
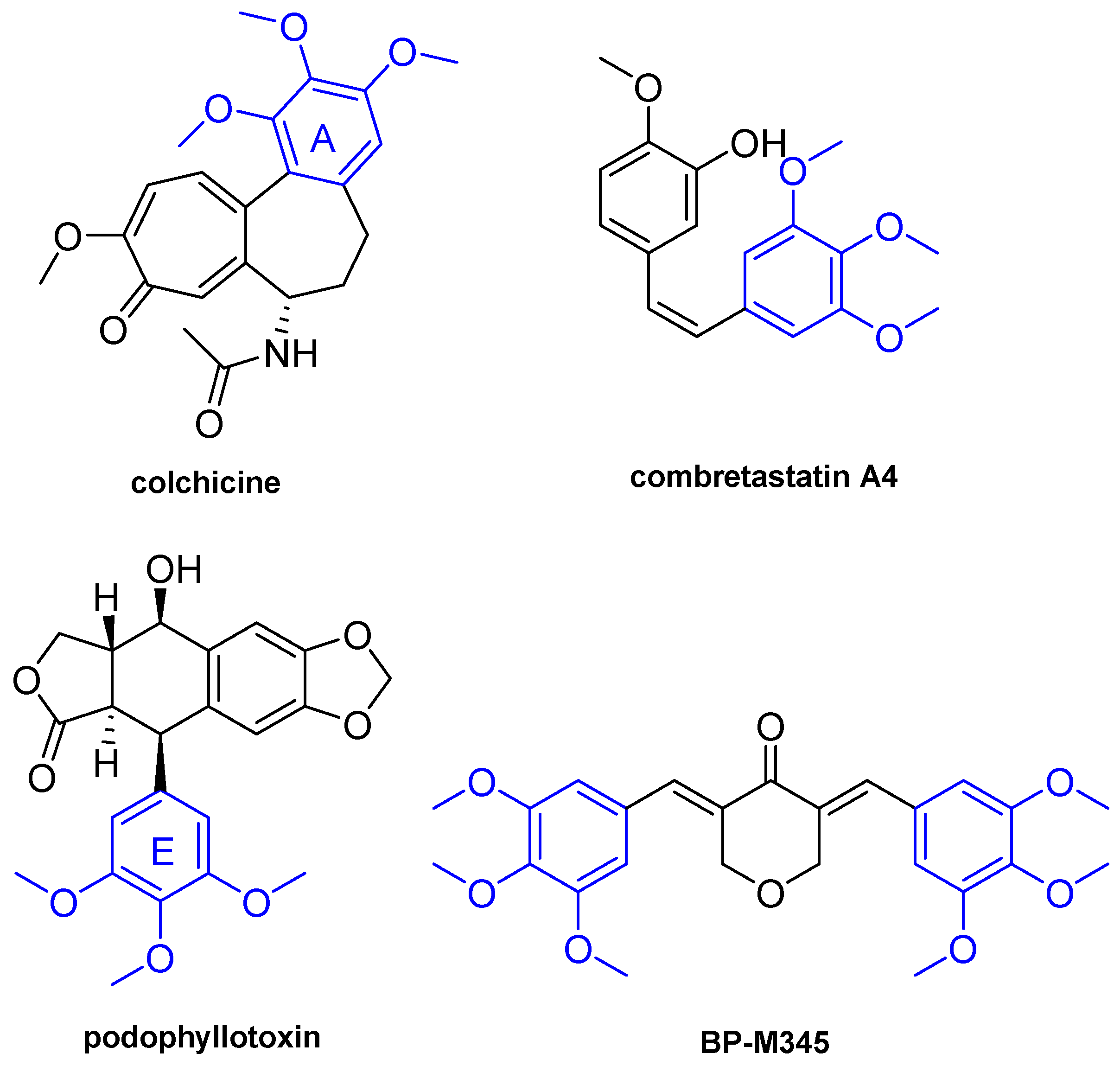
BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity

 ,
,  ,
,  , ,
, ,  ,
,  and
and 
Abstract
:
1. Introduction
2. Results
2.1. Compound BP-M345 Exhibits a Potent Tumor Growth Inhibitory Activity with High Selectivity Index
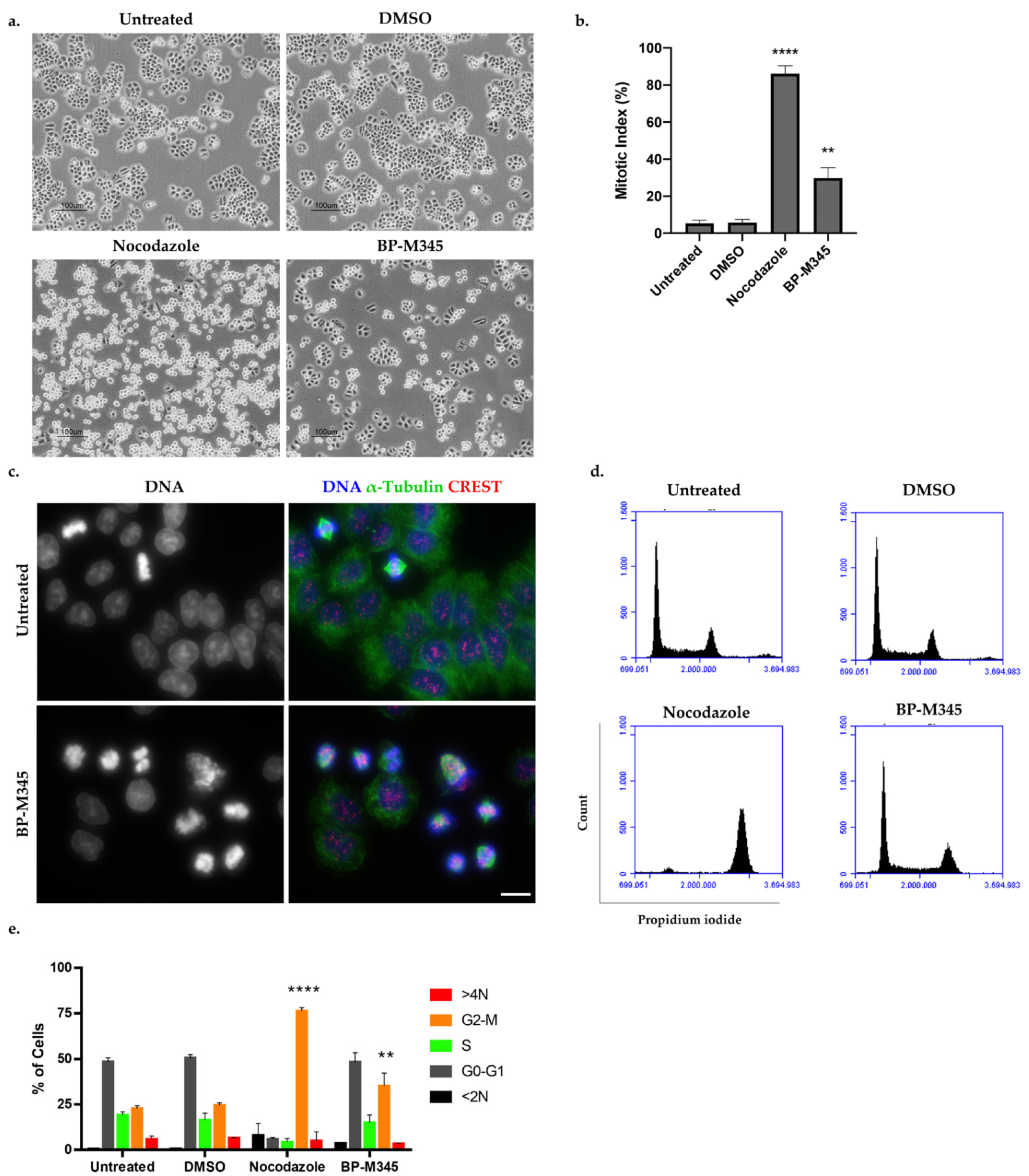
2.2. Compound BP-M345 Arrests Tumor Cells at Mitosis
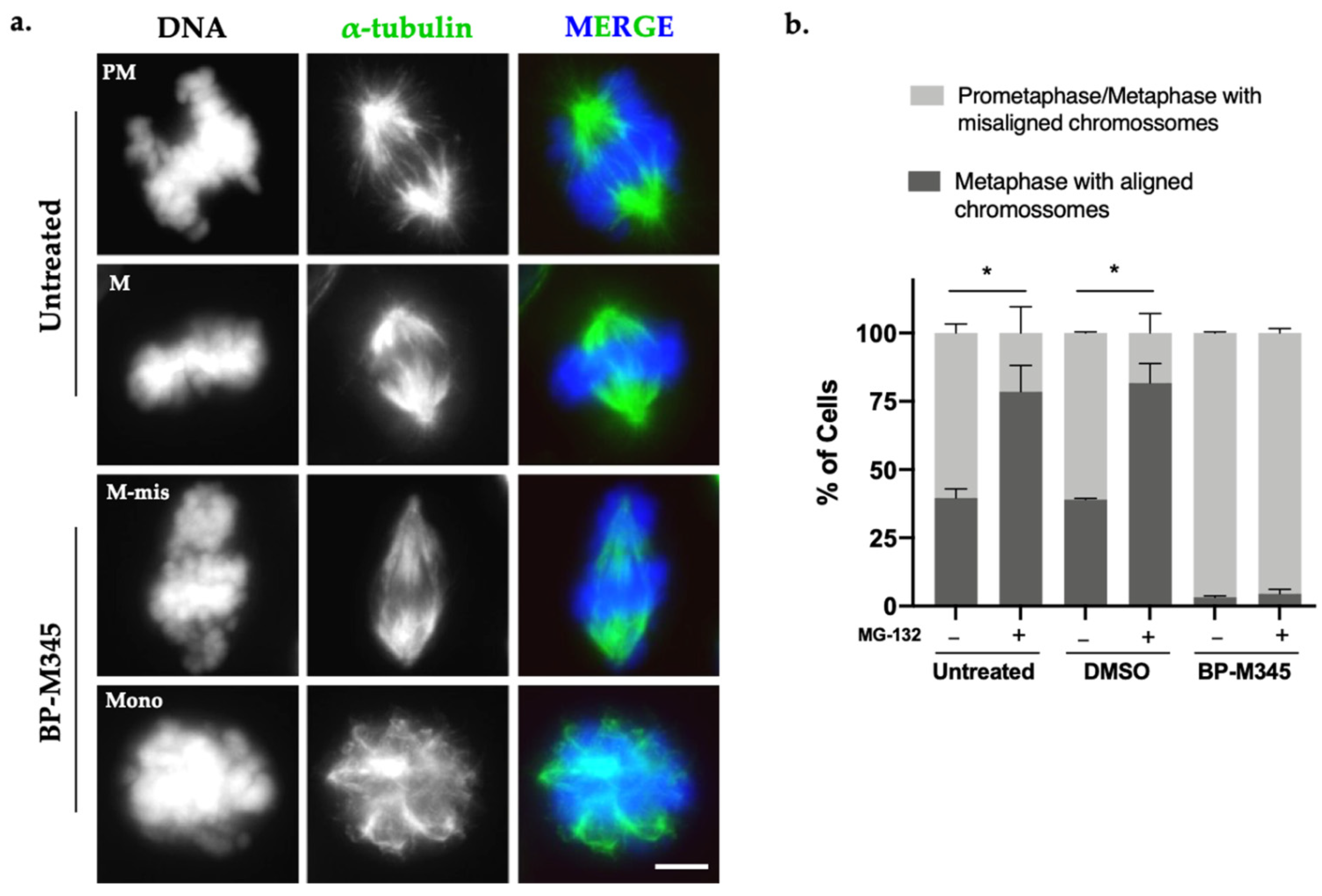
2.3. Compound BP-M345 Affects Mitotic Spindle Morphology and Disturbs Chromosome Congression
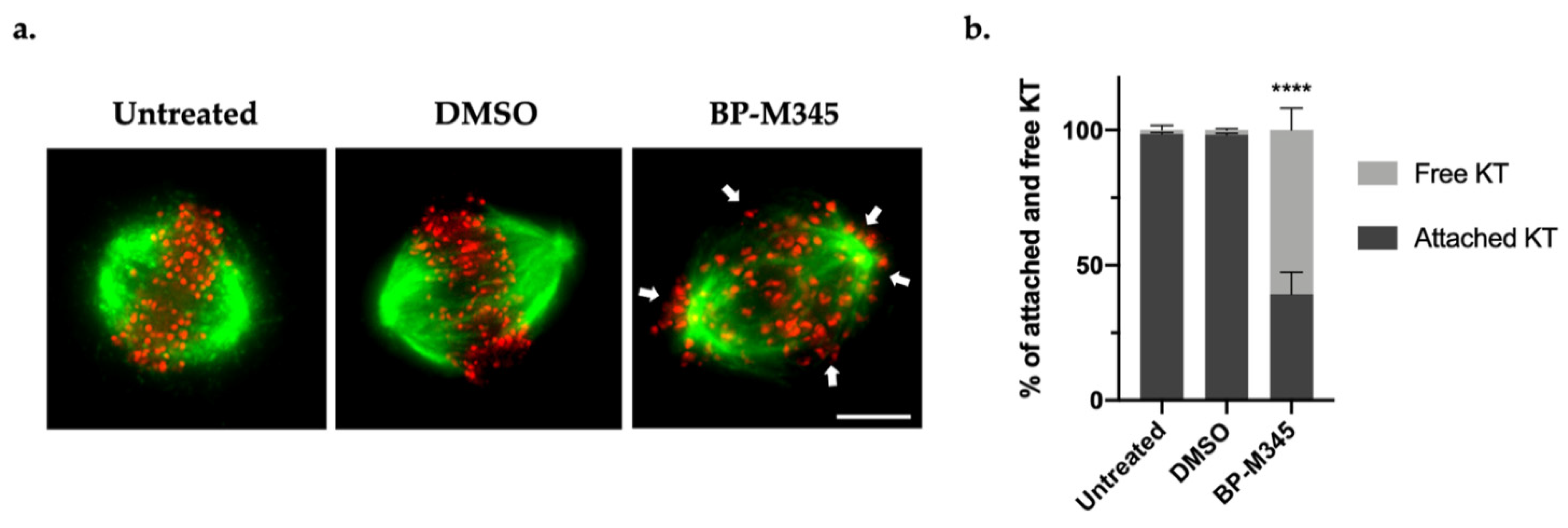
2.4. BP-M345 Interferes with the Stability of Kinetochore-Microtubule Attachments and Promotes SAC Activation
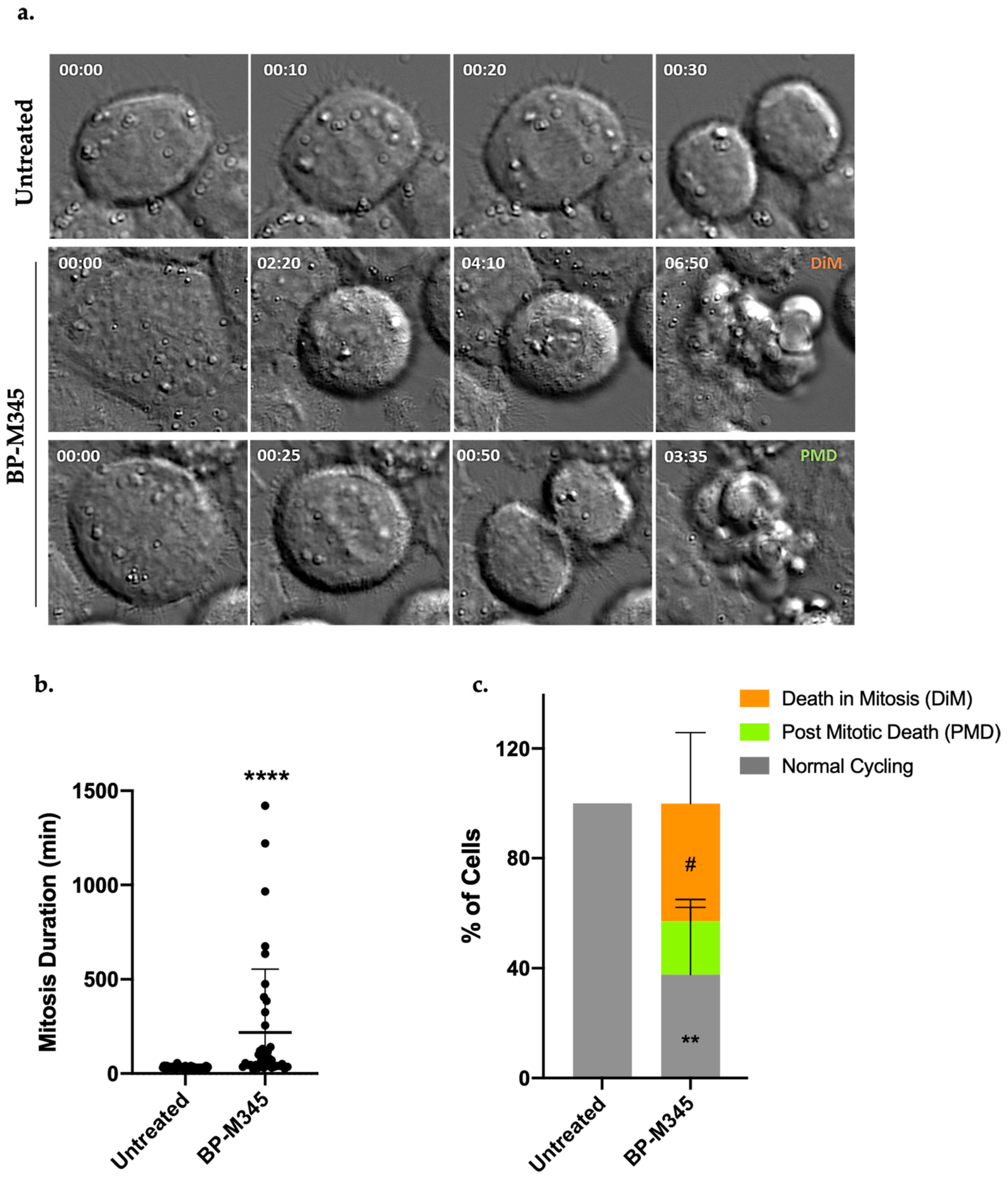
