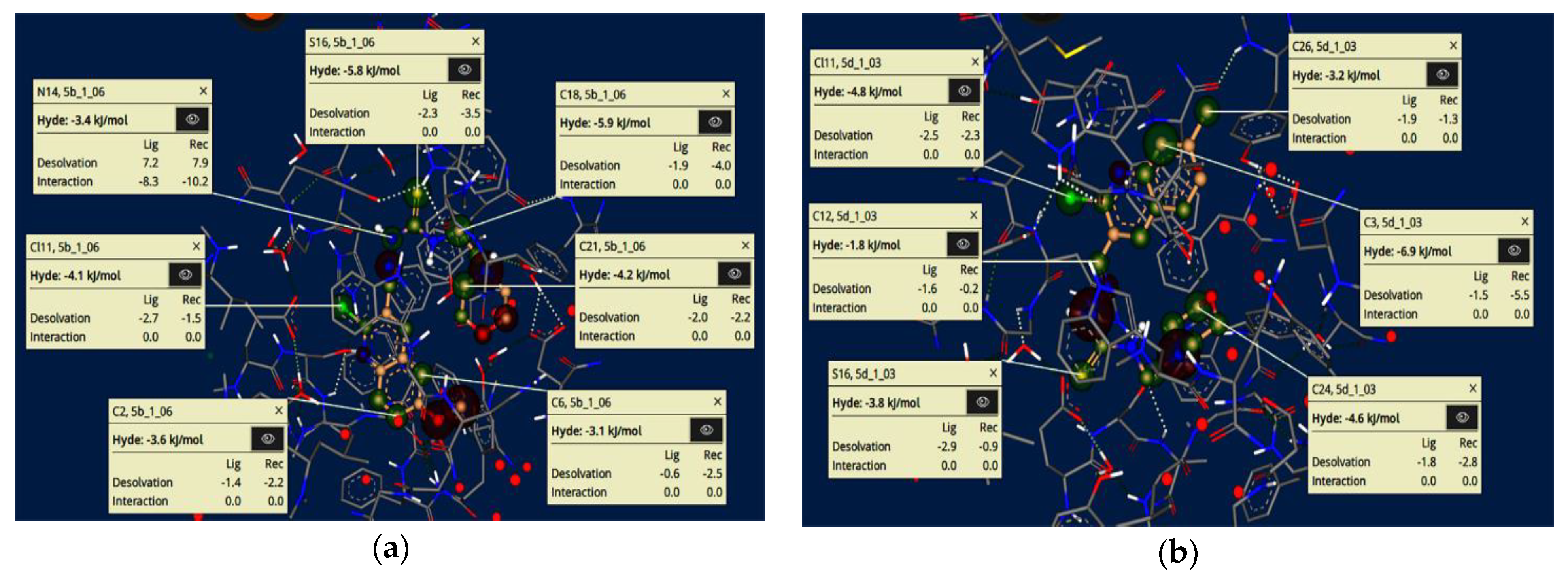
2.6. HYDE Assessment of Potent Inhibitors against Acetylcholinesterase
2.7. SeeSAR Visual Drug Design
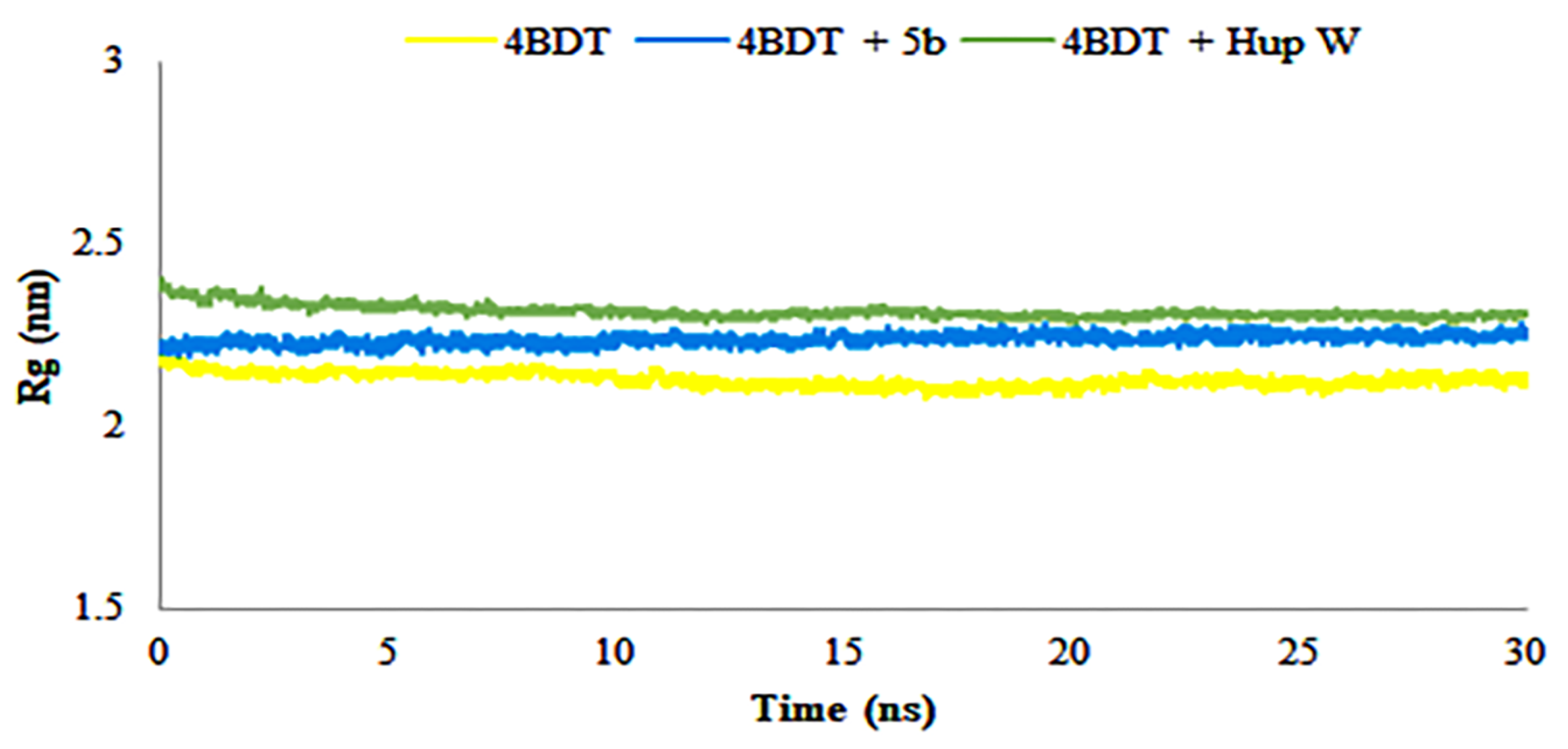
2.8. Molecular Dynamics Simulations
2.9. ADME Properties
3. Materials and Methods
3.1. General
3.2. Preparation of Acetanilides 2a–f
3.3. Preparation of 2-Chloroquinoline-3-carbaldehydes 3a–f
3.4. General Procedure for the Preparation of Quinoline-Thiosemicarbazones 5a–k
3.5. In Vitro Cholinesterase Inhibition Assay
3.6. Kinetics Studies
3.7. Molecular Docking Protocols
3.7.1. Structure Selection and Preparation
3.7.2. Compounds Preparation
3.7.3. Docking Studies
3.8. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sameem, B.; Saeedi, M.; Mahdavi, M.; Shafiee, A. A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 128, 332–345. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Albuquerque, H.M.; Cardoso, S.M.; Silva, A.M.; Silva, V.L. Dual-target compounds for Alzheimer’s disease: Natural and synthetic AChE and BACE-1 dual-inhibitors and their structure-activity relationship (SAR). Eur. J. Med. Chem. 2021, 221, 113492. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Singh, N.; Fišar, Z.; Ghosh, K.K. Progress in drug development for Alzheimer’s disease: An overview in relation to mitochondrial energy metabolism. Eur. J. Med. Chem. 2016, 121, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Kukreja, H.; Chugh, R.; Silakari, O.; Singh, D. Acetylcholinesterase inhibitors as Alzheimer therapy: From nerve toxins to neuroprotection. Eur. J. Med. Chem. 2013, 70, 165–188. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Wang, L.; Bharti; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef]
- Beato, A.; Gori, A.; Boucherle, B.; Peuchmaur, M.; Haudecoeur, R. β-Carboline as a Privileged Scaffold for Multitarget Strategies in Alzheimer’s Disease Therapy. J. Med. Chem. 2021, 64, 1392–1422. [Google Scholar] [CrossRef]
- Singh, Y.P.; Rai, H.; Singh, G.; Singh, G.K.; Mishra, S.; Kumar, S.; Srikrishna, S.; Modi, G. A review on ferulic acid and analogs based scaffolds for the management of Alzheimer’s disease. Eur. J. Med. Chem. 2021, 215, 113278. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef]
- Bortolami, M.; Pandolfi, F.; de Vita, D.; Carafa, C.; Messore, A.; Di Santo, R.; Feroci, M.; Costi, R.; Chiarotto, I.; Bagetta, D.; et al. New deferiprone derivatives as multi-functional cholinesterase inhibitors: Design, synthesis and in vitro evaluation. Eur. J. Med. Chem. 2020, 198, 112350. [Google Scholar] [CrossRef]
- Grutzendler, J.; Morris, J.C. Cholinesterase inhibitors for Alzheimer’s disease. Drugs 2001, 61, 41–52. [Google Scholar] [CrossRef]
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310. [Google Scholar] [CrossRef]
