
Identification of Novel Antagonists Targeting Cannabinoid Receptor 2 Using a Multi-Step Virtual Screening Strategy




Abstract
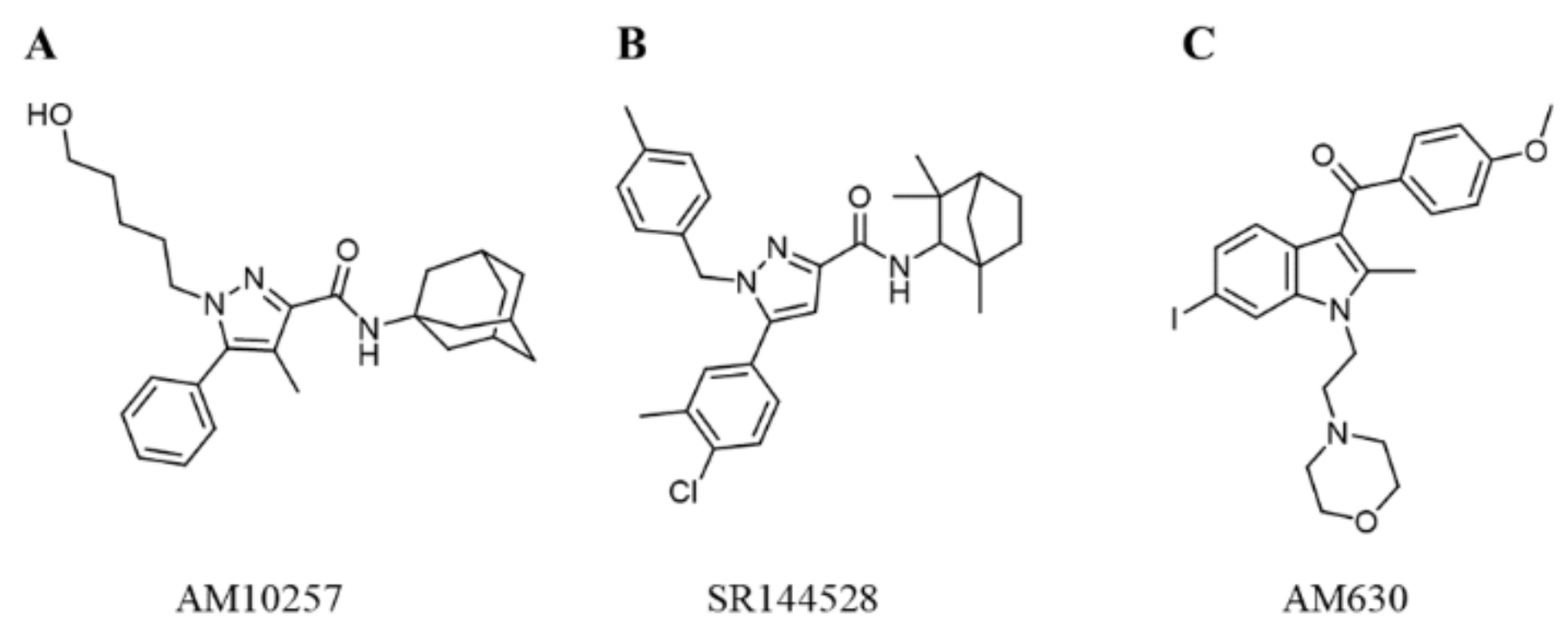
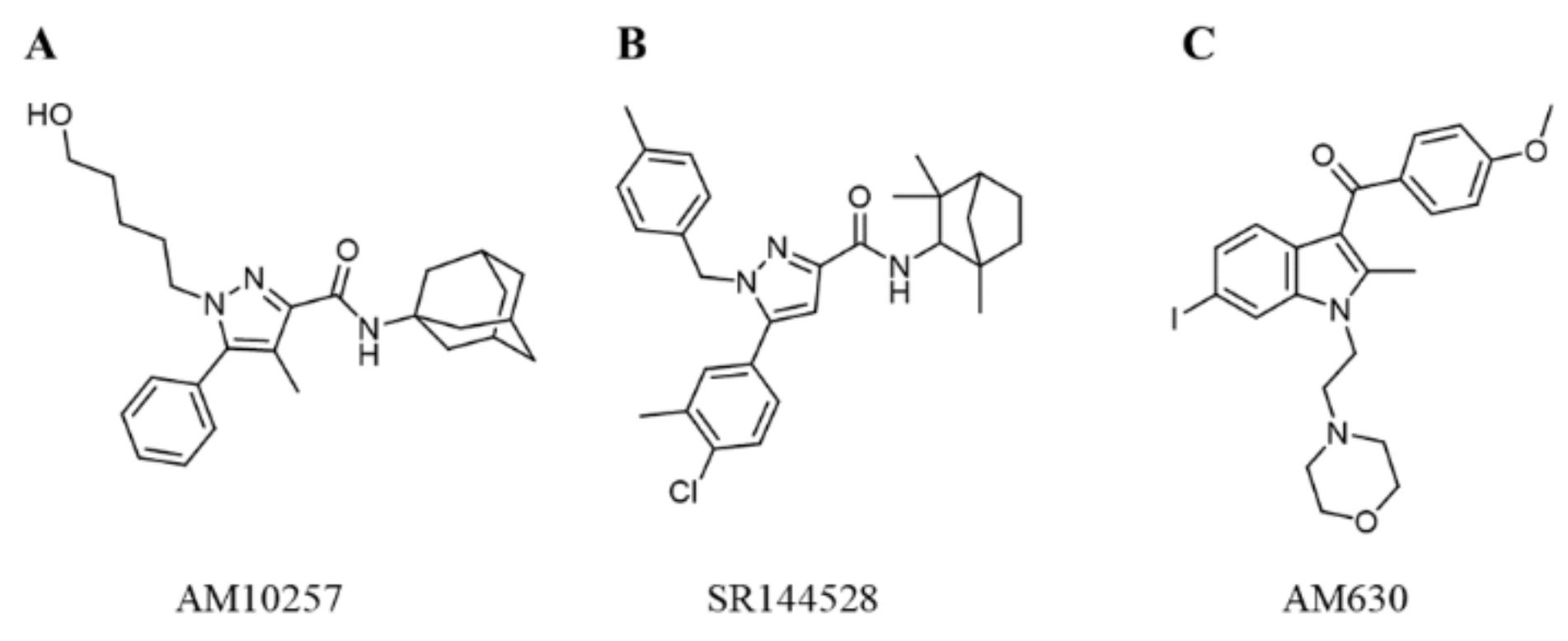
:1. Introduction
2. Results
2.1. Construction and Validation of DNN and CNN Models
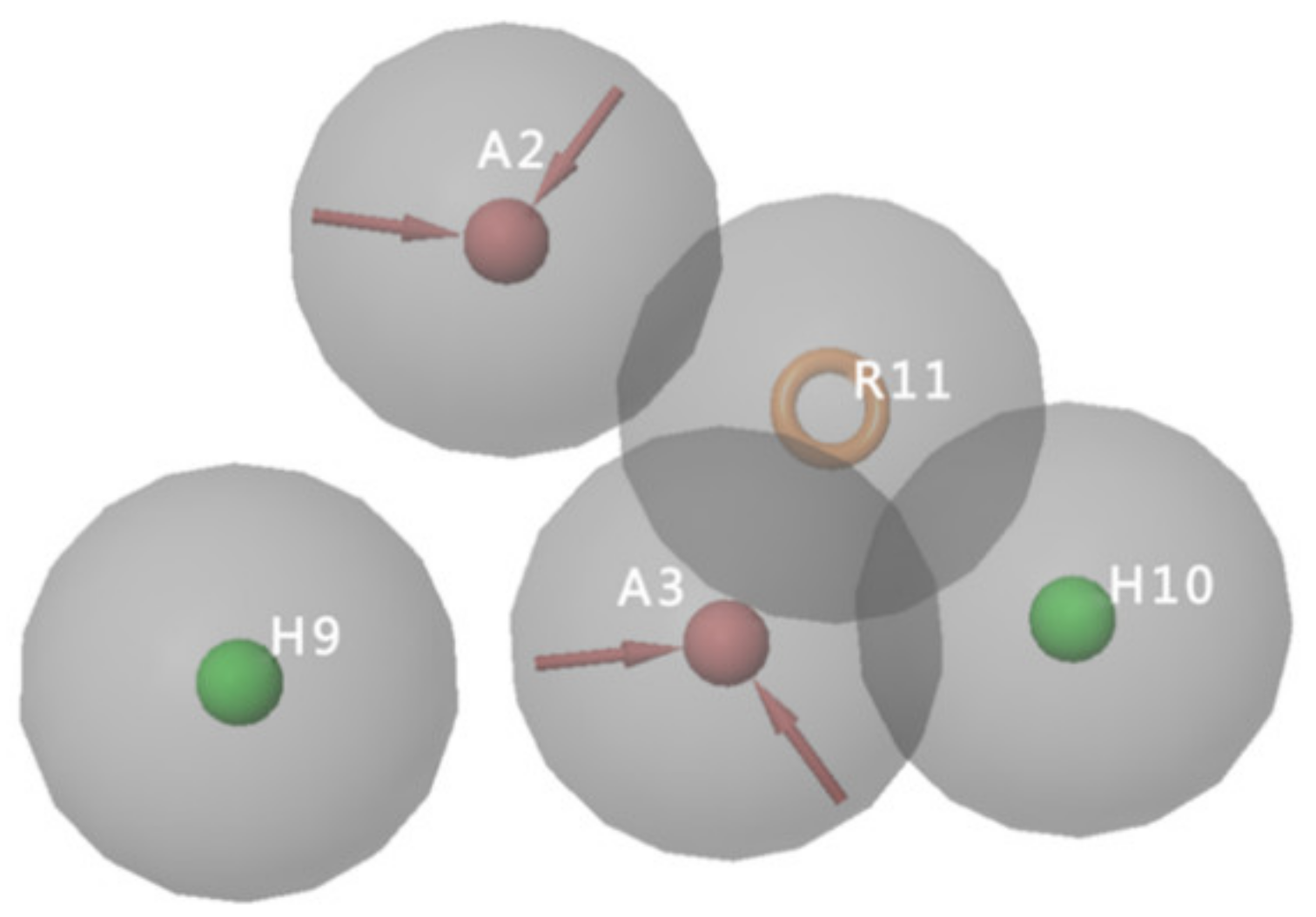
2.2. Generation of Pharmacophore Models
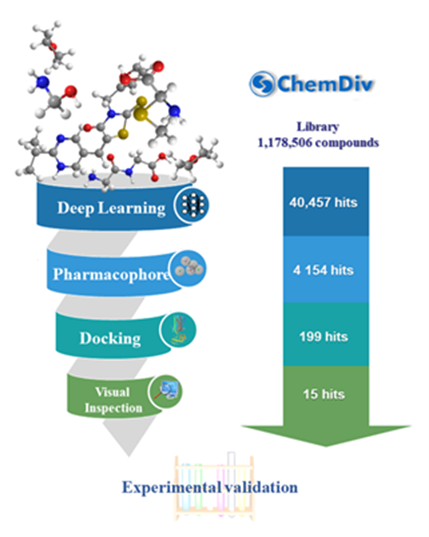
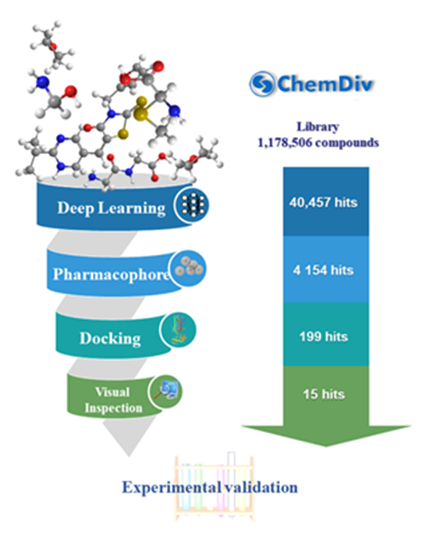
2.3. Virtual Screening
2.4. Biological Evaluation
2.4.1. CB2 Receptor Affinity Experiments
2.4.2. CB2 Receptor Functional Experiments
2.4.3. Selectivity of the Hit Compounds for the CB2 Receptor over the CB1 Receptor
2.5. Novelty Analysis of Hit Compounds
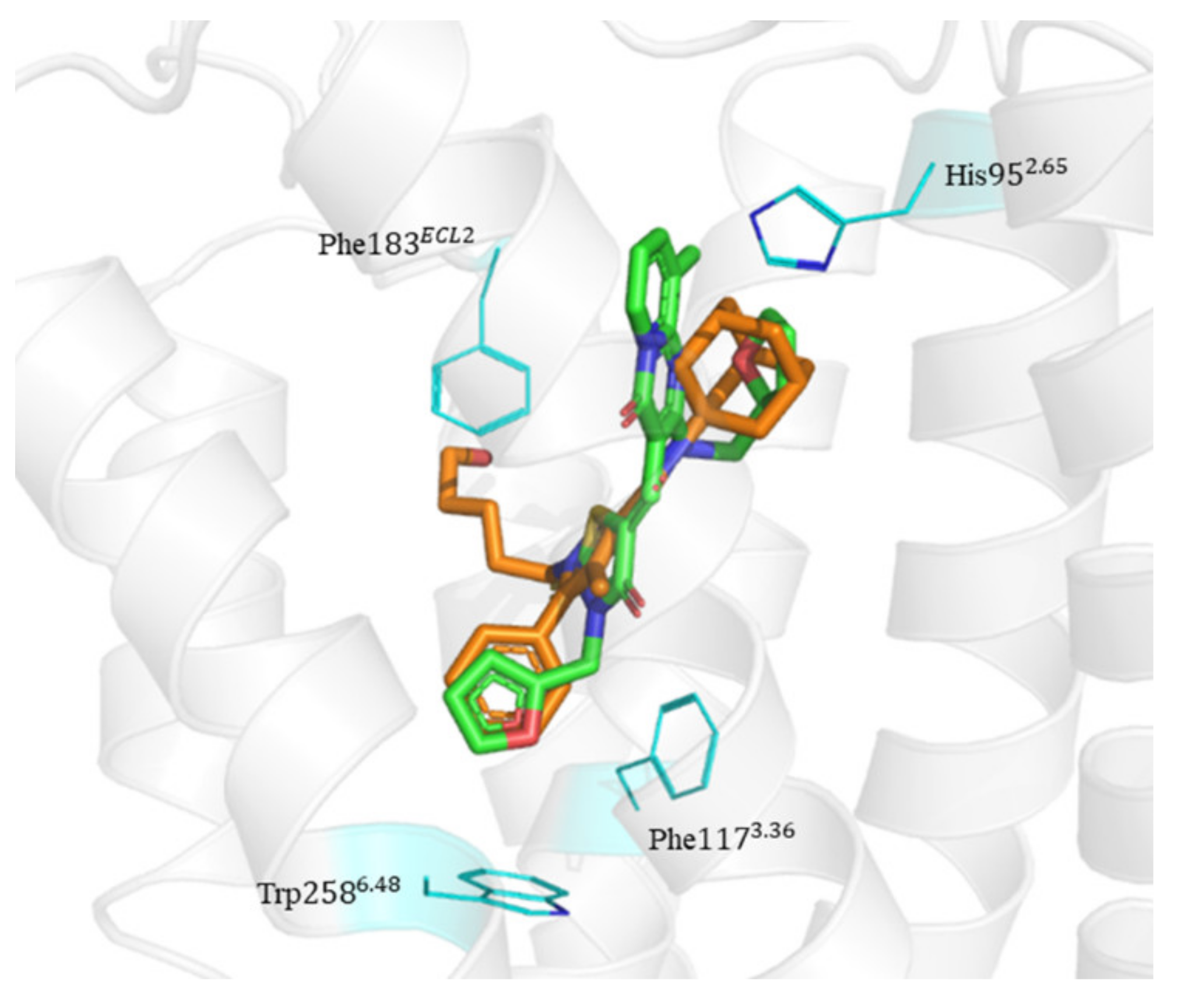
2.6. Binding Mode of Compounds 3, 7, 8, 12, and 15 in the CB2 Receptor
3. Materials and Methods
3.1. Data Sets
3.2. DNN and CNN Models
3.3. Pharmacophore Modeling
3.4. Molecular Docking
3.5. Radioligand Binding Assays
3.6. Functional Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hanlon, C.D.; Andrew, D.J. Outside-in signaling—A brief review of gpcr signaling with a focus on the drosophila gpcr family. J. Cell Sci. 2015, 128, 3533–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in gpcr drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Abood, M.E. Cb1 and cb2 receptor pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar] [PubMed]
- Atwood, B.K.; Mackie, K. Cb2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Bosier, B.; Muccioli, G.G.; Hermans, E.; Lambert, D.M. Functionally selective cannabinoid receptor signalling: Therapeutic implications and opportunities. Biochem. Pharmacol. 2010, 80, 1–12. [Google Scholar] [CrossRef]