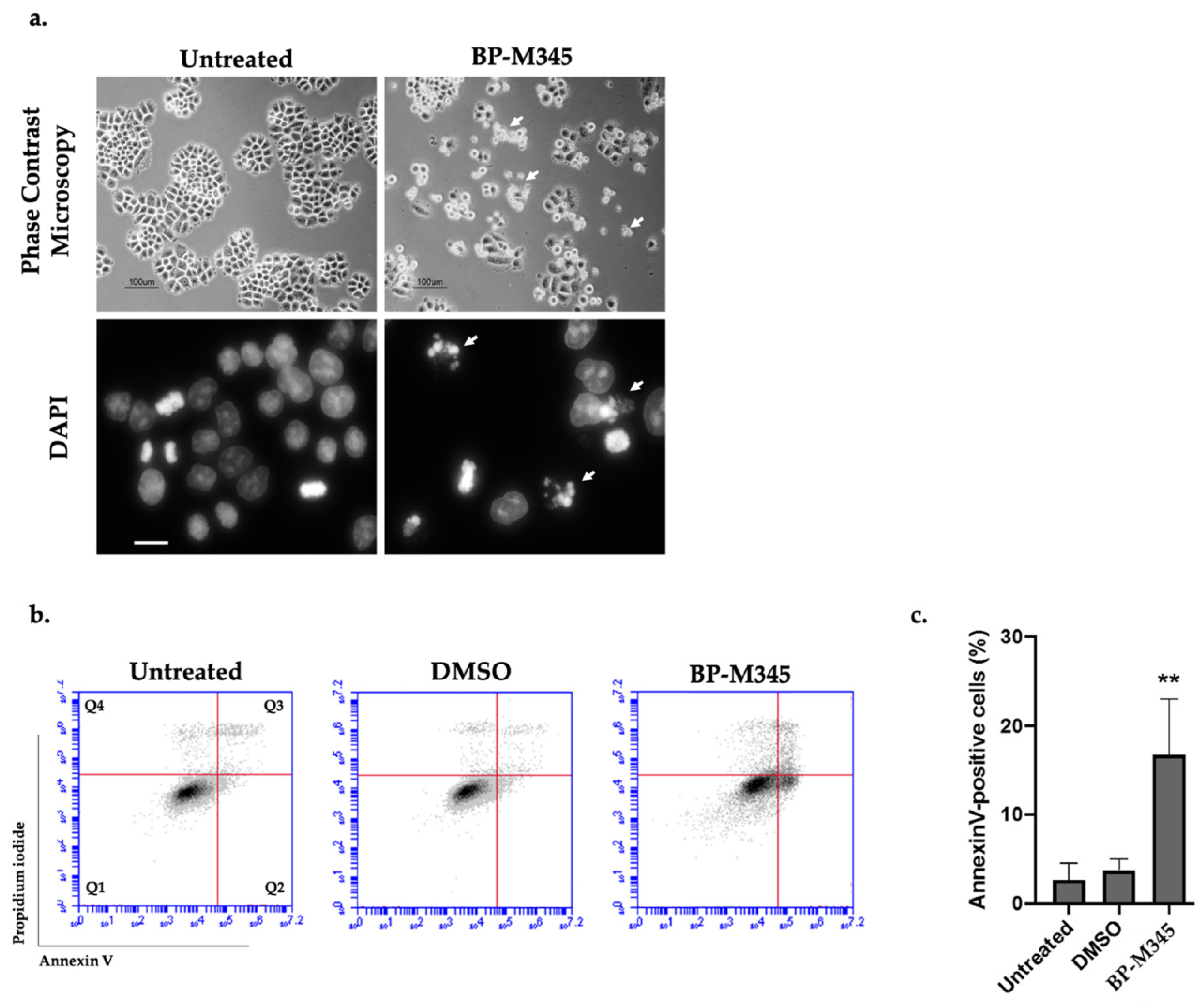
2.5. Fate of BP-M345-Treated Cells Arrested in Mitosis
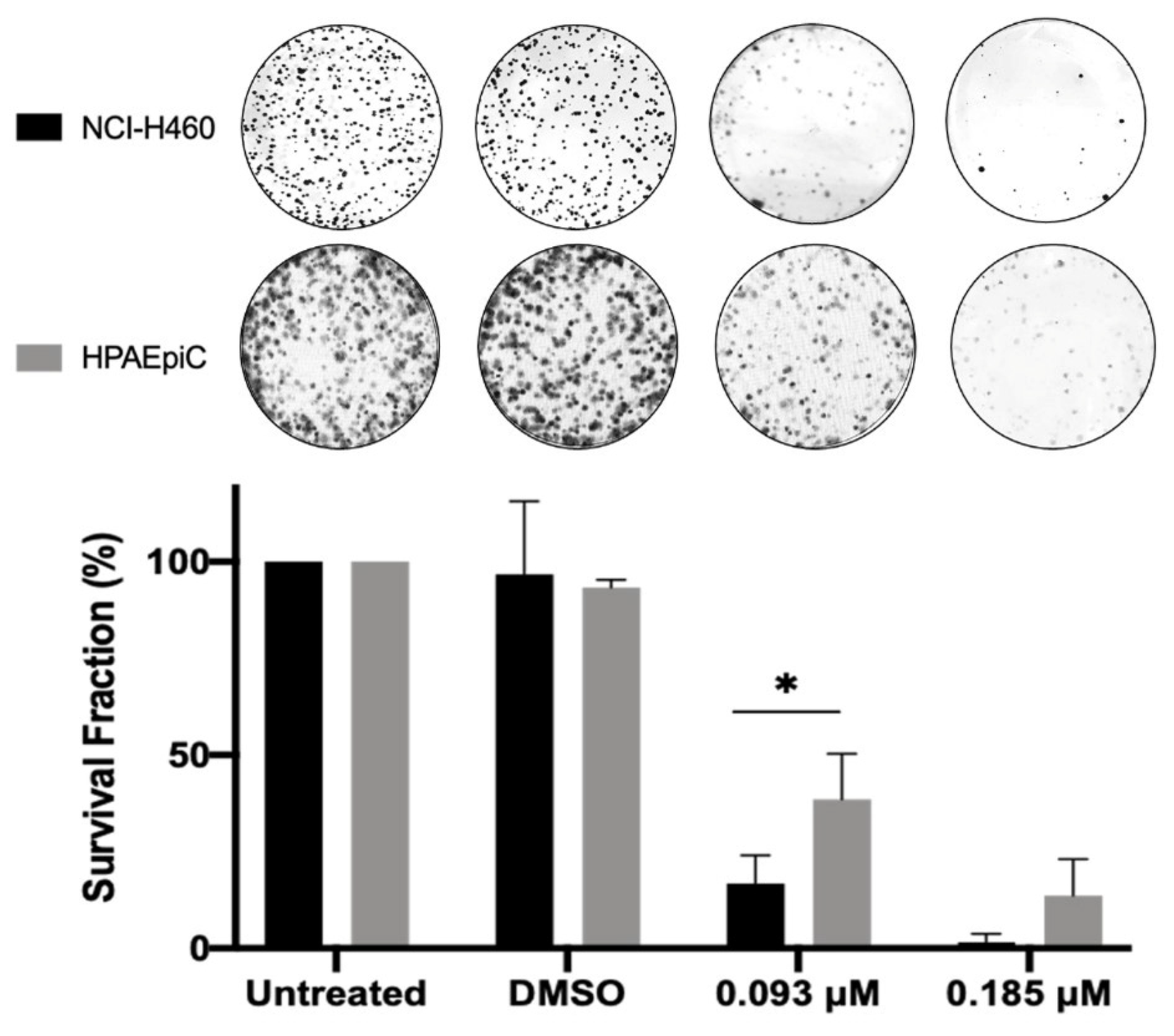
2.6. BP-M345 Exerts a Long-Term Inhibitory Effect on Tumor Cell Proliferation
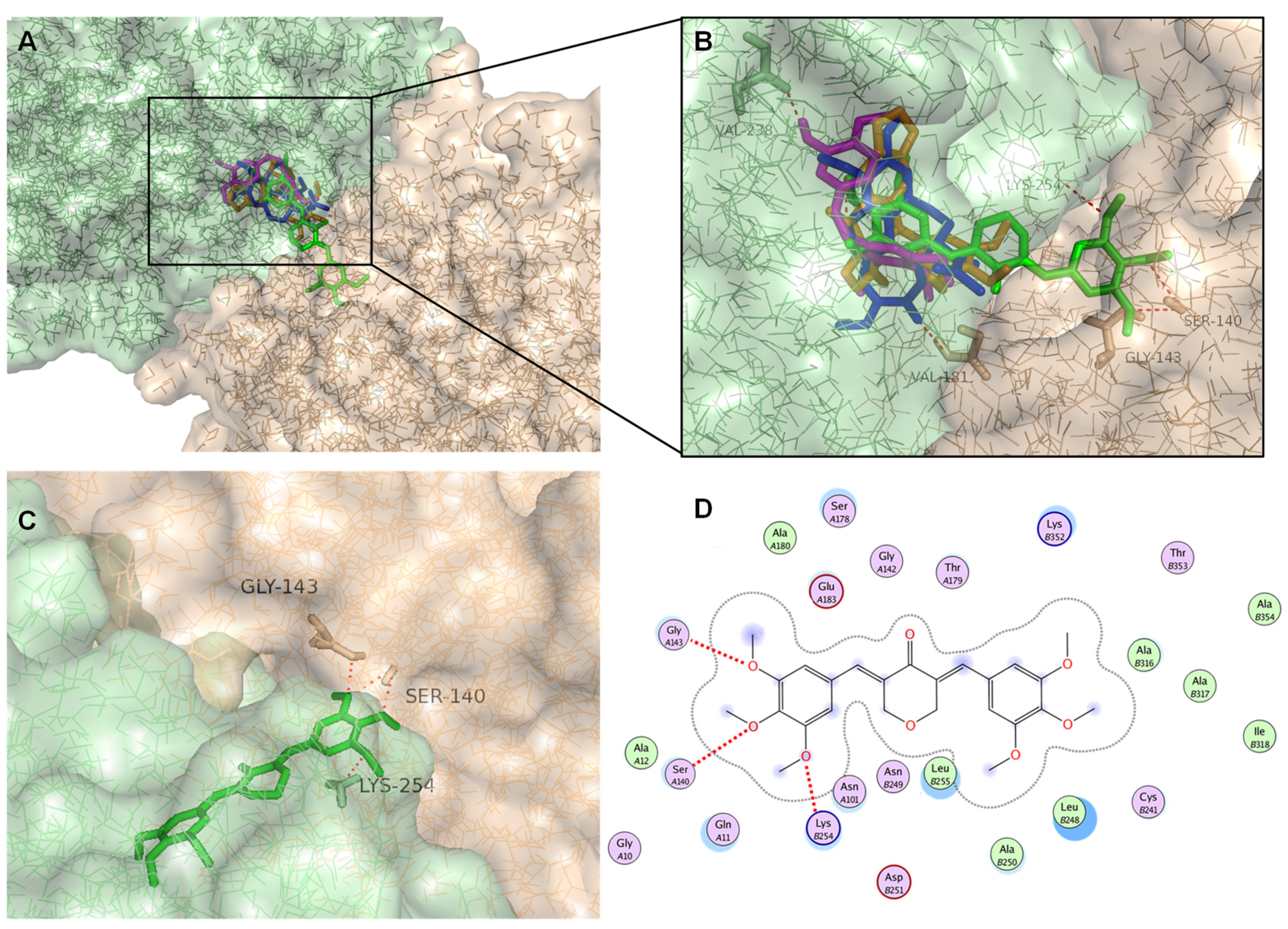
2.7. Docking Studies
3. Discussion
4. Material and Methods
4.1. Chemicals
4.2. Cell Lines and Culture Conditions
4.3. Sulforodamine B (SRB) Assay
4.4. Mitotic Index Determination
4.5. Flow Cytometry
4.6. Indirect Immunofluorescence
4.7. MG-132 Proteasome Inhibitor Assay
4.8. Cold Treatment Assay
4.9. Colony Formation Assay
4.10. Live-Cell Imaging
4.11. Image Acquisition and Processing
4.12. Statistical Analysis