- Jalili-Baleh, L.; Babaei, E.; Abdpour, S.; Bukhari, S.N.A.; Foroumadi, A.; Ramazani, A.; Sharifzadeh, M.; Abdollahi, M.; Khoobi, M. A review on flavonoid-based scaffolds as multi-target-directed ligands (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 152, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Gupta, H. Biological Activities of Quinoline Derivatives. Mini-Rev. Med. Chem. 2009, 9, 1648–1654. [Google Scholar] [CrossRef]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Kumar, S.; Drabu, S.; Kumar, R. Structural modifications of quinoline-based antimalarial agents: Recent developments. J. Pharm. Bioallied Sci. 2010, 2, 64–71. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; Loke, P.L.; Malone, J.F.; McRoberts, W.C.; Hamilton, J.T.G. Synthesis, structure and stereochemistry of quinoline alkaloids from Choisya ternata. Org. Biomol. Chem. 2007, 5, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Cretton, S.; Breant, L.; Pourrez, L.; Ambuehl, C.; Marcourt, L.; Ebrahimi, S.N.; Hamburger, M.; Perozzo, R.; Karimou, S.; Kaiser, M.; et al. Antitrypanosomal Quinoline Alkaloids from the Roots of Waltheria indica. J. Nat. Prod. 2014, 77, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Nainwal, L.M.; Tasneem, S.; Akhtar, W.; Verma, G.; Khan, M.F.; Parvez, S.; Shaquiquzzaman, M.; Akhter, M.; Alam, M.M. Green recipes to quinoline: A review. Eur. J. Med. Chem. 2018, 164, 121–170. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, R.; Ali, R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2014, 97, 871–910. [Google Scholar] [CrossRef]
- Hu, Y.-Q.; Gao, C.; Zhang, S.; Xu, L.; Xu, Z.; Feng, L.-S.; Wu, X.; Zhao, F. Quinoline hybrids and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2017, 139, 22–47. [Google Scholar] [CrossRef]
- Kaur, R.; Kumar, K. Synthetic and medicinal perspective of quinolines as antiviral agents. Eur. J. Med. Chem. 2021, 215, 113220. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; La Monica, G.; Bono, A.; Martorana, A. Quinoline anticancer agents active on DNA and DNA-interacting proteins: From classical to emerging therapeutic targets. Eur. J. Med. Chem. 2021, 220, 113555. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Shah, S.J.A.; Ejaz, S.A.; Ibrar, A.; Hameed, S.; Lecka, J.; Millán, J.L.; Sévigny, J.; Iqbal, J. Investigation of quinoline-4-carboxylic acid as a highly potent scaffold for the development of alkaline phosphatase inhibitors: Synthesis, SAR analysis and molecular modelling studies. RSC Adv. 2015, 5, 64404–64413. [Google Scholar] [CrossRef]
- Tomassoli, I.; Ismaili, L.; Pudlo, M.; Ríos, C.D.L.; Soriano, E.; Colmena, I.; Gandía, L.; Rivas, L.; Samadi, A.; Marco-Contelles, J.; et al. Synthesis, biological assessment and molecular modeling of new dihydroquinoline-3-carboxamides and dihydroquinoline-3-carbohydrazide derivatives as cholinesterase inhibitors, and Ca channel antagonists. Eur. J. Med. Chem. 2011, 46, 1–10. [Google Scholar] [CrossRef]
