
Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets


Abstract
:1. Introduction
2. Heterocyclic Compounds
2.1. N-Based Multi-Target Anticancer Heterocycles
2.1.1. Synthetic, Semi-Synthetic and Hybrid N-Heterocycles
2.1.2. Natural Products from Various Sources
2.1.3. Nitrogen-Heterocyclic Natural Products with Multitarget Inhibitory Properties
2.2. S-Based Heterocycles
2.3. O-Heterocyclic
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhiman, N.; Kaur, K.; Jaitak, V. Tetrazoles as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Bioorg. Med. Chem. 2020, 28, 115599. [Google Scholar] [CrossRef]
- Abbot, V.; Sharma, P.; Dhiman, S.; Noolvi, M.N.; Patel, H.M.; Bhardwaj, V. Small hybrid heteroaromatics: Resourceful biological tools in cancer research. RSC Adv. 2017, 7, 28313–28349. [Google Scholar] [CrossRef] [Green Version]
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. MedChemComm 2017, 8, 1742–1773. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Yee, E.M.H.; Pasquier, E.; Iskander, G.; Wood, K.; Black, D.S.; Kumar, N. Synthesis of novel isoflavene-propranolol hybrids as anti-tumor agents. Bioorg. Med. Chem. 2013, 21, 1652–1660. [Google Scholar] [CrossRef]
- Dorak, M.T.; Karpuzoglu, E. Gender differences in cancer susceptibility: An inadequately addressed issue. Front. Genet. 2012, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Edgren, G.; Liang, L.; Adami, H.O.; Chang, E.T. Enigmatic sex disparities in cancer incidence. Eur. J. Epidemiol. 2012, 27, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the role of angiogenesis in cancer ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Jeong, E.K.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Regulation of Tumor Progression by Programmed Necrosis. Oxidative Med. Cell. Longev. 2018, 2018, 3537471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomes, L.; Emberley, E.; Niu, Y.; Troup, S.; Pastorek, J.; Strange, K.; Harris, A.; Watson, P.H. Necrosis and hypoxia in invasive breast carcinoma. Breast Cancer Res. Treat. 2003, 81, 61–69. [Google Scholar] [CrossRef]
- Angiogenesis Inhibitors—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet (accessed on 14 February 2021).
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing angiogenesis, a key step in cancer vascularization, and treatment approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Sun, L.; Fan, G.; Shan, P.; Qiu, X.; Dong, S.; Liao, L.; Yu, C.; Wang, T.; Gu, X.; Li, Q.; et al. Regulation of energy homeostasis by the ubiquitin-independent REGγ 3 proteasome. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kania, E.; Pająk, B.; Orzechowski, A. Calcium homeostasis and ER stress in control of autophagy in cancer cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isoldi, M.; Visconti, M.; Castrucci, A. Anti-Cancer Drugs: Molecular Mechanisms of Action. Mini-Rev. Med. Chem. 2005, 5, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Singh, S.; Skvortsova, I.; Kumar, V. Promising Targets in Anti-cancer Drug Development: Recent Updates. Curr. Med. Chem. 2017, 24, 4729–4752. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.G.; Ferreira, L.L.G.; Andricopulo, A.D. Recent advances and perspectives in cancer drug design. Anais da Academia Brasileira de Ciências 2018, 1233–1250. [Google Scholar] [CrossRef] [Green Version]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ming, B.; Gong, G.H.; Wang, D.; Bao, G.L.; Yu, L.J. Current research on anti-breast cancer synthetic compounds. RSC Adv. 2018, 8, 4386–4416. [Google Scholar] [CrossRef] [Green Version]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Pharmacological profile of natural and synthetic compounds with rigid adamantane-based scaffolds as potential agents for the treatment of neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2020, 529, 1225–1241. [Google Scholar] [CrossRef]
- Ahuja, A.; Kim, J.H.; Kim, J.H.; Yi, Y.S.; Cho, J.Y. Functional role of ginseng-derived compounds in cancer. J. Ginseng Res. 2018, 42, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.M.L.; Chan, B.D.; Wong, W.Y.; Leung, T.W.; Qu, Z.; Huang, J.; Zhu, L.; Lee, C.S.; Chen, S.; Tai, W.C.S. Synthesis and Evaluation of Novel Anticancer Compounds Derived from the Natural Product Brevilin A. ACS Omega 2020, 5, 14586–14596. [Google Scholar] [CrossRef] [PubMed]
- Perks, C.M.; Keith, A.J.; Goodhew, K.L.; Savage, P.B.; Winters, Z.E.; Holly, J.M.P. Prolactin acts as a potent survival factor for human breast cancer cell lines. Br. J. Cancer 2004, 91, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozako, T.; Suzuki, T.; Yoshimitsu, M.; Arima, N.; Honda, S.I.; Soeda, S. Anticancer agents targeted to sirtuins. Molecules 2014, 19, 20295–20313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, E.S.; Kim, T.H.; Cho, Y.J.; Ryu, J.; Choi, J.J.; Lee, Y.Y.; Kim, T.J.; Choi, C.H.; Kim, W.Y.; Sa, J.K.; et al. Preclinical assessment of the VEGFR inhibitor axitinib as a therapeutic agent for epithelial ovarian cancer. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, M.; Cui, X.; Lv, D.; Jin, L.; Khan, M.; Ma, T. Brevilin A promotes oxidative stress and induces mitochondrial apoptosis in U87 glioblastoma cells. OncoTargets Ther. 2018, 11, 7031–7040. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Du, Y.; Nan, J.; Zhang, X.; Qin, X.; Wang, Y.; Hou, J.; Wang, Q.; Yang, J. Brevilin A, a Novel Natural Product, Inhibits Janus Kinase Activity and Blocks STAT3 Signaling in Cancer Cells. PLoS ONE 2013, 8, e63697. [Google Scholar] [CrossRef] [Green Version]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasir, J.K.; Vinod, L. Targeted Drug Delivery in Cancer Therapy. Technol. Cancer Res. Treat. 2005, 4, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Dratkiewicz, E.; Pietraszek-Gremplewicz, K.; Simiczyjew, A.; Mazur, A.J.; Nowak, D. Gefitinib or lapatinib with foretinib synergistically induce a cytotoxic effect in melanoma cell lines. Oncotarget 2018, 9, 18254–18268. [Google Scholar] [CrossRef] [Green Version]
- Segovia-Mendoza, M.; González-González, M.E.; Barrera, D.; Díaz, L.; García-Becerra, R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of her2-positive breast cancer: Preclinical and clinical evidence. Am. J. Cancer Res. 2015, 5, 2531–2561. [Google Scholar]
- Harris, J.D.; Quatman, C.E.; Manring, M.M.; Siston, R.A.; Flanigan, D.C. How to write a systematic review. Am. J. Sports Med. 2014, 42, 2761–2768. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Reprint-Preferred Reporting items for systematic reviews and meta-analyses: The PRISMA statement. Phys. Ther. 2009, 89, 873–880. [Google Scholar] [CrossRef]
- Marson, C.M. Saturated Heterocycles with Applications in Medicinal Chemistry. Adv. Heterocycl. Chem. 2016, 121, 13–33. [Google Scholar] [CrossRef]
- Paudel, A.; Hamamoto, H.; Panthee, S.; Kaneko, K.; Matsunaga, S.; Kanai, M.; Suzuki, Y.; Sekimizu, K. A novel spiro-heterocyclic compound identified by the silkworm infection model inhibits transcription in Staphylococcus aureus. Front. Microbiol. 2017, 8, 712. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.M.; Siddiqui, R.; Ong, S.K.; Shah, M.R.; Anwar, A.; Heard, P.J.; Khan, N.A. Identification and characterization of antibacterial compound(s) of cockroaches (Periplaneta americana). Appl. Microbiol. Biotechnol. 2017, 101, 253–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatoon, H.; Abdulmalek, E. Novel Synthetic Routes to Prepare Biologically Active Quinoxalines and Their Derivatives: A Synthetic Review for the Last Two Decades. Molecules 2021, 26, 1055. [Google Scholar] [CrossRef]
- Rashid, M.; Shrivastava, N.; Husain, A. Synthesis and SAR Strategy of Thiazolidinedione: A Novel Approach for Cancer Treatment. J. Chil. Chem. Soc. 2020, 65, 4817–4832. [Google Scholar] [CrossRef]
- Abdelsalam, M.A.; Aboulwafa, O.M.; Badawey, E.S.A.M.; El-Shoukrofy, M.S.; El-Miligy, M.M.; Gouda, N. Design and synthesis of some β-carboline derivatives as multi-target anticancer agents. Future Med. Chem. 2018, 10, 2791–2814. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Li, X.; Hou, J.; Ding, Y.; Zhang, J.; Xu, W.; Zhang, Y. Discovery of Multi-target Anticancer Agents Based on HDAC Inhibitor MS-275 and 5-FU. Med. Chem. 2016, 12, 30–36. [Google Scholar] [CrossRef]
- Ribatti, D. Judah Folkman, a pioneer in the study of angiogenesis. Angiogenesis 2008, 11, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.W.; Xu, X.H.; Feng, S.L.; Tang, Z.B.; Chen, S.W.; Hui, L. Synthesis of hybrid 4-deoxypodophyllotoxin-5-fluorouracil compounds that inhibit cellular migration and induce cell cycle arrest. Bioorg. Med. Chem. Lett. 2016, 26, 1561–1566. [Google Scholar] [CrossRef]
- Peng, F.W.; Xuan, J.; Wu, T.T.; Xue, J.Y.; Ren, Z.W.; Liu, D.K.; Wang, X.Q.; Chen, X.H.; Zhang, J.W.; Xu, Y.G.; et al. Design, synthesis and biological evaluation of N-phenylquinazolin-4-amine hybrids as dual inhibitors of VEGFR-2 and HDAC. Eur. J. Med. Chem. 2016, 109, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef]
- Elzahhar, P.A.; Abd El Wahab, S.M.; Elagawany, M.; Daabees, H.; Belal, A.S.F.; EL-Yazbi, A.F.; Eid, A.H.; Alaaeddine, R.; Hegazy, R.R.; Allam, R.M.; et al. Expanding the anticancer potential of 1,2,3-triazoles via simultaneously targeting Cyclooxygenase-2, 15-lipoxygenase and tumor-associated carbonic anhydrases. Eur. J. Med. Chem. 2020, 200, 112439. [Google Scholar] [CrossRef]
- Dragovich, T.; Laheru, D.; Dayyani, F.; Bolejack, V.; Smith, L.; Seng, J.; Burris, H.; Rosen, P.; Hidalgo, M.; Ritch, P.; et al. Phase II trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy (PCRT O4-001). Cancer Chemother. Pharmacol. 2014, 74, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.H.; Sojo, F.; Arvelo, F.; Calderón, C.; Morales, A.; López, S.E. Anticancer potential of new 3-nitroaryl-6-(N-methyl)piperazin-1,2,4-triazolo[3,4-a]phthalazines targeting voltage-gated K+ channel: Copper-catalyzed one-pot synthesis from 4-chloro-1-phthalazinyl-arylhydrazones. Bioorg. Chem. 2020, 101, 104031. [Google Scholar] [CrossRef]
- Özden, S.; Atabey, D.; Yildiz, S.; Göker, H. Synthesis and potent antimicrobial activity of some novel methyl or ethyl 1H-benzimidazole-5-carboxylates derivatives carrying amide or amidine groups. Bioorg. Med. Chem. 2005, 13, 1587–1597. [Google Scholar] [CrossRef]
- Romero-Castro, A.; León-Rivera, I.; Ávila-Rojas, L.C.; Navarrete-Vázquez, G.; Nieto-Rodríguez, A. Synthesis and preliminary evaluation of selected 2-aryl-5(6)-nitro- 1H-benzimidazole derivatives as potential anticancer agents. Arch. Pharm. Res. 2011, 34, 181–189. [Google Scholar] [CrossRef]
- Suk, F.M.; Liu, C.L.; Hsu, M.H.; Chuang, Y.T.; Wang, J.P.; Liao, Y.J. Treatment with a new benzimidazole derivative bearing a pyrrolidine side chain overcomes sorafenib resistance in hepatocellular carcinoma. Sci. Rep. 2019, 9, 17259. [Google Scholar] [CrossRef] [Green Version]