- Turu, G.; Hunyady, L. Signal transduction of the cb1 cannabinoid receptor. J. Mol. Endocrinol. 2010, 44, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid cb1 and cb2 receptors in immune cells. Acta Physiol. 2015, 214, 63–74. [Google Scholar] [CrossRef]
- Reggio, P.H. The Cannabinoid Receptors; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Mbvundula, E.C.; Rainsford, K.; Bunning, R.A. Cannabinoids in pain and inflammation. Inflammopharmacology 2004, 12, 99–114. [Google Scholar] [CrossRef]
- Guindon, J.; Hohmann, A. Cannabinoid cb2 receptors: A therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol. 2008, 153, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Feng, R.; Milcarek, C.A.; Xie, X.-Q. Antagonism of cannabinoid receptor 2 pathway suppresses il-6-induced immunoglobulin igm secretion. BMC Pharmacol. Toxicol. 2014, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhou, S.; Yang, P.; Tian, Y.; Feng, Z.; Xie, X.-Q.; Liu, Y. Targeted inhibition of the type 2 cannabinoid receptor is a novel approach to reduce renal fibrosis. Kidney Int. 2018, 94, 756–772. [Google Scholar] [CrossRef]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Qu, L. Crystal structure of the human cannabinoid receptor cb2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi-Carmona, M.; Barth, F.; Héaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Néliat, G.; Caput, D. Sr141716a, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi-Carmona, M.; Barth, F.; Millan, J.; Derocq, J.