- Pashaei, H.; Rouhani, A.; Nejabat, M.; Hadizadeh, F.; Mirzaei, S.; Nadri, H.; Maleki, M.F.; Ghodsi, R. Synthesis and molecular dynamic simulation studies of novel N-(1-benzylpiperidin-4-yl) quinoline-4-carboxamides as potential acetylcholinesterase inhibitors. J. Mol. Struct. 2021, 1244, 130919. [Google Scholar] [CrossRef]
- Mo, J.; Yang, H.; Chen, T.; Li, Q.; Lin, H.; Feng, F.; Liu, W.; Qu, W.; Guo, Q.; Chi, H.; et al. Design, synthesis, biological evaluation, and molecular modeling studies of quinoline-ferulic acid hybrids as cholinesterase inhibitors. Bioorganic Chem. 2019, 93, 103310. [Google Scholar] [CrossRef]
- Cai, R.; Wang, L.-N.; Fan, J.-J.; Geng, S.-Q.; Liu, Y.-M. New 4-N-phenylaminoquinoline derivatives as antioxidant, metal chelating and cholinesterase inhibitors for Alzheimer’s disease. Bioorganic Chem. 2019, 93, 103328. [Google Scholar] [CrossRef]
- Scarim, C.B.; Jornada, D.H.; Machado, M.G.M.; Ferreira, C.M.R.; Santos, J.L.; Chung, M.C. Thiazole, thio and semicarbazone derivatives against tropical infective diseases: Chagas disease, human African trypanosomiasis (HAT), leishmaniasis, and malaria. Eur. J. Med. Chem. 2018, 162, 378–395. [Google Scholar] [CrossRef]
- He, Z.; Qiao, H.; Yang, F.; Zhou, W.; Gong, Y.; Zhang, X.; Wang, H.; Zhao, B.; Ma, L.; Liu, H.-M.; et al. Novel thiosemicarbazone derivatives containing indole fragment as potent and selective anticancer agent. Eur. J. Med. Chem. 2019, 184, 111764. [Google Scholar] [CrossRef] [PubMed]
- Palanimuthu, D.; Poon, R.; Sahni, S.; Anjum, R.; Hibbs, D.; Lin, H.-Y.; Bernhardt, P.; Kalinowski, D.S.; Richardson, D.R. A novel class of thiosemicarbazones show multi-functional activity for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 139, 612–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrozek-Wilczkiewicz, A.; Malarz, K.; Rejmund, M.; Polanski, J.; Musiol, R. Anticancer activity of the thiosemicarbazones that are based on di-2-pyridine ketone and quinoline moiety. Eur. J. Med. Chem. 2019, 171, 180–194. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Wang, B.; Tao, Y.-Y.; Ma, Q.; Wang, H.-J.; He, Z.-X.; Wu, H.-P.; Li, Y.-H.; Zhao, B.; Ma, L.-Y.; et al. Thiosemicarbazone-based lead optimization to discover high-efficiency and low-toxicity anti-gastric cancer agents. Eur. J. Med. Chem. 2020, 199, 112349. [Google Scholar] [CrossRef]
- He, Z.-X.; Huo, J.-L.; Gong, Y.-P.; An, Q.; Zhang, X.; Qiao, H.; Yang, F.-F.; Jiao, L.-M.; Liu, H.-M.; Ma, L.-Y.; et al. Design, synthesis and biological evaluation of novel thiosemicarbazone-indole derivatives targeting prostate cancer cells. Eur. J. Med. Chem. 2020, 210, 112970. [Google Scholar] [CrossRef] [PubMed]
- de Siqueira, L.R.P.; Gomes, P.A.T.D.M.; Ferreira, L.P.D.L.; Rêgo, M.J.B.D.M.; Leite, A.C.L. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef]