- Bistrović, A.; Krstulović, L.; Harej, A.; Grbčić, P.; Sedić, M.; Koštrun, S.; Pavelić, S.K.; Bajić, M.; Raić-Malić, S. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2018, 143, 1616–1634. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Larosa, C.; Antwi, J.; Govindarajan, R.; Werbovetz, K.A. Imidazoles as potential anticancer agents: An update on recent studies. Molecules 2021, 26, 4213. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.B.; Huang, R.Z.; Fang, Y.L.; Liang, G.B.; Yang, C.H.; Ma, X.L.; Zhang, Y. Design, synthesis and pharmacological evaluation of novel 2-chloro-3-(1H-benzo[d]imidazol-2-yl)quinoline derivatives as antitumor agents: In vitro and in vivo antitumor activity, cell cycle arrest and apoptotic response. RSC Adv. 2018, 8, 24376–24385. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Donthiboina, K.; Anchi, P.; Gurram, S.; Sai Mani, G.; Lakshmi Uppu, J.; Godugu, C.; Shankaraiah, N.; Kamal, A. Synthesis and biological evaluation of substituted N-(2-(1H-benzo[d]imidazol-2-yl)phenyl)cinnamides as tubulin polymerization inhibitors. Bioorg. Chem. 2020, 103, 104191. [Google Scholar] [CrossRef] [PubMed]
- Li, A.L.; Yang, Y.Q.; Wang, W.Y.; Liu, Q.S.; Sun, Y.; Gu, W. Synthesis, cytotoxicity and apoptosis-inducing activity of novel 1H-benzo[d]imidazole derivatives of dehydroabietic acid. J. Chin. Chem. Soc. 2020, 67, 1668–1678. [Google Scholar] [CrossRef]
- Li, Y.; Tan, C.; Gao, C.; Zhang, C.; Luan, X.; Chen, X.; Liu, H.; Chen, Y.; Jiang, Y. Discovery of benzimidazole derivatives as novel multi-target EGFR, VEGFR-2 and PDGFR kinase inhibitors. Bioorg. Med. Chem. 2011, 19, 4529–4535. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Quan, D.; Chen, J.; Ding, J.; Zhao, J.; Lv, L.; Chen, J. Design, synthesis, and biological evaluation of 1-substituted -2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Liu, X.; Guan, Q.; Ding, N.; Wei, Q.; Tong, B.; Zhao, M.; Zhang, W.; Ma, L. 5-(3,4,5-trimethoxybenzoyl)-4-methyl-2-(p-tolyl) imidazol (BZML) targets tubulin and DNA to induce anticancer activity and overcome multidrug resistance in colorectal cancer cells. Chem. Biol. Interact. 2020, 315, 108886. [Google Scholar] [CrossRef]
- Zha, G.F.; Qin, H.L.; Youssif, B.G.M.; Amjad, M.W.; Raja, M.A.G.; Abdelazeem, A.H.; Bukhari, S.N.A. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur. J. Med. Chem. 2017, 135, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.K.; Hassan, A.H.E.; Sun Ahn, B.; Duk Park, K.; Joo Roh, E. Reprofiling of pyrimidine-based DAPK1/CSF1R dual inhibitors: Identification of 2,5-diamino-4-pyrimidinol derivatives as novel potential anticancer lead compounds. J. Enzym. Inhib. Med. Chem. 2020, 35, 311–324. [Google Scholar] [CrossRef]
- Ning, C.Q.; Lu, C.; Hu, L.; Bi, Y.J.; Yao, L.; He, Y.J.; Liu, L.F.; Liu, X.Y.; Yu, N.F. Macrocyclic compounds as anti-cancer agents: Design and synthesis of multi-acting inhibitors against HDAC, FLT3 and JAK2. Eur. J. Med. Chem. 2015, 95, 104–115. [Google Scholar] [CrossRef]
- Anonymous. Linifanib. Drugs R D 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Shi, Z.H.; Liu, F.T.; Tian, H.Z.; Zhang, Y.M.; Li, N.G.; Lu, T. Design, synthesis and structure-activity relationship of diaryl-ureas with novel isoxazol[3,4-b]pyridine-3-amino-structure as multi-target inhibitors against receptor tyrosine kinase. Bioorg. Med. Chem. 2018, 26, 4735–4744. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Pan, X.; Dai, B.; Sun, Y.; Li, C.; Zhang, J. Discovery of multi-target receptor tyrosine kinase inhibitors as novel anti-angiogenesis agents. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakyi, P.O.; Amewu, R.K.; Devine, R.N.O.A.; Ismaila, E.; Miller, W.A.; Kwofie, S.K. The Search for Putative Hits in Combating Leishmaniasis: The Contributions of Natural Products Over the Last Decade. Nat. Prod. Bioprospect. 2021, 11, 489–544. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Perdue, R.E.; Hartwel, J.L. Proceedings of the 16th Annual Meeting of the Society for Economic Botany—Plants and cancer. Cancer Treat. Rep. 1976, 60, 973–1214. [Google Scholar]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural Products as Sources of New Drugs over the Period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef]
- Li-Weber, M. New therapeutic aspects of flavones: The anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat. Rev. 2009, 35, 57–68. [Google Scholar] [CrossRef]
- Wen, C.; Wu, L.; Fu, L.; Zhang, X.; Zhou, H. Berberine enhances the anti-tumor activity of tamoxifen in drug-sensitive MCF-7 and drug-resistant MCF-7/TAM cells. Mol. Med. Rep. 2016, 14, 2250–2256. [Google Scholar] [CrossRef] [PubMed]
- Puthdee, N.; Seubwai, W.; Vaeteewoottacharn, K.; Boonmars, T.; Cha’on, U.; Phoomak, C.; Wongkham, S. Berberine induces cell cycle arrest in cholangiocarcinoma cell lines via inhibition of NF-κB and STAT3 pathways. Biol. Pharm. Bull. 2017, 40, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Kao, S.T.; Chen, G.W.; Chung, J.G. Berberine decreased N-acetylation of 2-aminofluorene through inhibition of N-acetyltransferase gene expression in human leukemia HL-60 cells. Anticancer Res. 2005, 25, 4149–4155. [Google Scholar]
- Jia, J.; Kang, X.; Liu, Y.; Zhang, J. Inhibition of human liver cancer cell growth by evodiamine involves apoptosis and deactivation of PI3K/AKT pathway. Appl. Biol. Chem. 2020, 63, 67. [Google Scholar] [CrossRef]
- Li, X.; Wu, S.; Dong, G.; Chen, S.; Ma, Z.; Liu, D.; Sheng, C. Natural Product Evodiamine with Borate Trigger Unit: Discovery of Potent Antitumor Agents against Colon Cancer. ACS Med. Chem. Lett. 2020, 11, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Qi, X.; Wang, Q.; Zhu, W.; Li, J. A novel multi-target inhibitor harboring selectivity of inhibiting EGFR T790M sparing wild-type EGFR. Am. J. Cancer Res. 2017, 7, 1884–1898. [Google Scholar]
- Bishayee, K.; Chakraborty, D.; Ghosh, S.; Boujedaini, N.; Khuda-Bukhsh, A.R. Lycopodine triggers apoptosis by modulating 5-lipoxygenase, and depolarizing mitochondrial membrane potential in androgen sensitive and refractory prostate cancer cells without modulating p53 activity: Signaling cascade and drug-DNA interaction. Eur. J. Pharmacol. 2013, 698, 110–121. [Google Scholar] [CrossRef]
- Robles, A.J.; Du, L.; Cichewicz, R.H.; Mooberry, S.L. Maximiscin Induces DNA Damage, Activates DNA Damage Response Pathways, and Has Selective Cytotoxic Activity against a Subtype of Triple-Negative Breast Cancer. J. Nat. Prod. 2016, 79, 1822–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crampton, S.L.; Adams, E.G.; Kuentzel, S.L.; Li, L.H.; Badiner, G.; Bhuyan1, B.K. Biochemical and Cellular Effects of Didemnins A and B. Cancer Res. 1984, 44, 1796–1801. [Google Scholar]
- Chun, H.G.; Davies, B.; Hoth, D.; Suffness, M.; Plowman, J.; Flora, K.; Grieshaber, C.; Leyland-Jones, B. Didemnin B—The first marine compound entering clinical trials as an antineoplastic agent. Investig. New Drugs 1986, 4, 279–284. [Google Scholar]
- Ahuja, D.; Vera, M.D.; SirDeshpande, B.V.; Morimoto, H.; Williams, P.G.; Joullié, M.M.; Toogood, P.L. Inhibition of protein synthesis by didemnin B: How EF-1α mediates inhibition of translocation. Biochemistry 2000, 39, 4339–4346. [Google Scholar] [CrossRef]
- Li, L.H.; Timmins, L.G.; Wallace, T.L.; Krueger, W.C.; Prairie, M.D.; Im, W.B. Mechanism of Action of Didemnin B, a Depsipeptide from the Sea. Cancer Lett. 1984, 23, 279–288. [Google Scholar] [CrossRef]
- Palumbo, A.; Castellano, I.; Napolitano, A. Ovothiol: A Potent Natural Antioxidant from Marine Organisms. Blue Biotechnol. 2018, 2, 583–610. [Google Scholar]
- Russo, G.L.; Russo, M.; Castellano, I.; Napolitano, A.; Palumbo, A. Ovothiol isolated from sea urchin oocytes induces autophagy in the Hep-G2 cell line. Mar. Drugs 2014, 12, 4069–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewley, C.A.; Detritus, C.; Faulkner, D.J. Microsclerodermins A and B. Antifungal Cyclic Peptides from the Lithistid Sponge Microscleroderma sp. J. Am. Chem. Soc. 1994, 116, 7631–7636. [Google Scholar] [CrossRef]
- Guzmán, E.A.; Maers, K.; Roberts, J.; Kemami-Wangun, H.V.; Harmody, D.; Wright, A.E. The marine natural product microsclerodermin A is a novel inhibitor of the nuclear factor kappa B and induces apoptosis in pancreatic cancer cells. Investig. New Drugs 2015, 33, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabowska, G.; Barg, E.; Wójcicka, A. Biological activity of naturally derived naphthyridines. Molecules 2021, 26, 4324. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Fedorov, S.N.; Shubina, L.K.; Kuzmich, A.S.; Bokemeyer, C.; Keller-Von Amsberg, G.; Honecker, F. Aaptamines from the marine sponge Aaptos sp. display anticancer activities in human cancer cell lines and modulate AP-1-, NF- B-, and p53-dependent transcriptional activity in mouse JB6 Cl41 cells. Biomed. Res. Int. 2014, 2014, 469309. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Cui, X.; Lee, D.S.; Sohn, J.H.; Yim, J.H.; Kim, Y.C.; Oh, H. Anti-Inflammatory Effect of Neoechinulin A from the Marine Fungus Eurotium sp. SF-5989 through the Suppression of NF-κB and p38 MAPK Pathways in Lipopolysaccharide-Stimulated RAW264.7 Macrophages. Molecules 2013, 18, 13245–13259. [Google Scholar] [CrossRef] [Green Version]
- Wijesekara, I.; Li, Y.X.; Vo, T.S.; Van Ta, Q.; Ngo, D.H.; Kim, S.K. Induction of apoptosis in human cervical carcinoma HeLa cells by neoechinulin A from marine-derived fungus Microsporum sp. Process. Biochem. 2013, 48, 68–72. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.; Lv, X.; Weng, Q.; Chen, M.; Cui, R.; Liang, G.; Ji, J. Piperlongumine, a novel TrxR1 inhibitor, induces apoptosis in hepatocellular carcinoma cells by ROS-mediated ER stress. Front. Pharmacol. 2019, 10, 1180. [Google Scholar] [CrossRef]
- Rawat, L.; Hegde, H.; Hoti, S.L.; Nayak, V. Piperlongumine Induces ROS Mediated Cell Death and Synergizes Paclitaxel in Human Intestinal Cancer Cells. Biomed. Pharmacother. 2020, 128, 110243. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kasukabe, T.; Kumakura, S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int. J. Oncol. 2018, 52, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
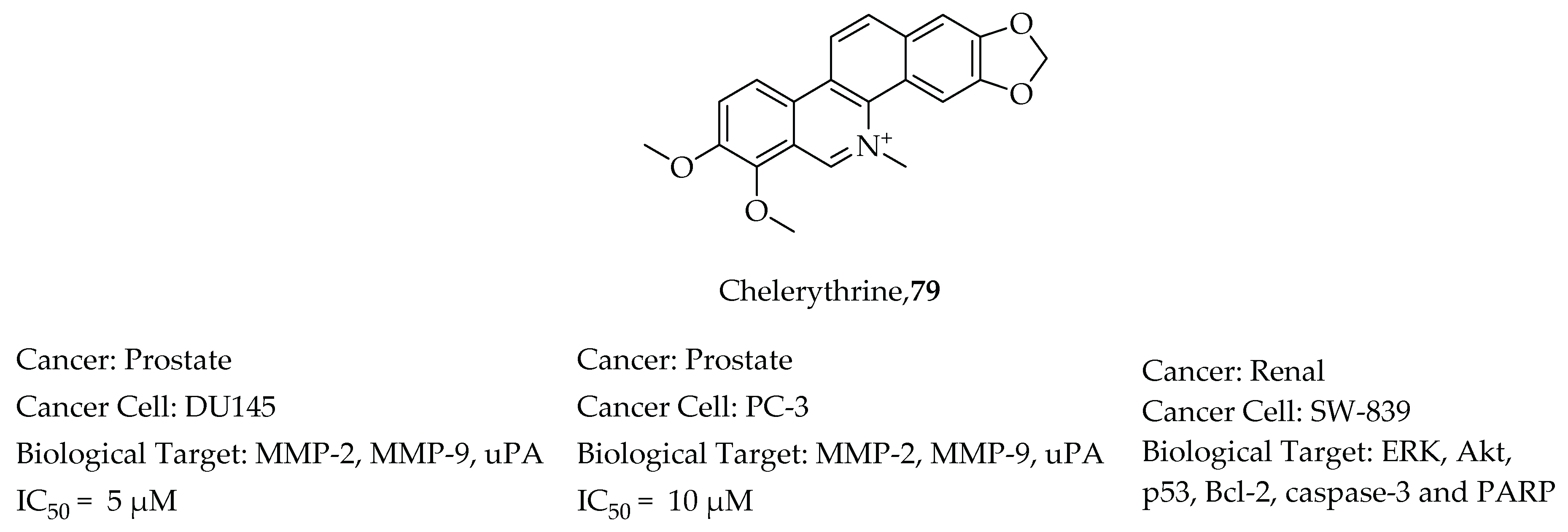
- Han, N.; Yang, Z.; Liu, Z.; Liu, H.; Yin, J. Research progress on natural benzophenanthridine alkaloids and their pharmacological functions: A review. Nat. Prod. Commun. 2016, 11, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.Y.; Meng, Q.H.; Chong, Y.; Jiao, Y.; Zhao, L.; Rosen, E.M.; Fan, S. Sanguinarine inhibits growth of human cervical cancer cells through the induction of apoptosis. Oncol. Rep. 2012, 28, 2264–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhami, V.M.; Aziz, M.H.; Reagan-Shaw, S.R.; Nihal, M.; Mukhtar, H.; Ahmad, N. Sanguinarine causes cell cyle blockade and apoptosis of human prostate carcinoma cells via modulation of cylin kinase inhibitor-cyclin-cyclin-dependent kinase machinery. Mol. Cancer Ther. 2004, 3, 933–940. [Google Scholar]
- Available online: https://www.ebi.ac.uk/chebi/searchId.do?chebiId=CHEBI:78373 (accessed on 1 September 2021).