-M.; Casellas, P.; Congy, C.; Oustric, D.; Sarran, M.; Bouaboula, M.; Calandra, B. Sr 144528, the first potent and selective antagonist of the cb2 cannabinoid receptor. J. Pharmacol. Exp. Ther. 1998, 284, 644–650. [Google Scholar]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at cb1 and cb2 cannabinoid receptors of l759633, l759656 and am630. Br. J. Pharmacol. 1999, 126, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. Chembl: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Markt, P.; Feldmann, C.; Rollinger, J.M.; Raduner, S.; Schuster, D.; Kirchmair, J.; Distinto, S.; Spitzer, G.M.; Wolber, G.; Laggner, C. Discovery of novel cb2 receptor ligands by a pharmacophore-based virtual screening workflow. J. Med. Chem. 2009, 52, 369–378. [Google Scholar] [CrossRef]
- Chen, J.-Z.; Wang, J.; Xie, X.-Q. Gpcr structure-based virtual screening approach for cb2 antagonist search. J. Chem. Inf. Model. 2007, 47, 1626–1637. [Google Scholar] [CrossRef]
- Hu, J.; Feng, Z.; Ma, S.; Zhang, Y.; Tong, Q.; Alqarni, M.H.; Gou, X.; Xie, X.-Q. Difference and influence of inactive and active states of cannabinoid receptor subtype cb2: From conformation to drug discovery. J. Chem. Inf. Model. 2016, 56, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Hou, S.; Wei, Y.; Li, D.; Lin, J. Discovery of novel dual adenosine a1/a2a receptor antagonists using deep learning, pharmacophore modeling and molecular docking. PLoS Comput. Biol. 2021, 17, e1008821. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional networks on graphs for learning molecular fingerprints. arXiv 2015, arXiv:1509.09292. [Google Scholar]
- Fillbrunn, A.; Dietz, C.; Pfeuffer, J.; Rahn, R.; Landrum, G.A.; Berthold, M.R. Knime for reproducible cross-domain analysis of life science data. J. Biotechnol. 2017, 261, 149–156. [Google Scholar] [CrossRef]