- Jawaria, R.; Hussain, M.; Ahmad, H.B.; Ashraf, M.; Hussain, S.; Naseer, M.M.; Khalid, M.; Hussain, M.A.; Al-Rashida, M.; Tahir, M.N.; et al. Probing ferrocene-based thiosemicarbazones and their transition metal complexes as cholinesterase inhibitors. Inorg. Chim. Acta 2020, 508, 119658. [Google Scholar] [CrossRef]
- Ishaq, M.; Taslimi, P.; Shafiq, Z.; Khan, S.; Salmas, R.E.; Zangeneh, M.M.; Saeed, A.; Zangeneh, A.; Sadeghian, N.; Asari, A.; et al. Synthesis, bioactivity and binding energy calculations of novel 3-ethoxysalicylaldehyde based thiosemicarbazone derivatives. Bioorganic Chem. 2020, 100, 103924. [Google Scholar] [CrossRef]
- Hashmi, S.; Khan, S.; Shafiq, Z.; Taslimi, P.; Ishaq, M.; Sadeghian, N.; Karaman, H.S.; Akhtar, N.; Islam, M.; Asari, A.; et al. Probing 4-(diethylamino)-salicylaldehyde-based thiosemicarbazones as multi-target directed ligands against cholinesterases, carbonic anhydrases and α-glycosidase enzymes. Bioorganic Chem. 2020, 107, 104554. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Hanif, M.; Hussain, M.T.; Khan, A.A.; Aslam, M.A.S.; Rama, N.H.; Iqbal, J. Synthesis, Acetylcholinesterase and Alkaline Phosphatase Inhibition of Some New 1,2,4-Triazole and 1,3,4-Thiadiazole Derivatives. Aust. J. Chem. 2012, 65, 1413–1419. [Google Scholar] [CrossRef]
- Khan, I.; Ibrar, A.; Zaib, S.; Ahmad, S.; Furtmann, N.; Hameed, S.; Simpson, J.; Bajorath, J.; Iqbal, J. Active compounds from a diverse library of triazolothiadiazole and triazolothiadiazine scaffolds: Synthesis, crystal structure determination, cytotoxicity, cholinesterase inhibitory activity, and binding mode analysis. Bioorganic Med. Chem. 2014, 22, 6163–6173. [Google Scholar] [CrossRef]
- Khan, I.; Zaib, S.; Ibrar, A.; Rama, N.H.; Simpson, J.; Iqbal, J. Synthesis, crystal structure and biological evaluation of some novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines. Eur. J. Med. Chem. 2014, 78, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Bakht, S.M.; Ibrar, A.; Abbas, S.; Hameed, S.; White, J.M.; Rana, U.A.; Zaib, S.; Shahid, M.; Iqbal, J. Exploration of a library of triazolothiadiazole and triazolothiadiazine compounds as a highly potent and selective family of cholinesterase and monoamine oxidase inhibitors: Design, synthesis, X-ray diffraction analysis and molecular docking studies. RSC Adv. 2015, 5, 21249–21267. [Google Scholar] [CrossRef]
- Ibrar, A.; Khan, A.; Ali, M.; Sarwar, R.; Mehsud, S.; Farooq, U.; Halimi, S.M.A.; Khan, I.; Al-Harrasi, A. Combined in Vitro and in Silico Studies for the Anticholinesterase Activity and Pharmacokinetics of Coumarinyl Thiazoles and Oxadiazoles. Front. Chem. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Larik, F.A.; Saeed, A.; Faisal, M.; Hamdani, S.; Jabeen, F.; Channar, P.A.; Mumtaz, A.; Khan, I.; Kazi, M.A.; Abbas, Q.; et al. Synthesis, inhibition studies against AChE and BChE, drug-like profiling, kinetic analysis and molecular docking studies of N-(4-phenyl-3-aroyl-2(3H)-ylidene) substituted acetamides. J. Mol. Struct. 2019, 1203, 127459. [Google Scholar] [CrossRef]