- Yang, B.; Zhang, D.; Qian, J.; Cheng, Y. Chelerythrine suppresses proliferation and metastasis of human prostate cancer cells via modulating MMP/TIMP/NF-κB system. Mol. Cell. Biochem. 2020, 474, 199–208. [Google Scholar] [CrossRef]
- Chen, X.M.; Zhang, M.; Fan, P.L.; Qin, Y.H.; Zhao, H.W. Chelerythrine chloride induces apoptosis in renal cancer HEK-293 and SW-839 cell lines. Oncol. Lett. 2016, 11, 3917–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, H.R.; Hui, W.H.; Ng, Y.L. An examination of the rutaceae of Hong Kong. Part II. The alkaloids, nitidine and oxynitidine, from Zanthoxylum nitidum. J. Chem. Soc. 1959, 1840–1845. [Google Scholar] [CrossRef]
- Torto, F.G.; Mensah, I.A. Alkaloids of Fagara macrophylla. Phytochemistry 1970, 9, 911–914. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; Mcphail, A.T. Plant Antitumor Agents. VI. The Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C.; Taylor, H. Plant antitumor agents, 27. Isolation, structure, and structure activity relationships of alkaloids from Fagara macrophylla. J. Nat. Prod. 1987, 50, 1095–1099. [Google Scholar] [CrossRef]
- Fish, F.; Waterman, P.G. Methanol-soluble quaternary alkaloids from African Fagara species. Phytochemistry 1972, 11, 3007–3014. [Google Scholar] [CrossRef]
- Bouquet, J.; Rivaud, M.; Chevalley, S.; Deharo, E.; Jullian, V.; Valentin, A. Biological activities of nitidine, a potential anti-malarial lead compound. Malar. J. 2012, 11, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gakunju, D.M.N.; Mberu, E.K.; Dossaji, S.F.; Gray, A.I.; Waigh, R.D.; Waterman, P.G.; Watkins, W.M. Potent antimalarial activity of the alkaloid nitidine, isolated from a Kenyan herbal remedy. Antimicrob. Agents Chemother. 1995, 39, 2606–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addae-Mensah, I.; Munenge, R.; Guantai, A.N. Comparative examination of two Zanthoxylum benzophenanthridine alkaloids for effects in rabbits. Phytother. Res. 1989, 3, 165–169. [Google Scholar] [CrossRef]
- Wang, L.K.; Hecht, S.M.; Johnson, R.K. Inhibition of Topoisomerase I Function by Nitidine and Fagaronine. Chem. Res. Toxicol. 1993, 6, 813–818. [Google Scholar] [CrossRef]
- Nyangulu, J.M.; Hargreaves, S.L.; Sharples, S.L.; Mackay, S.P.; Waigh, R.D.; Duval, O.; Mberu, E.K.; Watkins, W.M. Antimalarial benzo[c]phenanthridines. Bioorg. Med. Chem. Lett. 2005, 15, 2007–2010. [Google Scholar] [CrossRef]
- Pan, X.; Han, H.; Wang, L.; Yang, L.; Li, R.; Li, Z.; Liu, J.; Zhao, Q.; Qian, M.; Liu, M.; et al. Nitidine Chloride inhibits breast cancer cells migration and invasion by suppressing c-Src/FAK associated signaling pathway. Cancer Lett. 2011, 313, 181–191. [Google Scholar] [CrossRef]
- Liao, J.; Xu, T.; Zheng, J.X.; Lin, J.M.; Cai, Q.Y.; Yu, D.B.; Peng, J. Nitidine chloride inhibits hepatocellular carcinoma cell growth in vivo through the suppression of the JAK1/STAT3 signaling pathway. Int. J. Mol. Med. 2013, 32, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.R.; Zheng, Y.; Fang, L.Q.; Guan, Y.C.; Ma, A.Q.; Wu, J. Nano-Sized MIL-100(Fe) as a Carrier Material for Nitidine Chloride Reduces Toxicity and Enhances Anticancer Effects In Vitro. J. Inorg. Organomet. Polym. Mater. 2020, 30, 3388–3395. [Google Scholar] [CrossRef]
- Available online: https://www.wahooas.org/web-ooas/sites/default/files/publications/2185/west-african-herbal-pharmacopoeiaok.pdf (accessed on 1 October 2021).
- Dewick, P.M. Tumor inhibitors from plants. Trease Evans’ Pharmacogn. 1996, 14, 210–214. [Google Scholar] [CrossRef]
- Messmer, W.M.; Tin-wa, M.; Fong, H.H.S.; Bevelle, C.; Farnsworth, N.R.; Abraham, D.J.; Trojánek, J. Fagaronine, a new tumor inhibitor isolated from Fagara zanthoxyloides lam. (rutaceae). J. Pharm. Sci. 1972, 61, 1858–1859. [Google Scholar] [CrossRef]
- Barret, Y.; Sauvaire, Y. Fagaronine, a novel antileukaemic alkaloid. Phytother. Res. 1992, 6, 59–63. [Google Scholar] [CrossRef]
- Larsen, A.K.; Grondard, L.; Couprie, J.; Desoize, B.; Comoe, L.; Jardillier, J.C.; Riou, J.F. The antileukemic alkaloid fagaronine is an inhibitor of DNA topoisomerases I and II. Biochem. Pharmacol. 1993, 46, 1403–1412. [Google Scholar] [CrossRef]
- Torto, F.G.; Mensah, I.A.; Baxter, I. Fagaridine: A phenolic benzophenanthridine alkaloid from Fagara xanthoxyloides. Phytochemistry 1973, 12, 2315–2317. [Google Scholar] [CrossRef]
- Topcu, Z. DNA topoisomerases as targets for anticancer drugs. J. Clin. Pharm. Ther. 2001, 26, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef]
- Kupfahl, C.; Michalka, A.; Lass-Flo, C.; Fischer, G.; Haase, G.; Ruppert, T.; Geginat, G.; Hof, H. Gliotoxin production by clinical and environmental Aspergillus fumigatus strains. Int. J. Med. Microbiol. 2008, 298, 319–327. [Google Scholar] [CrossRef]
- Katz, R. United States. Eur. J. Polit. Res. Polit. Data Yearb. 2015, 54, 309–315. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Lee, J.S.; Qian, Z.J.; Li, Y.X.; Kim, K.N.; Heo, S.J.; Jeon, Y.J.; Park, W.S.; Choi, I.W.; Je, J.Y.; et al. Gliotoxin isolated from marine fungus Aspergillus sp. induces apoptosis of human cervical cancer and chondrosarcoma cells. Mar. Drugs 2014, 12, 69–87. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.; Lan, W.; Huang, C.; Lin, M.; Wang, Z.; Liang, W.; Iwamoto, A.; Yang, X.; Liu, H.; et al. Gliotoxin inhibits proliferation and induces apoptosis in colorectal cancer cells. Mar. Drugs 2015, 13, 6259–6273. [Google Scholar] [CrossRef] [Green Version]
- Abdelazeem, A.H.; El-Saadi, M.T.; Said, E.G.; Youssif, B.G.M.; Omar, H.A.; El-Moghazy, S.M. Novel diphenylthiazole derivatives with multi-target mechanism: Synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg. Chem. 2017, 75, 127–138. [Google Scholar] [CrossRef]
- Stiborová, M.; Poljaková, J.; Martínková, E.; Bořek-Dohalská, L.; Eckschlager, T.; Kizek, R.; Frei, E. Ellipticine cytotoxicity to cancer cell lines—A comparative study. Interdiscip. Toxicol. 2011, 4, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Ceramella, J.; Caruso, A.; Occhiuzzi, M.A.; Iacopetta, D.; Barbarossa, A.; Rizzuti, B.; Dallemagne, P.; Rault, S.; El-Kashef, H.; Saturnino, C.; et al. Benzothienoquinazolinones as new multi-target scaffolds: Dual inhibition of human Topoisomerase I and tubulin polymerization. Eur. J. Med. Chem. 2019, 181, 111583. [Google Scholar] [CrossRef]
- Aly, A.A.; Bräse, S.; Hassan, A.A.; Mohamed, N.K.; El-Haleem, L.E.A.; Nieger, M.; Morsy, N.M.; Alshammari, M.B.; Ibrahim, M.A.A.; Abdelhafez, E.M.N. Design, Synthesis, and Molecular Docking of Paracyclophanyl-Thiazole Hybrids as Novel CDK1 Inhibitors and Apoptosis Inducing Anti-Melanoma Agents. Molecules 2020, 25, 5569. [Google Scholar] [CrossRef] [PubMed]