- Godden, J.W.; Xue, L.; Bajorath, J. Combinatorial preferences affect molecular similarity/diversity calculations using binary fingerprints and tanimoto coefficients. J. Chem. Inf. Comput. Sci. 2000, 40, 163–166. [Google Scholar] [CrossRef]
- Sydow, D.; Wichmann, M.; Rodríguez-Guerra, J.; Goldmann, D.; Landrum, G.; Volkamer, A. Teachopencadd-knime: A teaching platform for computer-aided drug design using knime workflows. J. Chem. Inf. Model. 2019, 59, 4083–4086. [Google Scholar] [CrossRef] [Green Version]
- Cereto-Massagué, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. Decoyfinder: An easy-to-use python gui application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. Zinc—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the opls-aa force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Bastien, F.; Lamblin, P.; Pascanu, R.; Bergstra, J.; Goodfellow, I.; Bergeron, A.; Bouchard, N.; Warde-Farley, D.; Bengio, Y. Theano: New features and speed improvements. arXiv 2012, arXiv:1211.5590. [Google Scholar]
- Baldassi, C.; Malatesta, E.M.; Zecchina, R. Properties of the geometry of solutions and capacity of multilayer neural networks with rectified linear unit activations. Phys. Rev. Lett. 2019, 123, 170602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Jamal, S.; Scaria, V. Cheminformatic models based on machine learning for pyruvate kinase inhibitors of leishmania mexicana. BMC Bioinform. 2013, 14, 329. [Google Scholar] [CrossRef] [Green Version]
- DNN and CNN Models. Available online: https://github.com/Houshujing/DNN-CNN-models (accessed on 3 November 2021).