- Munir, R.; Zia-Ur-Rehman, M.; Murtaza, S.; Zaib, S.; Javid, N.; Awan, S.; Iftikhar, K.; Athar, M.; Khan, I. Microwave-Assisted Synthesis of (Piperidin-1-yl)quinolin-3-yl)methylene)hydrazinecarbothioamides as Potent Inhibitors of Cholinesterases: A Biochemical and In Silico Approach. Molecules 2021, 26, 656. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Recent advances in the structural library of functionalized quinazoline and quinazolinone scaffolds: Synthetic approaches and multifarious applications. Eur. J. Med. Chem. 2014, 76, 193–244. [Google Scholar] [CrossRef]
- Khan, I.; Ibrar, A.; Ahmed, W.; Saeed, A. Synthetic approaches, functionalization and therapeutic potential of quinazoline and quinazolinone skeletons: The advances continue. Eur. J. Med. Chem. 2014, 90, 124–169. [Google Scholar] [CrossRef]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: An update on the development of synthetic methods and pharmacological diversification. Bioorganic Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef]
- Arshad, F.; Khan, M.F.; Akhtar, W.; Alam, M.M.; Nainwal, L.M.; Kaushik, S.K.; Akhter, M.; Parvez, S.; Hasan, S.M.; Shaquiquzzaman, M. Revealing quinquennial anticancer journey of morpholine: A SAR based review. Eur. J. Med. Chem. 2019, 167, 324–356. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-Q.; Wang, Z.-C.; Qi, P.-F.; Li, G.; Zhu, H.-L. Design, synthesis and biological evaluation of 2-H pyrazole derivatives containing morpholine moieties as highly potent small molecule inhibitors of APC–Asef interaction. Eur. J. Med. Chem. 2019, 177, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Doan, P.; Karjalainen, A.; Chandraseelan, J.G.; Sandberg, O.; Yli-Harja, O.; Rosholm, T.; Franzen, R.; Candeias, N.R.; Kandhavelu, M. Synthesis and biological screening for cytotoxic activity of N-substituted indolines and morpholines. Eur. J. Med. Chem. 2016, 120, 296–303. [Google Scholar] [CrossRef]
- Marvadi, S.K.; Krishna, V.S.; Sriram, D.; Kantevari, S. Synthesis of novel morpholine, thiomorpholine and N-substituted piperazine coupled 2-(thiophen-2-yl)dihydroquinolines as potent inhibitors of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2018, 164, 171–178. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Z.-C.; Li, X.; Abbas, M.; Wu, S.-Y.; Ren, S.-Z.; Liu, Q.-X.; Liu, Y.; Chen, P.-W.; Duan, Y.-T.; et al. Design, synthesis and evaluation of novel diaryl-1,5-diazoles derivatives bearing morpholine as potent dual COX-2/5-LOX inhibitors and antitumor agents. Eur. J. Med. Chem. 2019, 169, 168–184. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Rhouati, S.; Tarnowski, B.; Robinson, A. A versatile new synthesis of quinolines and related fused pyridines, Part 5. The synthesis of 2-chloroquinoline-3-carbaldehydes. J. Chem. Soc. Perkin Trans. 