- Megally Abdo, N.Y.; Milad Mohareb, R.; Halim, P.A. Uses of cyclohexane-1,3-dione for the synthesis of 1,2,4-triazine derivatives as anti-proliferative agents and tyrosine kinases inhibitors. Bioorg. Chem. 2020, 97, 103667. [Google Scholar] [CrossRef] [PubMed]
- Pardhasaradhi, B.V.V.; Reddy, M.; Ali, A.M.; Kumari, A.L.; Khar, A. Differential cytotoxic effects of Annona squamosa seed extracts on human tumour cell lines: Role of reactive oxygen species and glutathione. J. Biosci. 2005, 30, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, Q.; Shi, Y.; Li, Y.; Wang, X.; Li, X.; Chen, Y.; Chen, J. Three new antitumor annonaceous acetogenins from the seeds of Annona squamosa. Nat. Prod. Res. 2017, 31, 2085–2090. [Google Scholar] [CrossRef]
- Youn, U.J.; Miklossy, G.; Chai, X.; Wongwiwatthananukit, S.; Toyama, O.; Songsak, T.; Turkson, J.; Chang, L.C. Bioactive sesquiterpene lactones and other compounds isolated from Vernonia cinerea. Fitoterapia 2014, 93, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Youn, U.J.; Park, E.J.; Kondratyuk, T.P.; Simmons, C.J.; Borris, R.P.; Tanamatayarat, P.; Wongwiwatthananukit, S.; Toyama, O.; Songsak, T.; Pezzuto, J.M.; et al. Anti-inflammatory sesquiterpene lactones from the flower of Vernonia cinerea. Bioorg. Med. Chem. Lett. 2012, 22, 5559–5562. [Google Scholar] [CrossRef]
- Jung, H.J.; Kim, Y.; Shin, J.Y.; Sohng, J.K.; Kwon, H.J. Antiangiogenic activity of herboxidiene via downregulation of vascular endothelial growth factor receptor-2 and hypoxia-inducible factor-1α. Arch. Pharm. Res. 2015, 38, 1728–1735. [Google Scholar] [CrossRef]
- Liu, Q.M.; Zhao, H.Y.; Zhong, X.K.; Jiang, J.G. Eclipta prostrata L. phytochemicals: Isolation, structure elucidation, and their antitumor activity. Food Chem. Toxicol. 2012, 50, 4016–4022. [Google Scholar] [CrossRef]
- Sarveswaran, S.; Gautam, S.C.; Ghosh, J. Wedelolactone, a medicinal plant-derived coumestan, induces caspase-dependent apoptosis in prostate cancer cells via downregulation of PKCε without inhibiting Akt. Int. J. Oncol. 2012, 41, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.J.; Woo, J.H.; Lee, J.S.; Jang, D.S.; Lee, K.T.; Choi, J.H. Eclalbasaponin II induces autophagic and apoptotic cell death in human ovarian cancer cells. J. Pharmacol. Sci. 2016, 132, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a Flavonoid with Potential for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Lee, E.J.; Kim, H.R.; Hwang, K.A. Molecular mechanisms of luteolin-7-o-glucoside-induced growth inhibition on human liver cancer cells: G2/m cell cycle arrest and caspase-independent apoptotic signaling pathways. BMB Rep. 2013, 46, 611–616. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Rasul, A.; Batool, R.; Sarfraz, I.; Hussain, G.; Mateen Tahir, M.; Qin, T.; Selamoglu, Z.; Ali, M.; Li, J.; et al. Potential Anticancer Properties and Mechanisms of Action of Formononetin. Biomed. Res. Int. 2019, 2019, 5854315. [Google Scholar] [CrossRef]
- García, V.; Lara-Chica, M.; Cantarero, I.; Sterner, O.; Calzado, M.A.; Muñoz, E. Galiellalactone induces cell cycle arrest and apoptosis through the ATM/ATR pathway in prostate cancer cells. Oncotarget 2016, 7, 4490–4506. [Google Scholar] [CrossRef] [Green Version]
- Don-Doncow, N.; Escobar, Z.; Johansson, M.; Kjellström, S.; Garcia, V.; Munoz, E.; Sterner, O.; Bjartell, A.; Hellsten, R. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J. Biol. Chem. 2014, 289, 15969–15978. [Google Scholar] [CrossRef] [Green Version]
- Bury, M.; Girault, A.; Megalizzi, V.; Spiegl-Kreinecker, S.; Mathieu, V.; Berger, W.; Evidente, A.; Kornienko, A.; Gailly, P.; Vandier, C.; et al. Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell Death Dis. 2013, 4, e561. [Google Scholar] [CrossRef] [Green Version]
- Mohyeldin, M.M.; Akl, M.R.; Siddique, A.B.; Hassan, H.M.; El Sayed, K.A. The marine-derived pachycladin diterpenoids as novel inhibitors of wild-type and mutant EGFR. Biochem. Pharmacol. 2017, 126, 51–68. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.J.; Lee, K.T.; Rho, J.R.; Choi, J.H. Phorbaketal A, isolated from the marine sponge Phorbas sp., exerts its anti-inflammatory effects via NF-κB inhibition and heme oxygenase-1 activation in lipopolysaccharide-stimulated macrophages. Mar. Drugs 2015, 13, 7005–7019. [Google Scholar] [CrossRef] [Green Version]
- Rho, J.R.; Hwang, B.S.; Sim, C.J.; Joung, S.; Lee, H.Y.; Kim, H.J. Phorbaketals A, B, and C, sesterterpenoids with a spiroketal of hydrobenzopyran moiety isolated from the marine sponge Phorbas sp. Org. Lett. 2009, 11, 5590–5593. [Google Scholar] [CrossRef]
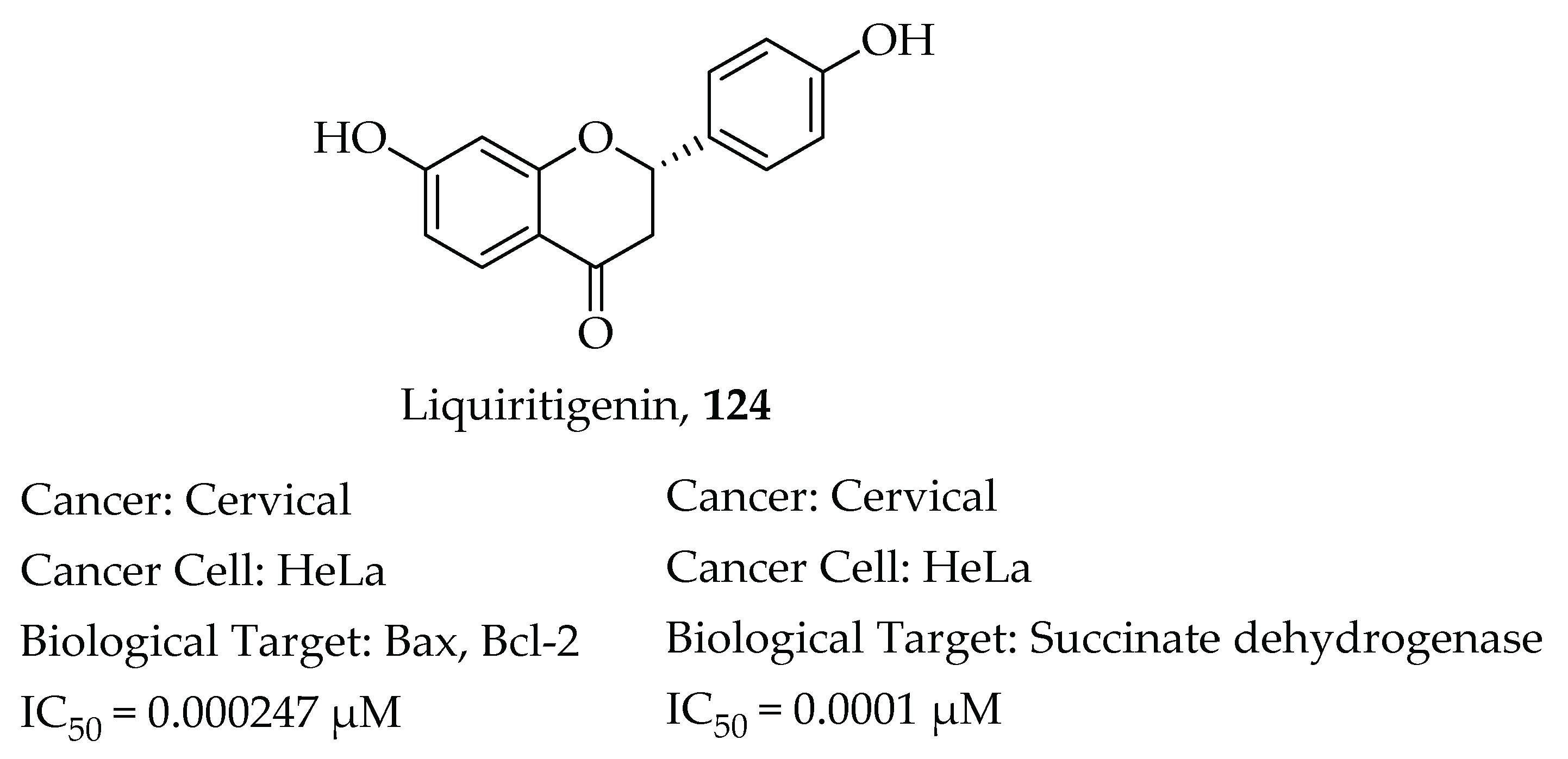
- Chen, H.; Zhang, X.; Feng, Y.; Rui, W.; Shi, Z.; Wu, L. Bioactive components of Glycyrrhiza uralensis mediate drug functions and properties through regulation of CYP450 enzymes. Mol. Med. Rep. 2014, 10, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.P.; Zhou, Y.J.; Liu, Y.; Cai, Y. qing Effect of liquiritigenin, a flavanone existed from Radix glycyrrhizae on pro-apoptotic in SMMC-7721 cells. Food Chem. Toxicol. 2009, 47, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xie, S.; Wang, Y.; Luo, K.; Wang, Y.; Cai, Y. Liquiritigenin inhibits tumor growth and vascularization in a mouse model of HeLa cells. Molecules 2012, 17, 7206–7216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayeka, P.A.; Bian, Y.; Mwitari, P.G.; Chu, X.; Zhang, Y.; Uzayisenga, R.; Otachi, E.O. Immunomodulatory and anticancer potential of Gan cao (Glycyrrhiza uralensis Fisch.) polysaccharides by CT-26 colon carcinoma cell growth inhibition and cytokine IL-7 upregulation in vitro. BMC Complement. Altern. Med. 2016, 16, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayeka, P.A.; Bian, Y.H.; Githaiga, P.M.; Zhao, Y. The immunomodulatory activities of licorice polysaccharides (Glycyrrhiza uralensis Fisch.) in CT 26 tumor-bearing mice. BMC Complement. Altern. Med. 2017, 17, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiugnanam, S.; Xu, L.; Ramaswamy, K.; Gnanasekar, M. Glycyrrhizin induces apoptosis in prostate cancer cell lines DU-145 and LNCaP. Oncol. Rep. 2008, 20, 1387–1392. [Google Scholar] [CrossRef] [Green Version]