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S. Rcsb protein data bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNN Model | Batch Size | SE | SP | Q+ | Q− | MCC | AUC |
|---|---|---|---|---|---|---|---|
| Model_D1 | 50 | 0.968 | 0.903 | 0.911 | 0.966 | 0.874 | 0.982 |
| Model_D2 | 100 | 0.975 | 0.897 | 0.906 | 0.972 | 0.875 | 0.98 |
| Model_D3 | 150 | 0.956 | 0.942 | 0.944 | 0.954 | 0.898 | 0.982 |
| Model_D4 | 200 | 0.956 | 0.923 | 0.926 | 0.953 | 0.879 | 0.982 |
| Model_D5 | 250 | 0.949 | 0.903 | 0.909 | 0.946 | 0.854 | 0.981 |
| Model_D6 | 300 | 0.975 | 0.942 | 0.945 | 0.973 | 0.917 | 0.986 |
| Model_D7 | 350 | 0.975 | 0.923 | 0.928 | 0.973 | 0.899 | 0.982 |
| CNN Model | Batch Size | SE | SP | Q+ | Q− | MCC | AUC |
|---|---|---|---|---|---|---|---|
| Model_C1 | 50 | 0.962 | 0.877 | 0.889 | 0.958 | 0.843 | 0.975 |
| Model_C2 | 100 | 0.93 | 0.877 | 0.886 | 0.925 | 0.809 | 0.971 |
| Model_C3 | 150 | 0.956 | 0.897 | 0.904 | 0.952 | 0.854 | 0.968 |
| Model_C4 | 200 | 0.937 | 0.89 | 0.897 | 0.932 | 0.828 | 0.952 |
| Model_C5 | 250 | 0.918 | 0.916 | 0.918 | 0.916 | 0.834 | 0.966 |
| Model_C6 | 300 | 0.937 | 0.884 | 0.892 | 0.932 | 0.822 | 0.96 |
| Model_C7 | 350 | 0.943 | 0.916 | 0.920 | 0.940 | 0.860 | 0.967 |
| Model_C8 | 400 | 0.93 | 0.897 | 0.902 | 0.927 | 0.828 | 0.963 |