1 1981, 1537–1543. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration energies in protein–ligand complexes: Methods behind the HYDE scoring function. J. Comput. Mol. Des. 2012, 27, 15–29. [Google Scholar] [CrossRef]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Khan, A.; Halim, S.A.; Khan, M.; Zaib, S.; Al-Yahyaei, B.E.M.; Al-Harrasi, A.; Ibrar, A. Utilization of the common functional groups in bioactive molecules: Exploring dual inhibitory potential and computational analysis of keto esters against α-glucosidase and carbonic anhydrase-II enzymes. Int. J. Biol. Macromol. 2020, 167, 233–244. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Mumtaz, A.; Shoaib, M.; Zaib, S.; Shah, M.S.; Bhatti, H.A.; Saeed, A.; Hussain, I.; Iqbal, J. Synthesis, molecular modelling and biological evaluation of tetrasubstituted thiazoles towards cholinesterase enzymes and cytotoxicity studies. Bioorganic Chem. 2018, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Protonate 3D, Chemical Computing Group. 2007. Available online: http://www.chemcomp.com/journal/proton.htm (accessed on 10 September 2021).
- Chemical Computing Group’s Molecular Operating Environment (MOE). MOE 2019. Available online: http://www.chemcomp.com/MOEMolecular_Operating_Environment.htm (accessed on 10 September 2021).
- LeadIT Version 2.3.2; BioSolveIT GmbH: Sankt Augustin, Germany, 2017; Available online: www.biosolveit.de/LeadIT (accessed on 10 September 2021).
- BIOVIA. Discovery Studio Client v19.1.0.18287. In Accelrys Discovery Studio; Accelrys Software Inc.: San Diego, CA, USA, 2019. [Google Scholar]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Insights on P-Glycoprotein’s Efflux Mechanism Obtained by Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Ozgeris, B.; Göksu, S.; Köse, L.P.; Gülçin, I.; Salmas, R.E.; Durdagi, S.; Tümer, F.; Supuran, C.T. Acetylcholinesterase and carbonic anhydrase inhibitory properties of novel urea and sulfamide derivatives incorporating dopaminergic 2-aminotetralin scaffolds. Bioorganic Med. Chem. 2016, 24, 2318–2329. [Google Scholar] [CrossRef]
- Mathew, B.; Haridas, A.; Uçar, G.; Baysal, I.; Adeniyi, A.A.; Soliman, M.; Joy, M.; Mathew, G.E.; Lakshmanan, B.; Jayaprakash, V. Exploration of chlorinated thienyl chalcones: A new class of monoamine oxidase-B inhibitors. Int. J. Biol. Macromol. 2016, 91, 680–695. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE, Version 5.1.19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Substituent | Acetylcholinesterase(AChE) | Butyrylcholinesterse(BChE) | ||
|---|---|---|---|---|---|
| R1 | R2 | R3 | IC50 ± SEM (μM)/%inhibition | ||
| 5a | H | H | ethylmorpholine | 2.95 ± 0.24 | 3.32% |
| 5b | OMe | H | ethylmorpholine | 0.12 ± 0.02 | 8.12% |
| 5c | H | OMe | ethylmorpholine | 5.53 ± 0.11 | 1.33% |