- Chueh, F.S.; Hsiao, Y.T.; Chang, S.J.; Wu, P.P.; Yang, J.S.; Lin, J.J.; Chung, J.G.; Lai, T.Y. Glycyrrhizic acid induces apoptosis in WEHI-3 mouse leukemia cells through the caspase- and mitochondria-dependent pathways. Oncol. Rep. 2012, 28, 2069–2076. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Y.; Chu, Y.L.; Jiang, Z.B.; Chen, X.M.; Zhang, X.; Zeng, X. Glycyrrhizin suppresses lung adenocarcinoma cell growth through inhibition of thromboxane synthase. Cell. Physiol. Biochem. 2014, 33, 375–388. [Google Scholar] [CrossRef]
- Niwa, K.; Lian, Z.; Onogi, K.; Yun, W.U.; Tang, L.; Mori, H.; Tamaya, T. Preventive effects of glycyrrhizin on estrogen-related endometrial carcinogenesis in mice. Oncol. Rep. 2007, 17, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Cathcart, M.C.; Reynolds, J.V.; O’Byrne, K.J.; Pidgeon, G.P. The role of prostacyclin synthase and thromboxane synthase signaling in the development and progression of cancer. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Kang, H.E.; Lee, M.G.; Hwang, S.J.; Kim, S.C.; Lee, C.H.; Kim, S.G. Liquiritigenin, a flavonoid aglycone from licorice, has a choleretic effect and the ability to induce hepatic transporters and phase-II enzymes. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mersereau, J.E.; Levy, N.; Staub, R.E.; Baggett, S.; Zogric, T.; Chow, S.; Ricke, W.A.; Tagliaferri, M.; Cohen, I.; Bjeldanes, L.F.; et al. Liquiritigenin is a plant-derived highly selective estrogen receptor agonist. Mol. Cell. Endocrinol. 2008, 283, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Green, S.E. In vitro Comparison of Estrogenic Activities of Popular Women’s Health Botanicals. Ph.D. Thesis, University of Illinois Chicago, Chicago, IL, USA, 2015. [Google Scholar]
- Liu, C.; Wang, Y.; Xie, S.; Zhou, Y.; Ren, X.; Li, X.; Cai, Y. Liquiritigenin induces mitochondria-mediated apoptosis via cytochrome c release and caspases activation in heLa cells. Phytother. Res. 2011, 25, 277–283. [Google Scholar] [CrossRef] [PubMed]
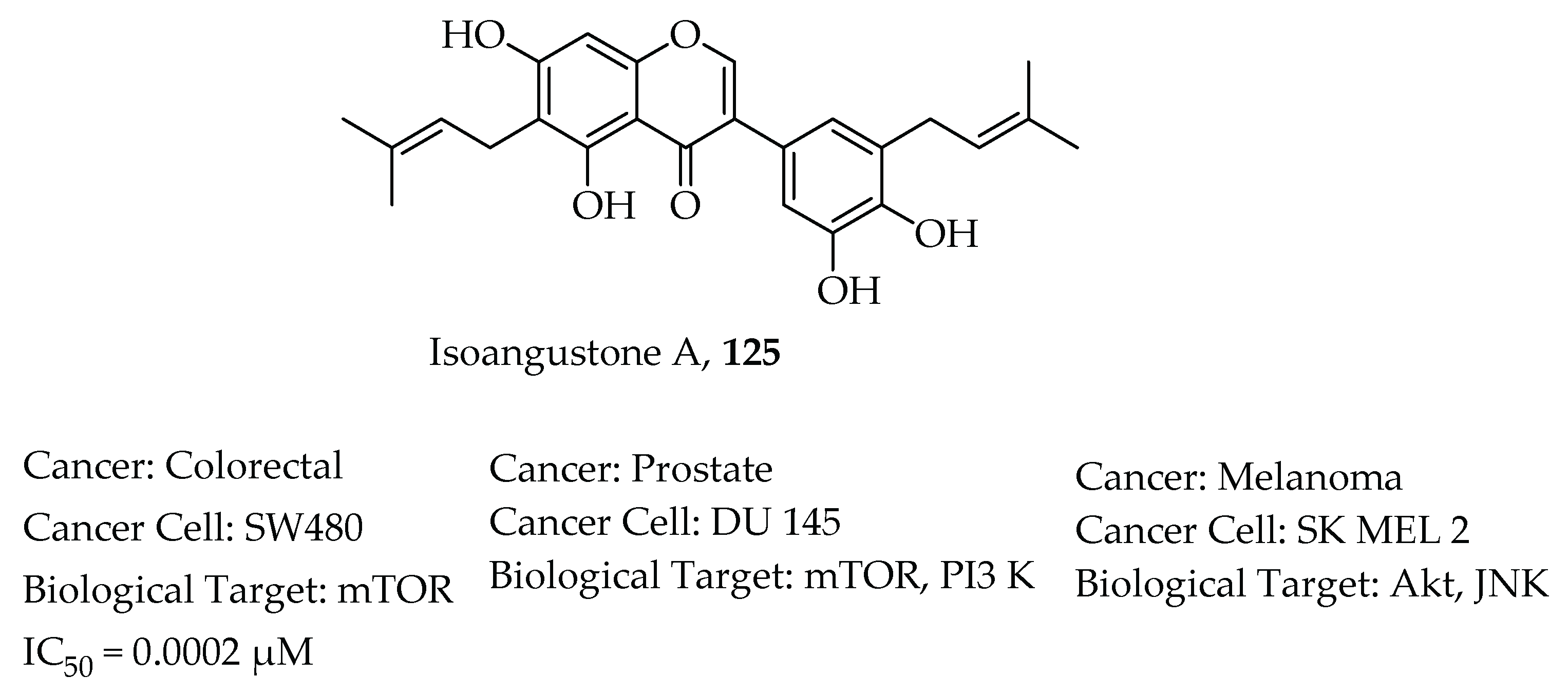
- Huang, W.; Tang, S.; Qiao, X.; Ma, W.; Ji, S.; Wang, K.; Ye, M.; Yu, S. Isoangustone A induces apoptosis in SW480 human colorectal adenocarcinoma cells by disrupting mitochondrial functions. Fitoterapia 2014, 94, 36–47. [Google Scholar] [CrossRef]
- Tang, S.; Cai, S.; Ji, S.; Yan, X.; Zhang, W.; Qiao, X.; Zhang, H.; Ye, M.; Yu, S. Isoangustone A Induces Autophagic Cell Death in Colorectal Cancer Cells by Activating AMPK Signaling. Fitoterapia 2021, 152, 104935. [Google Scholar] [CrossRef]
- Song, N.R.; Lee, E.; Byun, S.; Kim, J.E.; Mottamal, M.; Park, J.H.Y.; Lim, S.S.; Bode, A.M.; Lee, H.J.; Lee, K.W.; et al. Isoangustone A, a novel licorice compound, inhibits cell proliferation by targeting PI3K, MKK4, and MKK7 in human melanoma. Cancer Prev. Res. 2013, 6, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Seon, M.R.; Lim, S.S.; Choi, H.J.; Park, S.Y.; Cho, H.J.; Kim, J.K.; Kim, J.; Kwon, D.Y.; Park, J.H.Y. Isoangustone a present in hexane/ethanol extract of Glycyrrhiza uralensis induces apoptosis in DU145 human prostate cancer cells via the activation of DR4 and intrinsic apoptosis pathway. Mol. Nutr. Food Res. 2010, 54, 1329–1339. [Google Scholar] [CrossRef]
- Fraunfelder, F.T. Herbal medicine and dietary supplement induced ocular side effects. Clin. Ocul. Toxicol. 2008, 307–313. [Google Scholar] [CrossRef]
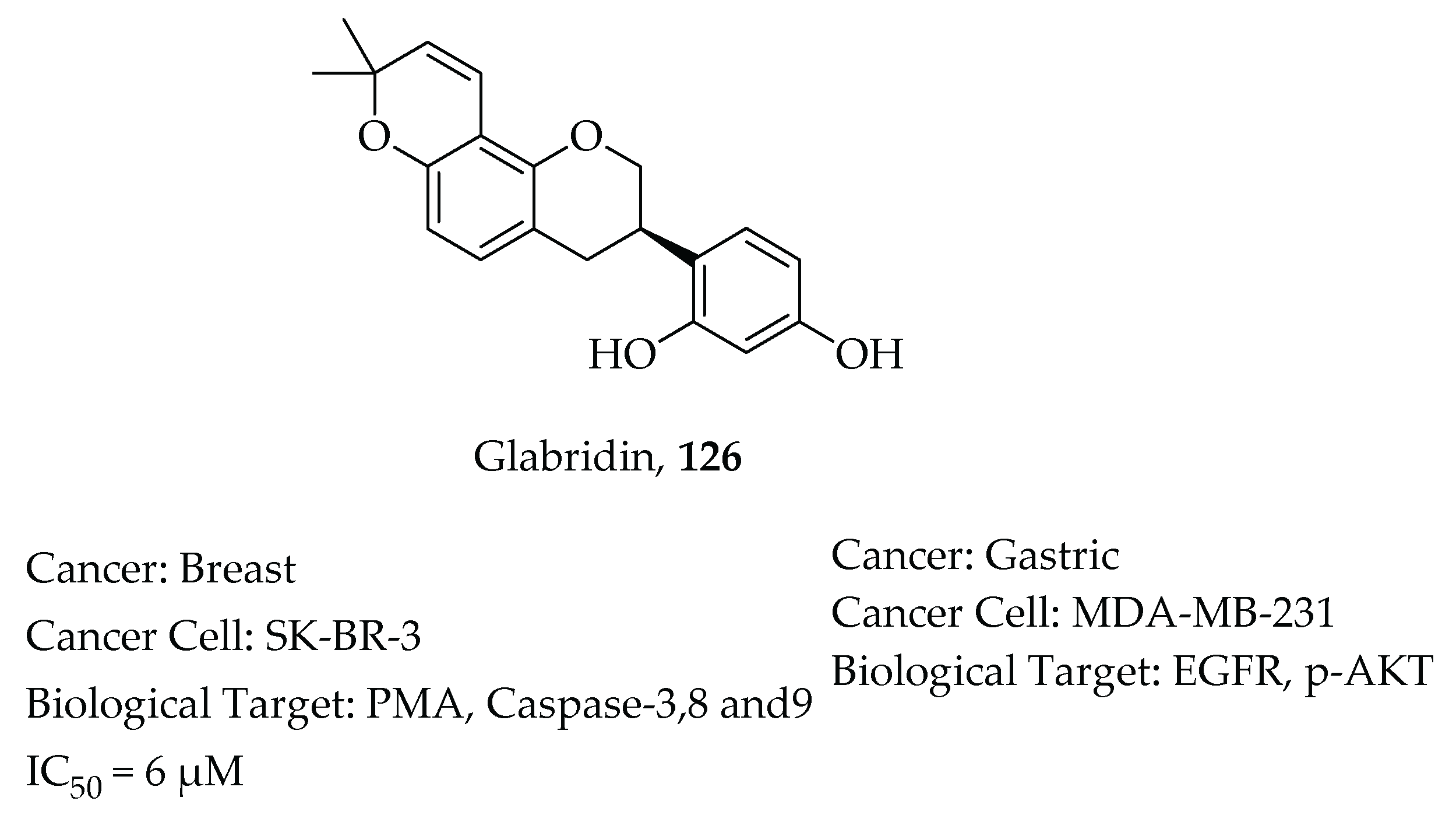
- Hsu, Y.L.; Wu, L.Y.; Hou, M.F.; Tsai, E.M.; Lee, J.N.; Liang, H.L.; Jong, Y.J.; Hung, C.H.; Kuo, P.L. Glabridin, an isoflavan from licorice root, inhibits migration, invasion and angiogenesis of MDA-MB-231 human breast adenocarcinoma cells by inhibiting focal adhesion kinase/Rho signaling pathway. Mol. Nutr. Food Res. 2011, 55, 318–327. [Google Scholar] [CrossRef]
- Kim, A.; Ma, J.Y. Isoliquiritin Apioside Suppresses in vitro Invasiveness and Angiogenesis of Cancer Cells and Endothelial Cells. Front. Pharmacol. 2018, 9, 1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.D.; Zhang, Y.; Pa, X.; Tan, R.X.; Cheng, C.H. Inhibition of xanthine oxidase by liquiritigenin and isoliquiritigenin isolated from Sinofranchetia chinensis. Cell. Mol. Life Sci. 2000, 57, 500–505. [Google Scholar] [CrossRef]
- Ji, S.; Tang, S.; Li, K.; Li, Z.; Liang, W.; Qiao, X.; Wang, Q.; Yu, S.; Ye, M. Licoricidin inhibits the growth of SW480 human colorectal adenocarcinoma cells in vitro and in vivo by inducing cycle arrest, apoptosis and autophagy. Toxicol. Appl. Pharmacol. 2017, 326, 25–33. [Google Scholar] [CrossRef]
- Park, S.Y.; Kwon, S.J.; Lim, S.S.; Kim, J.K.; Lee, K.W.; Park, J.H.Y. Licoricidin, an active compound in the hexane/ethanol extract of Glycyrrhiza uralensis, inhibits lung metastasis of 4T1 murine mammary carcinoma cells. Int. J. Mol. Sci. 2016, 17, 934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, S.; Liu, J.; Lu, Y.; Li, D. Licoricidin enhances gemcitabine-induced cytotoxicity in osteosarcoma cells by suppressing the Akt and NF-κB signal pathways. Chem. Biol. Interact. 2018, 290, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Xuan, S.H.; Park, S.N. Licoricidin, an isoflavonoid isolated from Glycyrrhiza uralensis Fisher, prevents UVA-induced photoaging of human dermal fibroblasts. Int. J. Cosmet. Sci. 2017, 39, 133–140. [Google Scholar] [CrossRef]
- Shin, E.M.; Kim, S.; Merfort, I.; Kim, Y.S. Glycyrol induces apoptosis in human Jurkat T cell lymphocytes via the Fas-FasL/caspase-8 pathway. Planta Med. 2011, 77, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Katiyar, A.; Agrawal, R.C.; Sharma, V.; Katiyar, A. Glycyrrhiza Glabra: Chemistry and Pharmacological Activity 4. Springer Int. Publ. AG 2018, 4, 87–100. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, D.; Gao, Z.; Kan, H.; Qiu, F.; Chen, L.; Li, H. Licocoumarone induces BxPC-3 pancreatic adenocarcinoma cell death by inhibiting DYRK1A. Chem. Biol. Interact. 2020, 316, 108913. [Google Scholar] [CrossRef]
- Watanabe, M.; Hayakawa, S.; Isemura, M.; Kumazawa, S.; Nakayama, T.; Mori, C.; Kawakami, T. Identification of licocoumarone as an apoptosis-inducing component in licorice. Biol. Pharm. Bull. 2002, 25, 1388–1390. [Google Scholar] [CrossRef] [Green Version]
- Ardalani, H.; Avan, A.; Ghayour-Mobarhan, M. Podophyllotoxin: A novel potential natural anticancer agent. Avicenna J. Phytomed. 2017, 7, 285. [Google Scholar] [PubMed]
- Wei, J.; Chen, J.; Ju, P.; Ma, L.; Chen, L.; Ma, W.; Zheng, T.; Yang, G.; Wang, Y.X. Synthesis and biological evaluation of 4β-n-acetylamino substituted podophyllotoxin derivatives as novel anticancer agents. Front. Chem. 2019, 7, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaled, M.; Belaaloui, G.; Jiang, Z.Z.; Zhu, X.; Zhang, L.Y. Antitumor effect of Deoxypodophyllotoxin on human breast cancer xenograft transplanted in BALB/c nude mice model. J. Infect. Chemother. 2016, 22, 692–696. [Google Scholar] [CrossRef]