| Hypothesis | PhaseHypoScore | EF1% | BEDROC (α-160.9) | ROC | AUAC | Total Actives | Ranked Actives | Matches |
|---|---|---|---|---|---|---|---|---|
| AAHHR_1 | 0.89 | 10.20 | 0.39 | 0.71 | 0.71 | 29 | 28 | 4 of 5 |
| AAHHR_2 | 0.89 | 10.20 | 0.34 | 0.71 | 0.71 | 29 | 28 | 4 of 5 |
| AHHHR_1 | 0.86 | 6.80 | 0.32 | 0.67 | 0.67 | 29 | 27 | 4 of 5 |
| AAHHR_4 | 0.85 | 13.59 | 0.47 | 0.68 | 0.68 | 29 | 28 | 4 of 5 |
| AAHHR_3 | 0.83 | 6.80 | 0.20 | 0.72 | 0.72 | 29 | 28 | 4 of 5 |
| HHHR_1 | 0.82 | 3.40 | 0.22 | 0.43 | 0.66 | 29 | 13 | 4 of 4 |
| HHHR_2 | 0.81 | 10.20 | 0.30 | 0.43 | 0.67 | 29 | 13 | 4 of 4 |
| AHHHR_2 | 0.79 | 6.80 | 0.31 | 0.68 | 0.69 | 29 | 27 | 4 of 5 |
| AHHR_6 | 0.78 | 10.20 | 0.42 | 0.73 | 0.77 | 29 | 24 | 4 of 4 |
| AHHR_1 | 0.77 | 13.59 | 0.39 | 0.64 | 0.70 | 29 | 22 | 4 of 4 |
| AHHR_2 | 0.76 | 3.40 | 0.14 | 0.52 | 0.59 | 29 | 21 | 4 of 4 |
| AHHHR_3 | 0.75 | 10.20 | 0.36 | 0.54 | 0.68 | 29 | 17 | 4 of 5 |
| AHHHR_4 | 0.73 | 6.80 | 0.27 | 0.65 | 0.69 | 29 | 23 | 4 of 5 |
| AHHR_7 | 0.72 | 0 | 0.01 | 0.60 | 0.68 | 29 | 21 | 4 of 4 |
| AHHR_4 | 0.68 | 0 | 0.01 | 0.51 | 0.62 | 29 | 18 | 4 of 4 |
| AHHR_3 | 0.68 | 0 | 0.00 | 0.55 | 0.65 | 29 | 19 | 4 of 4 |
| AAHR_1 | 0.67 | 0 | 0.01 | 0.56 | 0.65 | 29 | 19 | 4 of 4 |
| AHHHR_5 | 0.67 | 10.20 | 0.44 | 0.63 | 0.65 | 29 | 24 | 4 of 5 |