4.13. Virtual Screening and Docking Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett. 2019, 440, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Florian, S.; Mitchison, T.J. Anti-Microtubule Drugs. Methods Mol. Biol. 2016, 1413, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.; Barbosa, J.; Nascimento, A.V.; Faria, J.; Reis, R.; Bousbaa, H. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011, 44, 391–400. [Google Scholar] [CrossRef]
- Lara-Gonzalez, P.; Pines, J.; Desai, A. Spindle assembly checkpoint activation and silencing at kinetochores. Semin. Cell Dev. Biol. 2021, 117, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.M.A.; Reis, R.M.; Bolanos-Garcia, V.M.; Florindo, C.; Tavares, Á.A.; Bousbaa, H. Dynein-dependent transport of spindle assembly checkpoint proteins off kinetochores toward spindle poles. FEBS Lett. 2014, 588, 3265–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musacchio, A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Karahalil, B.; Yardım-Akaydin, S.; Nacak Baytas, S. An overview of microtubule targeting agents for cancer therapy. Arch. Ind. Hyg. Toxicol. 2019, 70, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic slippage: An old tale with a new twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoigne, K.E.; Taylor, S.S. Cancer Cells Display Profound Intra- and Interline Variation following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Kanakkanthara, A. Beyond the Paclitaxel and Vinca Alkaloids: Next Generation of Plant-Derived Microtubule-Targeting Agents with Potential Anticancer Activity. Cancers 2020, 12, 1721. [Google Scholar] [CrossRef]
- Cao, Y.-N.; Zheng, L.-L.; Wang, D.; Liang, X.-X.; Gao, F.; Zhou, X.-L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef]
- Luduena, R.F.; Roach, M.C. Tubulin sulfhydryl groups as probes and targets for antimitotic and antimicrotubule agents. Pharmacol. Ther. 1991, 49, 133–152. [Google Scholar] [CrossRef]