- Joubouhi, C.; Mabou, F.D.; Foning Tebou, P.L.; Ngnokam, D.; Harakat, D.; Voutquenne-Nazabadioko, L. Five new iridoïd dimers from the fruits of Canthium subcordatum DC (syn. Psydrax subcordata DC). Phytochem. Lett. 2015, 13, 348–354. [Google Scholar] [CrossRef]
- Hussain, H.; Nazir, M.; Green, I.R.; Saleem, M.; Raza, M.L. Therapeutic potential of iridoid derivatives: Patent review. Inventions 2019, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Essien, E.E.; Newby, J.S.; Walker, T.M.; Setzer, W.N.; Ekundayo, O. Characterization and Antimicrobial Activity of Volatile Constituents from Fresh Fruits of Alchornea cordifolia and Canthium subcordatum. Medicines 2016, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Hirata, T.; Ikeda, T.; Fujikawa, T.; Nishibe, S. The chemistry and bioactivity of eucommia ulmoides oliver leaves. Stud. Nat. Prod. Chem. 2014, 41, 225–260. [Google Scholar] [CrossRef]
- Kim, B.C.; Kim, H.G.; Lee, S.A.; Lim, S.; Park, E.H.; Kim, S.J.; Lim, C.J. Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of mitochondrial pathway. Biochem. Pharmacol. 2005, 70, 1398–1407. [Google Scholar] [CrossRef]
- Ko, H.; Kim, J.M.; Kim, S.J.; Shim, S.H.; Ha, C.H.; Chang, H.I. Induction of apoptosis by genipin inhibits cell proliferation in AGS human gastric cancer cells via Egr1/p21 signaling pathway. Bioorg. Med. Chem. Lett. 2015, 25, 4191–4196. [Google Scholar] [CrossRef]
- Yang, X.; Yao, J.; Luo, Y.; Han, Y.; Wang, Z.; Du, L. P38 MAP kinase mediates apoptosis after genipin treatment in non-small-cell lung cancer H1299 cells via a mitochondrial apoptotic cascade. J. Pharmacol. Sci. 2013, 121, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Hsu, J.D.; Wang, C.J. Inhibition of 12-o-tetradecanoylphorbol-13-acetate-caused tumor promotion in benzo[a]pyrene-initiated CD-1 mouse skin by geniposide. Anticancer Res. 1995, 15, 411–416. [Google Scholar] [PubMed]
- Liu, J.H.; Yin, F.; Guo, L.X.; Deng, X.H.; Hu, Y.H. Neuroprotection of geniposide against hydrogen peroxide induced PC12 cells injury: Involvement of PI3 kinase signal pathway. Acta Pharmacol. Sin. 2009, 30, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, T.S.O.; Abe, M.; Hershenson, M.B.; Gomes, I.; Rosner, M.R. Thapsigargin Activates Src, Raf-l, and MAP Kinase. Cancer Res. 1997, 57, 3168–3173. [Google Scholar] [PubMed]
- Oryan, A.; Bemani, E.; Bahrami, S. Emerging role of amiodarone and dronedarone, as antiarrhythmic drugs, in treatment of leishmaniasis. Acta Trop. 2018, 185, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2-ATPase by thapsigargin analogs induces cell death via ER Ca2 depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Lee, D.I.; Rong, R.; Yu, M.; Luo, X.; Klein, M.; El-Deiry, W.S.; Huang, Y.; Hussain, A.; Sheikh, M.S. Endoplasmic reticulum calcium pool depletion-induced apoptosis is coupled with activation of the death receptor 5 pathway. Oncogene 2002, 21, 2623–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasaian, J.; Mohammadi, A. Biological activities of farnesiferol C: A review. J. Asian Nat. Prod. Res. 2018, 20, 27–35. [Google Scholar] [CrossRef]
- Hasanzadeh, D.; Mahdavi, M.; Dehghan, G.; Charoudeh, H.N. Farnesiferol C induces cell cycle arrest and apoptosis mediated by oxidative stress in MCF-7 cell line. Toxicol. Rep. 2017, 4, 420–426. [Google Scholar] [CrossRef]
- Kahraman, H. The Importance of L-Rhamnose Sugar Mini Review. Biomed. J. Tech. Res. 2019, 21, 15906–15908. [Google Scholar] [CrossRef]
- Tomsik, P.; Soukup, T.; Cermakova, E.; Micuda, S.; Niang, M.; Sucha, L.; Rezacova, M. L-rhamnose and L-fucose suppress cancer growth in mice. Cent. Eur. J. Biol. 2011, 6, 1–9. [Google Scholar] [CrossRef]
- Yau, T.; Dan, X.; Ng, C.C.W.; Ng, T.B. Lectins with potential for anti-cancer therapy. Molecules 2015, 20, 3791–3810. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Xia, T.S.; Wang, Y.F.; Zhou, W.; Liang, X.Q.; Xue, J.Q.; Shi, L.; Wang, Y.; Ding, Q. The apoptotic effect of D Rhamnose β-hederin, a novel oleanane-type triterpenoid saponin on breast cancer cells. PLoS ONE 2014, 9, 90848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitahiro, Y.; Koike, A.; Sonoki, A.; Muto, M.; Ozaki, K.; Shibano, M. Anti-inflammatory activities of Ophiopogonis Radix on hydrogen peroxide-induced cellular senescence of normal human dermal fibroblasts. J. Nat. Med. 2018, 72, 905–914. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Q.; Jiang, Y.; Li, F.; Xin, H. Effects of ophiopogonin B on the proliferation and apoptosis of SGC-7901 human gastric cancer cells. Mol. Med. Rep. 2016, 13, 4981–4986. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Dong, Q.; Ding, H. Ophiopogonin B induces reactive oxygen species-dependent apoptosis through the Hippo pathway in nasopharyngeal carcinoma. Mol. Med. Rep. 2021, 24, 534. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; De Cicco, P.; Ianaro, A. New drugs from the sea: Pro-apoptotic activity of sponges and algae derived compounds. Mar. Drugs 2019, 17, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chen, S.; Xu, S.; Yu, X.; Ma, D.; Hu, X.; Cao, X. In Vivo induction of apoptosis by fucoxanthin, a marine carotenoid, associated with down-regulating STAT3/EGFR signaling in sarcoma 180 (S180) xenografts-bearing mice. Mar. Drugs 2012, 10, 2055–2068. [Google Scholar] [CrossRef] [Green Version]
- Rokkaku, T.; Kimura, R.; Ishikawa, C.; Yasumoto, T.; Senba, M.; Kanaya, F.; Mori, N. Anticancer effects of marine carotenoids, fucoxanthin and its deacetylated product, fucoxanthinol, on osteosarcoma. Int. J. Oncol. 2013, 43, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Hosokawa, M.; Kasajima, H.; Hatanaka, K.; Kudo, K.; Shimoyama, N.; Miyashita, K. Anticancer effects of fucoxanthin and fucoxanthinol on colorectal cancer cell lines and colorectal cancer tissues. Oncol. Lett. 2015, 10, 1463–1467. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. Fucoxanthin and Its Metabolite Fucoxanthinol in Cancer Prevention and Treatment. Mar. Drugs 2015, 13, 4784–4798. [Google Scholar] [CrossRef] [Green Version]
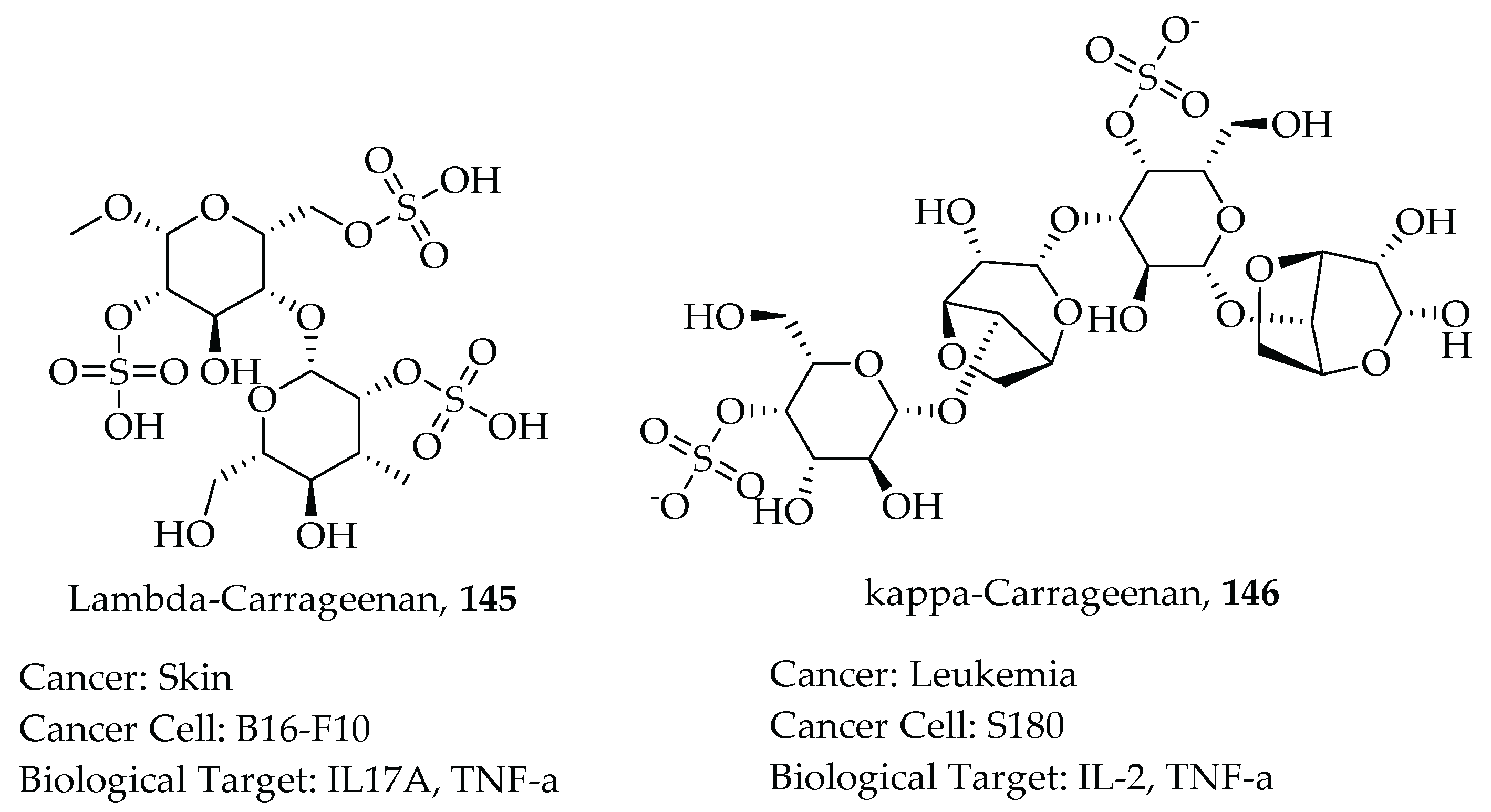
- Yuan, H.; Song, J.; Li, X.; Li, N.; Dai, J. Immunomodulation and antitumor activity of κ-carrageenan oligosaccharides. Cancer Lett. 2006, 243, 228–234. [Google Scholar] [CrossRef]
- Available online: www.new-ag.info/00-2/focuson/focuson2.html (accessed on 1 October 2021).