| AHHR_5 | 0.67 | 6.80 | 0.41 | 0.49 | 0.68 | 29 | 15 | 4 of 4 |
| AHHHR_6 | 0.66 | 6.80 | 0.31 | 0.62 | 0.64 | 29 | 25 | 4 of 5 |
| Compound Number | Compound ID | Chemical Structures | Binding Affinities pKi | cAMP Assay pIC50 | LogP | Tc |
|---|---|---|---|---|---|---|
| 1 | G748-0093 | 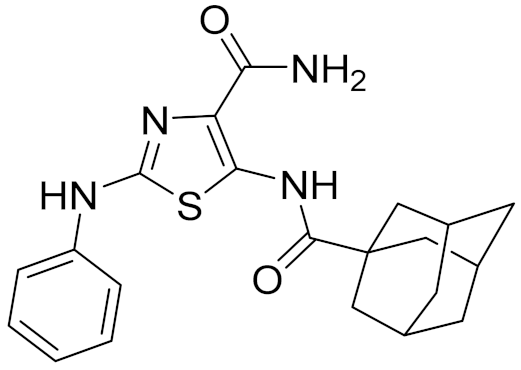 | 5.80 | <4.70 | 3.73 | |
| 2 | 8018-1162 | 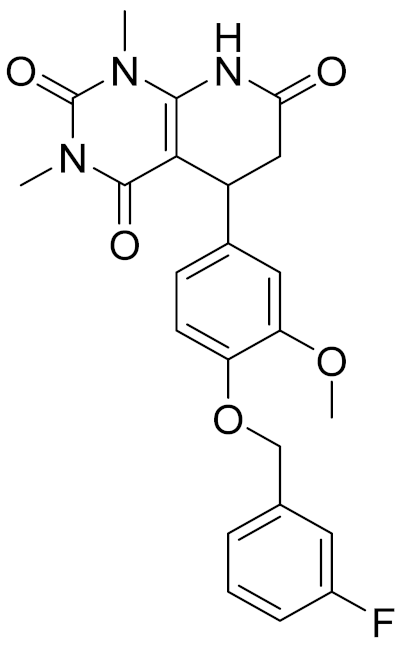 | <4 | - | 3.53 | |
| 3 | C200-3916 | 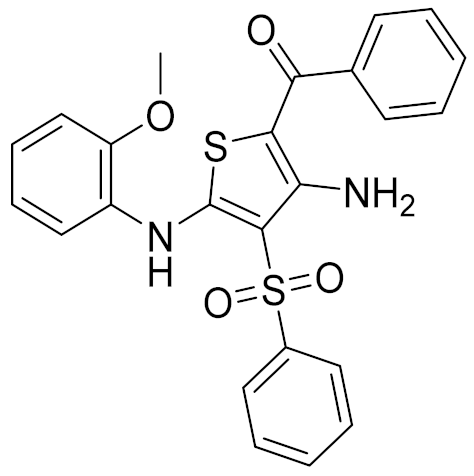 | 5.31 | 6.47 | 4.91 | 0.32 |
| 4 | C688-1110 | 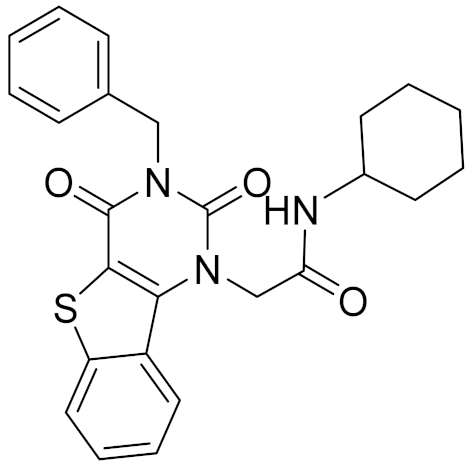 | <4 | - | 5.58 | |
| 5 | E196-0346 | 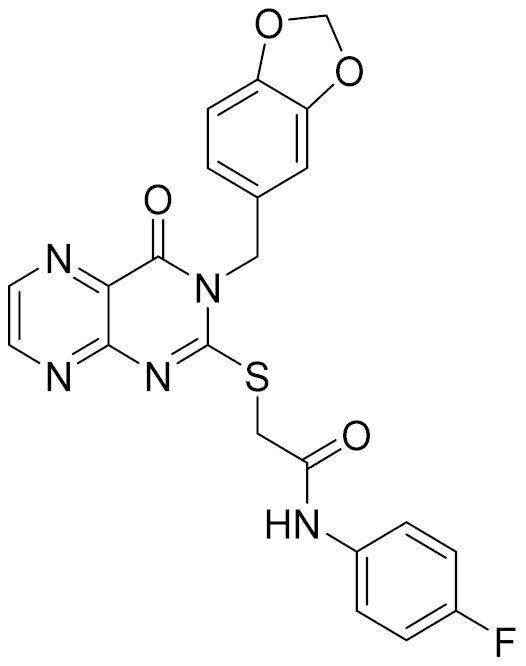 | 4.42 | - | 3.01 | |
| 6 | C728-0838 | 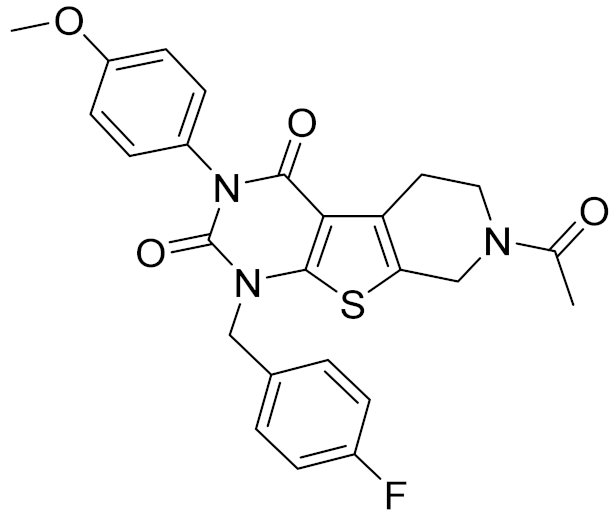 | 5.15 | <4.70 | 3.07 | |
| 7 | C728-0198 |  | 5.18 | 5.46 | 4.74 | 0.28 |
| 8 | 4428-0510 | 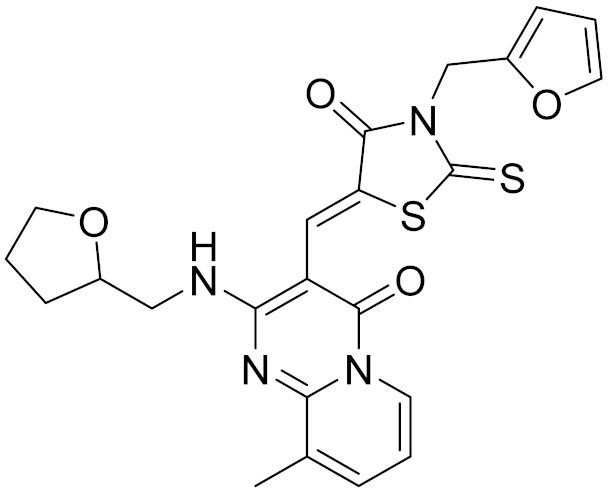 | 6.66 | 6.93 | 4.55 | 0.37 |
| 9 | C566-1034 | 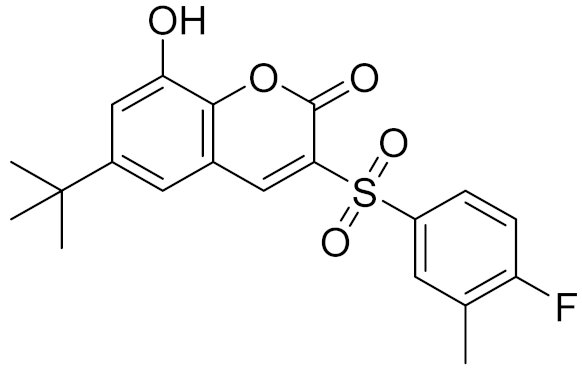 | 4.63 | - | 3.36 | |
| 10 | C241-0788 | 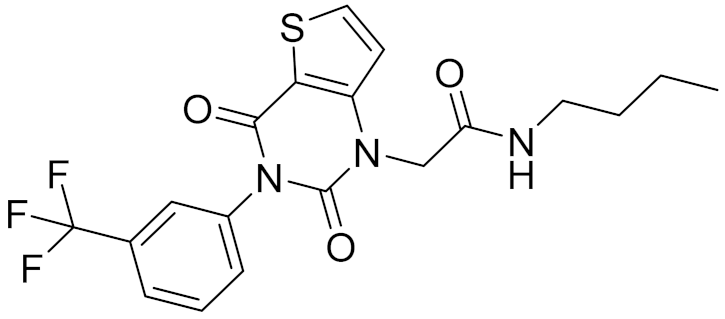 | 4.75 | - | 3.80 | |
| 11 | E146-1216 | 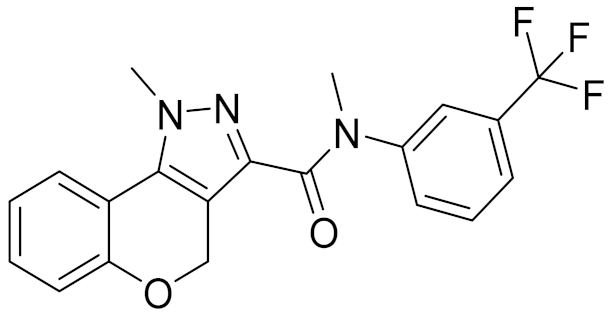 | 4.86 | - | 3.93 | |
| 12 | E196-0403 | 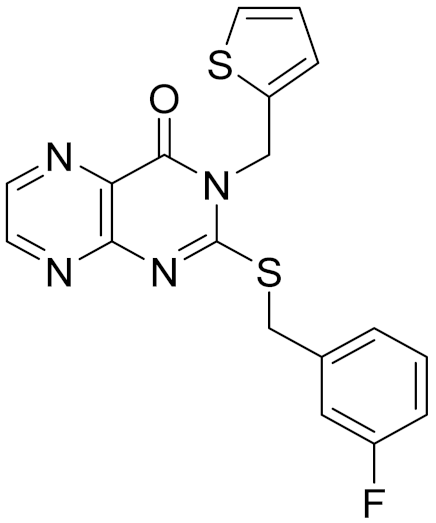 | 5.45 | 5.25 | 3.09 | 0.26 |
| 13 | E538-0230 | 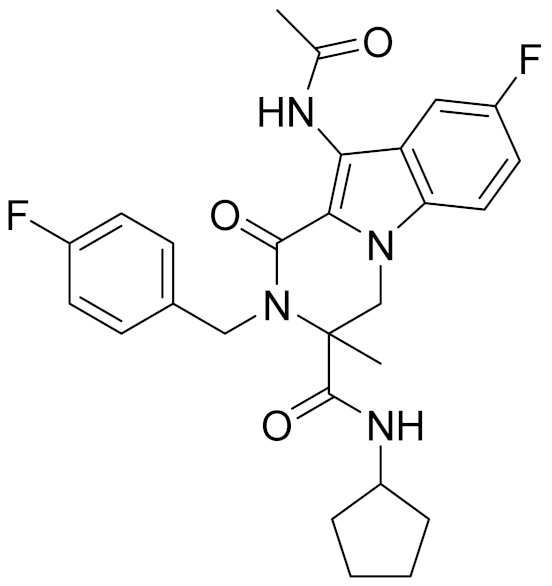 | 4.48 | - | 4.34 | |