| 5d | H | Me | ethylmorpholine | 0.55 ± 0.01 | 1.52% |
| 5e | H | Cl | ethylmorpholine | 10.5 ± 0.16 | 1.88% |
| 5f | H | H | Ph | 23.2 ± 1.28 | 4.47% |
| 5g | OMe | H | Ph | 34.2 ± 1.02 | 11.3 ± 0.67 |
| 5h | Me | H | Ph | 49.3 ± 2.49 | 32.6% |
| 5i | H | OMe | Ph | 60.9 ± 6.57 | 24.0% |
| 5j | H | Me | Ph | 46.5 ± 3.12 | 16.8% |
| 5k | H | Cl | Ph | 47.1 ± 1.45 | 20.3% |
| Galantamine | ― | ― | ― | 0.62 ± 0.01 | 0.87 ± 0.03 |
| Compounds | Binding interactions | |||
|---|---|---|---|---|
| Ligand Atom | Receptor Atom | Interaction Type | Distance (Å) | |
| 5b | O26 | GLY122 | H-bond | 2.82 |
| phenyl ring | TRP86 | π-π T-shaped & π-sulfur | 6.23 & 4.34 | |
| Cl11 | TYR337 | π-lone pair | 2.80 | |
| S16 | TRP439 | π-sulfur | 4.68 | |
| N20 | GLU452 | Attractive charges | 4.93 | |
| S16 | TYR449 | π-sulfur | 5.86 | |
| 5d | N20 | GLU452 | salt bridge | 2.69 |
| N20 | TYR449 | π-cation | 4.78 | |
| S16, phenyl ring & N20 | TRP86 | H-bond, π-π T-shaped & π-cation | 3.30, 5.43 & 5.26 | |
| Cl11 | TRP439 | π-alkyl | 5.43 | |
| Methyl & Cl11 | TYR337 | π-alkyl | 5.14 & 3.32 | |
| Methyl | TYR124 | π-alkyl | 5.37 | |
| Methyl | TYR341 | π-alkyl | 3.76 | |
| Hup W | O1 | GLY122 | H-bond | 2.96 |
| O1 | SER203 | H-bond | 2.33 | |
| Quinoline ring | TRP86 | π-π Stacked & π-alkyl | 4.41, 5.30, 4.00, 3.69 & 4.18, 4.87 | |
| Cl1 | PRO446 | Alkyl | 4.52 | |
| Cl1 | TYR337 | π-π Stacked & π-alkyl, π-donor | 4.47, 2.54 & 4.45, 4.01 | |
| Cl1 | MET443 | Alkyl | 4.89 | |
| Cl1 | TRP439 | π-alkyl | 3.46 & 3.80 | |
| Cl1 | TYR449 | π-alkyl | 5.38 | |
| Compound | Docking Score by FlexX | Pose Rank | Binding Free Energy ΔG (kJ/mol) |
|---|---|---|---|
| 5a | −22.12 | 1 | −17 |
| 5b | −26.58 | 2 | −25 |
| 5c | −21.56 | 2 | −16 |
| 5d | −23.01 | 3 | −18 |
| 5e | −19.67 | 2 | −16 |
| 5f | −14.55 | 4 | −12 |
| 5g | −14.62 | 3 | −10 |
| 5h | −13.69 | 1 | −8 |
| 5i | −12.58 | 2 | −9 |
| 5j | −14.24 | 1 | −10 |
| 5k | −13.99 | 2 | −9 |
| Huprine W | −16.29 | 1 | −23 |
| Properties. | Compounds | |
|---|---|---|
| 5b | 5d | |
| Physicochemical Properties | ||
| Molecular weight (g/mol) | 407.92 | 391.92 |
| No. of heavy atoms | 27 | 26 |
| No. of aromatic heavy atoms | 10 | 10 |
| Fraction Csp3 | 0.39 | 0.39 |
| No. of rotatable bonds | 8 | 7 |
| No. of H-bond acceptors | 5 | 4 |
| No. of H-bond donors | 2 | 2 |
| Molar refractivity | 114.72 | 113.20 |
| TPSA (Å2) | 103.10 | 93.87 |
| Lipophilicity | ||
| Log Po/w (iLOGP) | 3.65 | 3.21 |
| Log Po/w (XLOGP3) | 2.47 | 2.86 |
| Log Po/w (WLOGP) | 1.65 | 1.95 |
| Log Po/w (MLOGP) | 0.97 | 1.50 |
| Log Po/w (SILICOS-IT) | 3.96 | 4.41 |
| Consensus Log Po/w | 2.54 | 2.79 |
| Water Solubility | ||
| Log S (ESOL) | −3.67 | −3.89 |
| Solubility (mg/mL; mol/L) | 8.70 × 10−2; 2.13 × 10−4 | 5.00 × 10−2; 1.28 × 10−4 |
| Class | Soluble | Soluble |
| Log S (ALi) | −4.28 | −4.49 |