- Lee, V.; Tobey, J.; Castro, K.; Crawford, B. Marine Biodiversity Assets and Threats Assessment Gambia-Senegal Sustainable Fisheries Project; Coastal Resources Center, University of Rhode Island: Kingston, RI, USA, 2009. [Google Scholar]
- Necas, J.; Bartosikova, L. Carrageenan: A review. Vet. Med. 2013, 58, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Al-Nahdi, Z.M.; Al-Alawi, A.; Al-Marhobi, I. The Effect of Extraction Conditions on Chemical and Thermal Characteristics of Kappa-Carrageenan Extracted from Hypnea bryoides. J. Mar. Biol. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, V.D.; Maheriya, P.M.; Jani, G.K.; Solanki, H.K. Carrageenan: A natural seaweed polysaccharide and its applications. Carbohydr. Polym. 2014, 105, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Shao, B.; Nie, W.; Wei, X.W.; Li, Y.L.; Wang, B.L.; He, Z.Y.; Liang, X.; Ye, T.H.; Wei, Y.Q. Antitumor and Adjuvant Activity of λ-carrageenan by Stimulating Immune Response in Cancer Immunotherapy. Sci. Rep. 2015, 5, 11062. [Google Scholar] [CrossRef] [PubMed]
- Luthuli, S.; Wu, S.; Cheng, Y.; Zheng, X.; Wu, M.; Tong, H. Therapeutic effects of fucoidan: A review on recent studies. Mar. Drugs 2019, 17, 487. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.Y.; Lin, T.Y.; Lu, M.K.; Leng, P.J.; Tsao, S.M.; Wu, Y.C. Fucoidan induces Toll-like receptor 4-regulated reactive oxygen species and promotes endoplasmic reticulum stress-mediated apoptosis in lung cancer. Sci. Rep. 2017, 7, 44990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Teruya, K.; Eto, H.; Shirahata, S. Induction of apoptosis by low-molecular-weight fucoidan through calcium- and caspase-dependent mitochondrial pathways in MDA-MB-231 breast cancer cells. Biosci. Biotechnol. Biochem. 2013, 77, 235–242. [Google Scholar] [CrossRef]
- Adrian, T.E.; Collin, P. The anti-cancer effects of frondoside A. Mar. Drugs 2018, 16, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menchinskaya, E.S.; Pislyagin, E.A.; Kovalchyk, S.N.; Davydova, V.N.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I.; Aminin, D.L. Antitumor activity of cucumarioside A2-2. Chemotherapy 2014, 59, 181–191. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Menchinskaya, E.S.; Venz, S.; Rast, S.; Amann, K.; Hauschild, J.; Otte, K.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; et al. The marine triterpene glycoside frondoside A exhibits activity in vitro and in vivo in prostate cancer. Int. J. Cancer 2016, 138, 2450–2465. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Shastina, V.V.; Shin, S.W.; Xu, Q.; Park, J.I.; Rasskazov, V.A.; Avilov, S.A.; Fedorov, S.N.; Stonik, V.A.; Kwak, J.Y. Differential effects of triterpene glycosides, frondoside A and cucumarioside A2-2 isolated from sea cucumbers on caspase activation and apoptosis of human leukemia cells. FEBS Lett. 2009, 583, 697–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janakiram, N.B.; Mohammed, A.; Zhang, Y.; Choi, C.I.; Woodward, C.; Collin, P.; Steele, V.E.; Rao, C.V. Chemopreventive effects of Frondanol A5, a Cucumaria frondosa extract, against rat colon carcinogenesis and inhibition of human colon cancer cell growth. Cancer Prev. Res. 2010, 3, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Roginsky, A.B.; Ding, X.Z.; Woodward, C.; Ujiki, M.B.; Singh, B.; Bell, R.H.; Collin, P.; Adrian, T.E. Anti-pancreatic cancer effects of a polar extract from the edible sea cucumber, cucumaria frondosa. Pancreas 2010, 39, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Liu, Z.D.; Xue, Y.; Wang, J.F.; Li, H.; Tang, Q.J.; Wang, Y.M.; Dong, P.; Xue, C.H. Ds-echinoside A, a new triterpene glycoside derived from sea cucumber, exhibits antimetastatic activity via the inhibition of NF-κB-dependent MMP-9 and VEGF expressions. J. Zhejiang Univ. Sci. B 2011, 12, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Zhang, X.; Tian, F.; Yi, Y.; Xu, Q.; Li, L.; Tong, L.; Lin, L.; Ding, J. Philinopside A, a novel marine-derived compound possessing dual anti-angiogenic and anti-tumor effects. Int. J. Cancer 2005, 114, 843–853. [Google Scholar] [CrossRef]
- Tian, F.; Zhu, C.H.; Zhang, X.W.; Xie, X.; Xin, X.L.; Yi, Y.H.; Lin, L.P.; Geng, M.Y.; Ding, J. Philinopside E, a new sulfated saponin from sea cucumber, blocks the interaction between kinase insert domain-containing receptor (KDR) and αvβ3 integrin via binding to the extracellular domain of KDR. Mol. Pharmacol. 2007, 72, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Dinku, W.; Isaksson, J.; Rylandsholm, F.G.; Bouř, P.; Brichtová, E.; Choi, S.U.; Lee, S.H.; Jung, Y.S.; No, Z.S.; Svendsen, J.S.M.; et al. Anti-proliferative activity of a novel tricyclic triterpenoid acid from Commiphora africana resin against four human cancer cell lines. Appl. Biol. Chem. 2020, 63, 1–11. [Google Scholar] [CrossRef]
- Zhang, S.L.; Li, L.; Yi, Y.H.; Sun, P. Philinopsides E and F, two new sulfated triterpene glycosides from the sea cucumber Pentacta quadrangularis. Nat. Prod. Res. 2006, 20, 399–407. [Google Scholar] [CrossRef]
- Malyarenko, O.S.; Dyshlovoy, S.A.; Kicha, A.A.; Ivanchina, N.V.; Malyarenko, T.V.; Carsten, B.; Gunhild, V.A.; Stonik, V.A.; Ermakova, S.P. The Inhibitory Activity of Luzonicosides from the Starfish Echinaster luzonicus against Human Melanoma Cells. Mar. Drugs 2017, 15, 227. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, K.; Yamagishi, T.; Ichihara, T.; Nakaike, S.; Iguchi, K.; Yamada, Y.; Fukumoto, H.; Yoneda, T.; Samata, K.; Ikeya, H.; et al. Mechanism of action of aragusterol A (YTA0040), a potent anti-tumor marine steroid targeting the G1 phase of the cell cycle. Int. J. Cancer 2000, 88, 810–819. [Google Scholar] [CrossRef]
- Wu, S.Y.; Sung, P.J.; Chang, Y.L.; Pan, S.L.; Teng, C.M. Heteronemin, a spongean sesterterpene, induces cell apoptosis and autophagy in human renal carcinoma cells. Biomed. Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; Jaspars, M.; Dicato, M.; Diederich, M. Heteronemin, a spongean sesterterpene, inhibits TNFα-induced NF-κB activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol. 2010, 79, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Huang, M.Y.; Zhang, L.L.; Feng, Z.L.; Jiang, X.M.; Yuan, L.W.; Huang, R.Y.; Liu, B.; Yu, H.; Wang, Y.T.; et al. Nagilactone E increases PD-L1 expression through activation of c-Jun in lung cancer cells. Chin. J. Nat. Med. 2020, 18, 517–525. [Google Scholar] [CrossRef]
- Zhang, L.L.; Guo, J.; Jiang, X.M.; Chen, X.P.; Wang, Y.T.; Li, A.; Lin, L.G.; Li, H.; Lu, J.J. Identification of nagilactone E as a protein synthesis inhibitor with anticancer activity. Acta Pharmacol. Sin. 2020, 41, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Jiang, X.M.; Huang, M.Y.; Feng, Z.L.; Chen, X.; Wang, Y.; Li, H.; Li, A.; Lin, L.G.; Lu, J.J. Nagilactone E suppresses TGF-β1-induced epithelial–mesenchymal transition, migration and invasion in non-small cell lung cancer cells. Phytomedicine 2019, 52, 32–39. [Google Scholar] [CrossRef]
- Tang, S.A.; Zhou, Q.; Guo, W.Z.; Qiu, Y.; Wang, R.; Jin, M.; Zhang, W.; Li, K.; Yamori, T.; Dan, S.; et al. In vitro antitumor activity of stellettin B, a triterpene from marine sponge Jaspis stellifera, on human glioblastoma cancer SF295 cells. Mar. Drugs 2014, 12, 4200–4213. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Crambescidin 800, isolated from the marine sponge monanchora viridis, induces cell cycle arrest and apoptosis in triple-negative breast cancer cells. Mar. Drugs 2018, 16, 53. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Kim, G.D.; Jeon, J.E.; Shin, J.; Lee, S.K. Antimetastatic effect of halichondramide, a trisoxazole macrolide from the marine sponge chondrosia corticata, on human prostate cancer cells via modulation of epithelial-to-mesenchymal transition. Mar. Drugs 2013, 11, 2472–2485. [Google Scholar] [CrossRef]