| 14 | C796-1142 | 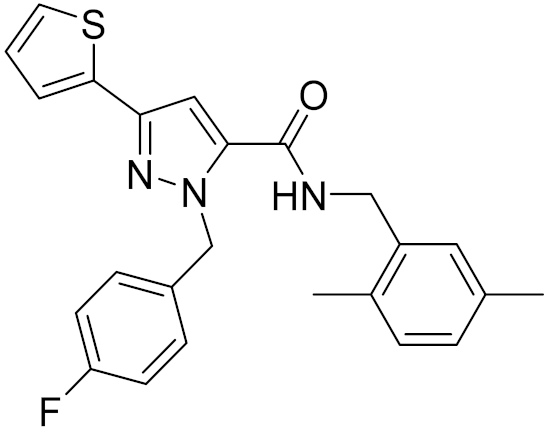 | 4.75 | - | 7.58 | |
| 15 | C796-1158 | 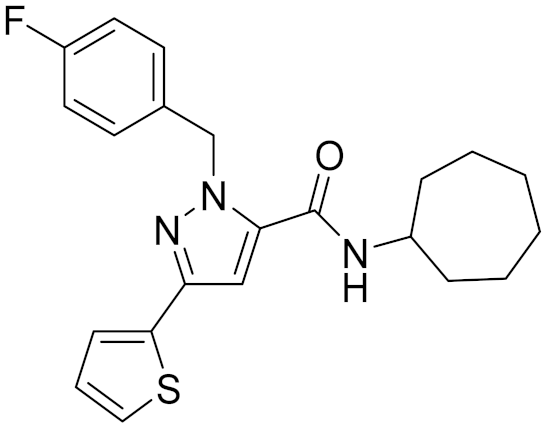 | 5.76 | 5.55 | 7.61 | 0.55 |
| - | WIN-55212-2 | 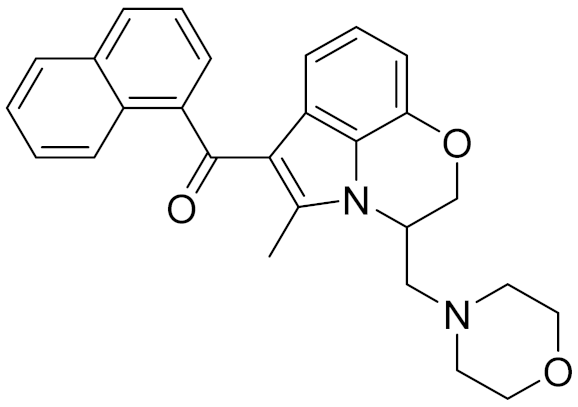 | 8.27 | - | 5.13 | - |
| - | SR144528 | 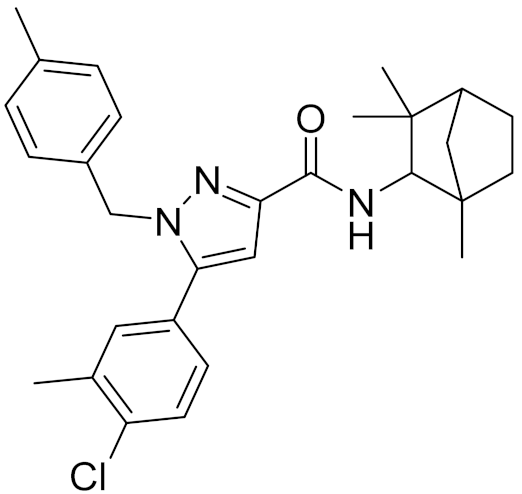 | - | 7.49 | 10.17 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Hou, S.; Liu, Y.; Li, D.; Lin, J. Identification of Novel Antagonists Targeting Cannabinoid Receptor 2 Using a Multi-Step Virtual Screening Strategy. Molecules 2021, 26, 6679. https://doi.org/10.3390/molecules26216679
Wang M, Hou S, Liu Y, Li D, Lin J. Identification of Novel Antagonists Targeting Cannabinoid Receptor 2 Using a Multi-Step Virtual Screening Strategy. Molecules. 2021; 26(21):6679. https://doi.org/10.3390/molecules26216679
Chicago/Turabian StyleWang, Mukuo, Shujing Hou, Ye Liu, Dongmei Li, and Jianping Lin. 2021. "Identification of Novel Antagonists Targeting Cannabinoid Receptor 2 Using a Multi-Step Virtual Screening Strategy" Molecules 26, no. 21: 6679. https://doi.org/10.3390/molecules26216679
APA StyleWang, M., Hou, S., Liu, Y., Li, D., & Lin, J. (2021). Identification of Novel Antagonists Targeting Cannabinoid Receptor 2 Using a Multi-Step Virtual Screening Strategy. Molecules, 26(21), 6679. https://doi.org/10.3390/molecules26216679