| Solubility(mg/ml; mol/l) | 2.14 × 10−2; 5.26 × 10−5 | 1.27 × 10−2; 3.23 × 10−5 |
| Class | Moderately soluble | Moderately soluble |
| Log S (SILICOS-IT) | −5.70 | −5.98 |
| Solubility (mg/ml; mol/l) | 8.06 × 10−4; 1.98 × 10−6 | 4.13 × 10−4; 1.05 × 10−6 |
| Class | Moderately soluble | Moderately soluble |
| Pharmacokinetics | ||
| GI absorption | High | High |
| BBB permeant | No | No |
| P-gp substrate | Yes | Yes |
| CYP1A2 inhibitor | Yes | Yes |
| CYP2C19 inhibitor | Yes | Yes |
| CYP2C9 inhibitor | Yes | Yes |
| CYP2D6 inhibitor | No | Yes |
| CYP3A4 inhibitor | Yes | Yes |
| Log Kp (skin permeation) (cm/s) | −7.03 | −6.66 |
| Druglikeness | ||
| Lipinski | Yes; 0 violation | Yes; 0 violation |
| Ghose | Yes; 0 violation | Yes; 0 violation |
| Veber | Yes; 0 violation | Yes; 0 violation |
| Egan | Yes; 0 violation | Yes; 0 violation |
| Muegge | Yes; 0 violation | Yes; 0 violation |
| Bioavailability score | 0.55 | 0.55 |
| Medicinal Chemistry | ||
| PAINS | 0 alert | 0 alert |
| Brenk | 3 alerts: 2-halo_pyridine, imine_1, thiocarbonyl_group | 3 alerts: 2-halo_pyridine, imine_1, thiocarbonyl_group |
| Leadlikeness | No; 2 Violations: MW > 350, Rotors > 7 | No; 1 Violation MW > 350 |
| Synthetic accessibility | 3.16 | 3.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaib, S.; Munir, R.; Younas, M.T.; Kausar, N.; Ibrar, A.; Aqsa, S.; Shahid, N.; Asif, T.T.; Alsaab, H.O.; Khan, I. Hybrid Quinoline-Thiosemicarbazone Therapeutics as a New Treatment Opportunity for Alzheimer’s Disease‒Synthesis, In Vitro Cholinesterase Inhibitory Potential and Computational Modeling Analysis. Molecules 2021, 26, 6573. https://doi.org/10.3390/molecules26216573
Zaib S, Munir R, Younas MT, Kausar N, Ibrar A, Aqsa S, Shahid N, Asif TT, Alsaab HO, Khan I. Hybrid Quinoline-Thiosemicarbazone Therapeutics as a New Treatment Opportunity for Alzheimer’s Disease‒Synthesis, In Vitro Cholinesterase Inhibitory Potential and Computational Modeling Analysis. Molecules. 2021; 26(21):6573. https://doi.org/10.3390/molecules26216573
Chicago/Turabian StyleZaib, Sumera, Rubina Munir, Muhammad Tayyab Younas, Naghmana Kausar, Aliya Ibrar, Sehar Aqsa, Noorma Shahid, Tahira Tasneem Asif, Hashem O. Alsaab, and Imtiaz Khan. 2021. "Hybrid Quinoline-Thiosemicarbazone Therapeutics as a New Treatment Opportunity for Alzheimer’s Disease‒Synthesis, In Vitro Cholinesterase Inhibitory Potential and Computational Modeling Analysis" Molecules 26, no. 21: 6573. https://doi.org/10.3390/molecules26216573
APA StyleZaib, S., Munir, R., Younas, M. T., Kausar, N., Ibrar, A., Aqsa, S., Shahid, N., Asif, T. T., Alsaab, H. O., & Khan, I. (2021). Hybrid Quinoline-Thiosemicarbazone Therapeutics as a New Treatment Opportunity for Alzheimer’s Disease‒Synthesis, In Vitro Cholinesterase Inhibitory Potential and Computational Modeling Analysis. Molecules, 26(21), 6573. https://doi.org/10.3390/molecules26216573