- Bae, S.Y.; Kim, G.D.; Jeon, J.E.; Shin, J.; Lee, S.K. Anti-proliferative effect of (19Z)-halichondramide, a novel marine macrolide isolated from the sponge Chondrosia corticata, is associated with G2/M cell cycle arrest and suppression of mTOR signaling in human lung cancer cells. Toxicol. In Vitro 2013, 27, 694–699. [Google Scholar] [CrossRef]
- Suna, H.; Aoki, S.; Setiawan, A.; Kobayashi, M. Crambescidin 800, a pentacyclic guanidine alkaloid, protects a mouse hippocampal cell line against glutamate-induced oxidative stress. J. Nat. Med. 2007, 61, 288–295. [Google Scholar] [CrossRef]
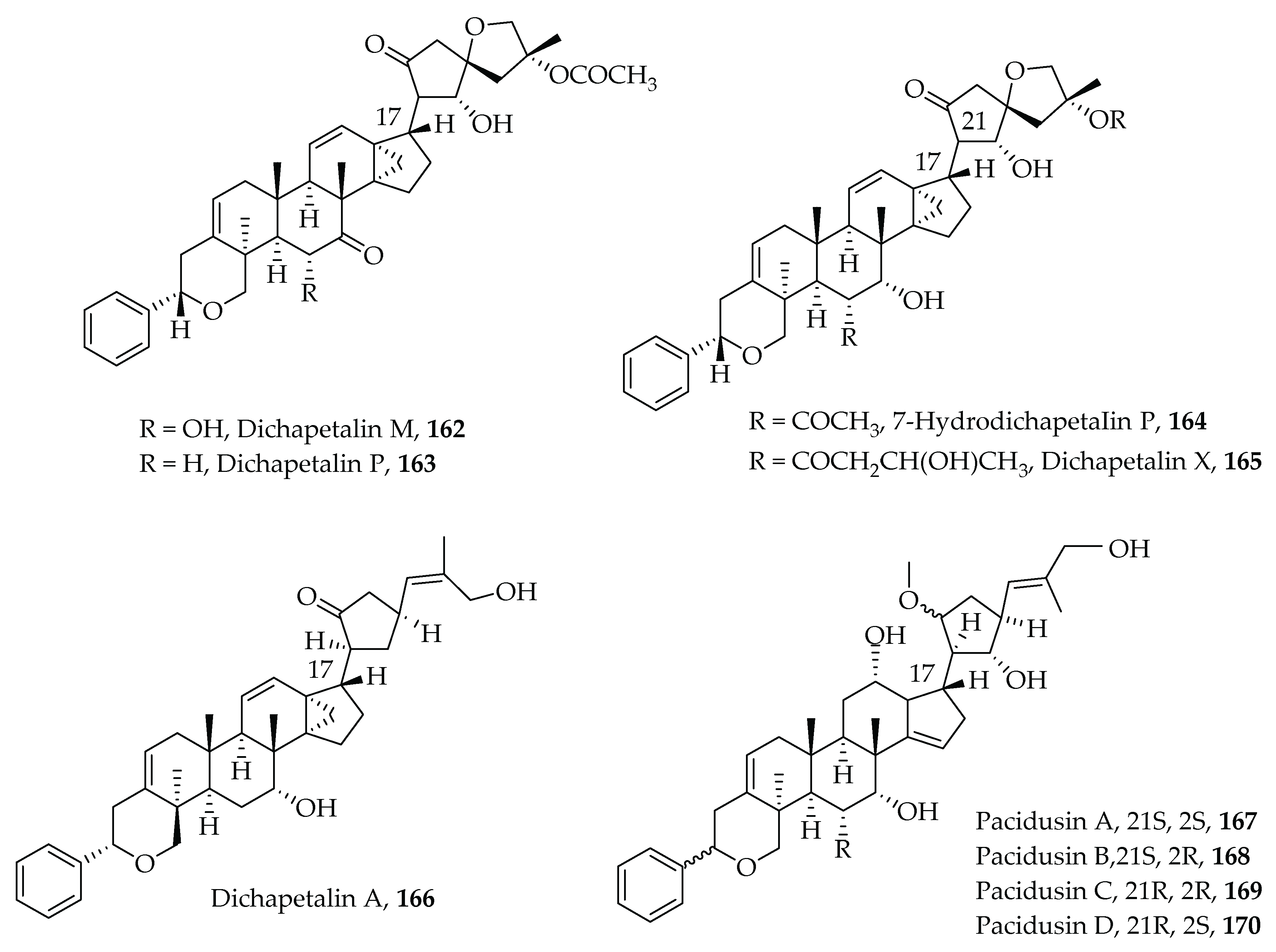
- Achenbach, H.; Waibel, R.; Asonka, S.A.; Addae-Mensah, I.; Oppong, I.V. Dichapetalin A, a novel plant constituent from Dichapetalum madagascariense with potential antineoplastic activity. Nat. Prod. Lett. 1995, 7, 93–100. [Google Scholar] [CrossRef]
- Addae-Mensah, I.; Waibel, R.; Asunka, S.A.; Oppong, I.V.; Achenbach, H. The dichapetalins—A new class of triterpenoids. Phytochemistry 1996, 43, 649–656. [Google Scholar] [CrossRef]
- Osei-Safo, D.; Chama, M.A.; Addae-Mensah, I.; Waibel, R.; Asomaning, W.A.; Oppong, I.V. Dichapetalin M from Dichapetalum madagascariensis. Phytochem. Lett. 2008, 1, 147–150. [Google Scholar] [CrossRef]
- Tuchinda, P.; Kornsakulkarn, J.; Pohmakotr, M.; Kongsaeree, P.; Prabpai, S.; Yoosook, C.; Kasisit, J.; Napaswad, C.; Sophasan, S.; Reutrakul, V. Dichapetalin-type triterpenoids and lignans from the aerial parts of Phyllanthus acutissima. J. Nat. Prod. 2008, 71, 655–663. [Google Scholar] [CrossRef]
- Long, C.; Aussagues, Y.; Molinier, N.; Marcourt, L.; Vendier, L.; Samson, A.; Poughon, V.; Mutiso, P.B.C.; Ausseil, F.; Sautel, F.; et al. Dichapetalins from Dichapetalum species and their cytotoxic properties. Phytochemistry 2013, 94, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Osei-Safo, D.; Dziwornu, G.A.; Appiah-Opong, R.; Chama, M.A.; Tuffour, I.; Waibel, R.; Amewu, R.; Addae-Mensah, I.; Newman, D.J. Constituents of the Roots of Dichapetalum pallidum and Their Anti-Proliferative Activity. Molecules 2017, 22, 532. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.-C.; Zhu, H.-T.; Yang, W.-N.; Wang, D.; Yang, C.-R.; Zhang, Y.-J. New cytotoxic dichapetalins in the leaves of Phyllanthus acidus: Identification, quantitative analysis, and preliminary toxicity assessment. Bioorg. Chem. 2021, 114, 105125. [Google Scholar] [CrossRef]
- Blackwell, M. The fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef]
- Jakubczyk, D.; Dussart, F. Selected Fungal Natural Products with Antimicrobial Properties. Molecules 2020, 25, 911. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Yates, C.; Gary, B.D.; McClellan, S.; Tan, M.; Xi, Y.; Reed, E.; Piazza, G.A.; Owen, L.B.; Dean-Colomb, W. Panepoxydone targets NF-κB and FOXM1 to inhibit proliferation, induce apoptosis and reverse epithelial to mesenchymal transition in breast cancer. PLoS ONE 2014, 9, e98370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rether, J.; Erkel, G.; Anke, T.; Sterner, O. Inhibition of inducible TNF-α expression by oxaspirodion, a novel spiro-compound from the ascomycete Chaetomium subspirale. Biol. Chem. 2004, 385, 829–834. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Uranchimeg, B.; Cardellina, J.H.; Meragelman, T.L.; Matsunaga, S.; Fusetani, N.; Del Bufalo, D.; Shoemaker, R.H.; Melillo, G. Induction of apoptosis in human cancer cells by candidaspongiolide, a novel sponge polyketide. J. Natl. Cancer Inst. 2008, 100, 1233–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitson, E.L.; Pluchino, K.M.; Hall, M.D.; McMahon, J.B.; McKee, T.C. New candidaspongiolides, tedanolide analogues that selectively inhibit melanoma cell growth. Org. Lett. 2011, 13, 3518–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Lu, Z.; Meng, L.; Wei, S.; Hong, K.; Zhu, W.; Huang, C. The novel agent ophiobolin O induces apoptosis and cell cycle arrest of MCF-7 cells through activation of MAPK signaling pathways. Bioorg. Med. Chem. Lett. 2012, 22, 579–585. [Google Scholar] [CrossRef]
- Lv, C.; Qin, W.; Zhu, T.; Wei, S.; Hong, K.; Zhu, W.; Chen, R.; Huang, C. Ophiobolin O isolated from Aspergillus ustus induces G1 arrest of MCF-7 cells through interaction with AKT/GSK3β/cyclin D1 signaling. Mar. Drugs 2015, 13, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.Y.; Zhou, T.; Zhao, Y.Y.; Chen, L.; Gong, M.W.; Xia, Q.W.; Ying, M.G.; Zheng, Q.H.; Zhang, Q.Q. Antitumor effects and related mechanisms of Penicitrinine A, a novel alkaloid with a unique spiro skeleton from the marine fungus Penicillium citrinum. Mar. Drugs 2015, 13, 4733–4753. [Google Scholar] [CrossRef] [PubMed]
- Dewangan, J.; Srivastava, S.; Rath, S.K. Salinomycin: A new paradigm in cancer therapy. Tumor Biol. 2017, 39, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.Y.; Kim, S.H.; Yu, S.N.; Park, S.K.; Choi, H.D.; Yu, H.S.; Ji, J.H.; Seo, Y.K.; Ahn, S.C. Salinomycin enhances doxorubicin-induced cytotoxicity in multidrug resistant MCF-7/MDR human breast cancer cells via decreased efflux of doxorubicin. Mol. Med. Rep. 2015, 12, 1898–1904. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hao, Y.; Li, Y.; Zheng, Y.; Dai, J.; Zhong, F.; Wei, W.; Fang, Z. Salinomycin induces autophagic cell death in salinomycin-sensitive melanoma cells through inhibition of autophagic flux. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Lee, S.J.; Moon, G.S.; Jung, K.H.; Kim, W.J.; Moon, S.K. c-Jun N-terminal kinase 1 is required for cordycepin-mediated induction of G2/M cell-cycle arrest via p21WAF1 expression in human colon cancer cells. Food Chem. Toxicol. 2010, 48, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.C.; Hwang, J.H.; Jo, E.; Kim, Y.R.; Kim, D.J.; Lee, K.B.; Park, S.J.; Jang, I.S. Cordycepin induces apoptosis by caveolin-1-mediated JNK regulation of Foxo3a in human lung adenocarcinoma. Oncotarget 2017, 8, 12211–12224. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Tabanca, N.; Kiss, R. Assessing the anticancer effects associated with food products and/or nutraceuticals using in vitro and in vivo preclinical development-related pharmacological tests. Semin. Cancer Biol. 2017, 46, 14–32. [Google Scholar] [CrossRef] [PubMed]
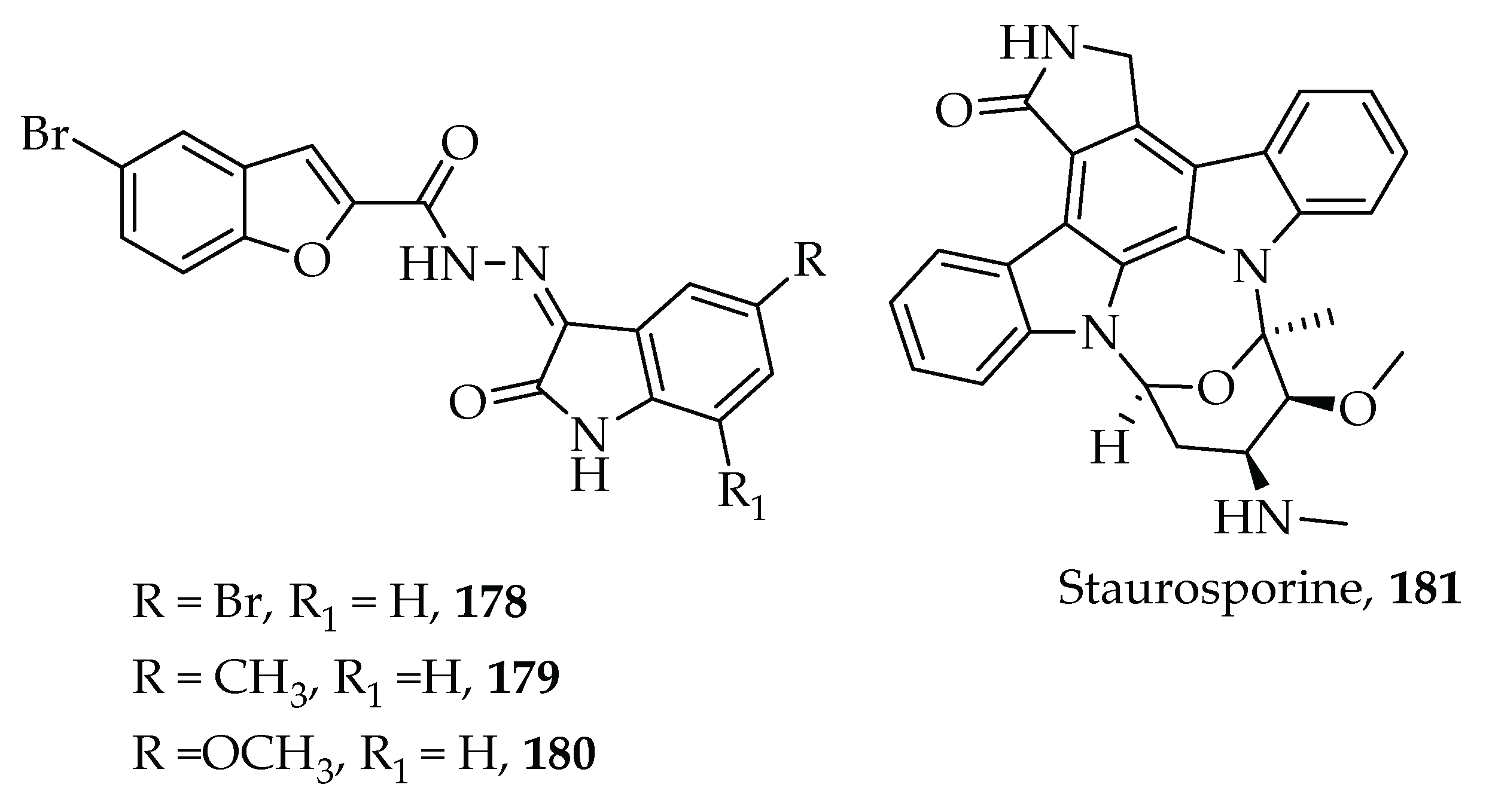
- Eldehna, W.M.; Al-Rashood, S.T.; Al-Warhi, T.; Eskandrani, R.O.; Alharbi, A.; Kerdawy, A.M. El Novel oxindole/benzofuran hybrids as potential dual CDK2/GSK-3β inhibitors targeting breast cancer: Design, synthesis, biological evaluation, and in silico studies. J. Enzyme Inhib. Med. Chem. 2021, 36, 270–285. [Google Scholar] [CrossRef] [PubMed]















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | Cpd | c-Met | c-Kit | Flt-3 | VEGFR-2 | EGFR | PDGFR | Pim-1 |
|---|---|---|---|---|---|---|---|---|
 | 94 | 3.42 | 0.32 | 0.24 | 0.63 | 0.42 | 0.57 | >10,000 |
 | 95 96 | 0.24 0.35 | 0.23 3.08 | 0.19 3.16 | 0.59 5.73 | 1.03 2.92 | 0.36 2.73 | 420 260 |
 | 97 98 99 | 0.27 0.32 0.48 | 0.68 0.36 1.17 | 0.47 0.29 1.34 | 0.83 0.64 0.93 | 5.06 0.52 2.53 | 2.27 0.38 1.03 | 230 400 >10,000 |
 | 100 101 102 103 | 0.42 0.68 0.49 0.42 | 0.62 0.52 0.16 0.63 | 0.49 0.21 0.24 0.51 | 0.26 0.53 0.37 1.08 | 0.38 0.80 0.49 0.74 | 0.41 0.46 0.22 0.80 | 360 290 >10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amewu, R.K.; Sakyi, P.O.; Osei-Safo, D.; Addae-Mensah, I. Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets. Molecules 2021, 26, 7134. https://doi.org/10.3390/molecules26237134
Amewu RK, Sakyi PO, Osei-Safo D, Addae-Mensah I. Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets. Molecules. 2021; 26(23):7134. https://doi.org/10.3390/molecules26237134
Chicago/Turabian StyleAmewu, Richard Kwamla, Patrick Opare Sakyi, Dorcas Osei-Safo, and Ivan Addae-Mensah. 2021. "Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets" Molecules 26, no. 23: 7134. https://doi.org/10.3390/molecules26237134
APA StyleAmewu, R. K., Sakyi, P. O., Osei-Safo, D., & Addae-Mensah, I. (2021). Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets. Molecules, 26(23), 7134. https://doi.org/10.3390/molecules26237134