- Zheng, Q.T.; Yang, Z.H.; Yu, L.Y.; Ren, Y.Y.; Huang, Q.X.; Liu, Q.; Ma, X.Y.; Chen, Z.K.; Wang, Z.B.; Zheng, X. Synthesis and antioxidant activity of curcumin analogs. J. Asian Nat. Prod. Res. 2017, 19, 489–503. [Google Scholar] [CrossRef]
- Arshad, L.; Haque, M.A.; Abbas Bukhari, S.N.; Jantan, I. An overview of structure–activity relationship studies of curcumin analogs as antioxidant and anti-inflammatory agents. Future Med. Chem. 2017, 9, 605–626. [Google Scholar] [CrossRef]
- Moreira, J.; Saraiva, L.; Pinto, M.M.; Cidade, H. Diarylpentanoids with antitumor activity: A critical review of structure-activity relationship studies. Eur. J. Med. Chem. 2020, 192, 112177. [Google Scholar] [CrossRef]
- Chen, H.; Yang, X.; Lu, K.; Lu, C.; Zhao, Y.; Zheng, S.; Li, J.; Huang, Z.; Huang, Y.; Zhang, Y.; et al. Inhibition of high glucose-induced inflammation and fibrosis by a novel curcumin derivative prevents renal and heart injury in diabetic mice. Toxicol. Lett. 2017, 278, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Saka, C. Analytical Strategies for the Determination of Norepinephrine Reuptake Inhibitors in Pharmaceutical Formulations and Biological Fluids. Crit. Rev. Anal. Chem. 2016, 46, 40–66. [Google Scholar] [CrossRef]
- Din, Z.U.; Santos, A.D.; Trapp, M.A.; Lazarin-Bidóia, D.; Garcia, F.P.; Peron, F.; Nakamura, C.V.; Rodrigues-Filho, E. Curcumin inspired synthesis of unsymmetrical diarylpentanoids with highly potent anti-parasitic activities: In silico studies and DFT-based stereochemical calculation. Medchemcomm 2016, 7, 820–831. [Google Scholar] [CrossRef]
- Ao, G.Z.; Zhou, M.Z.; Li, Y.Y.; Li, S.N.; Wang, H.N.; Wan, Q.W.; Li, H.Q.; Hu, Q.H. Discovery of novel curcumin derivatives targeting xanthine oxidase and urate transporter 1 as anti-hyperuricemic agents. Bioorganic Med. Chem. 2017, 25, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Du, Z.; Xue, G.; Chen, Q.; Lu, Y.; Zheng, X.; Conney, A.H.; Zhang, K. Synthesis and biological evaluation of unsymmetrical curcumin analogues as tyrosinase inhibitors. Molecules 2013, 18, 3948–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, S.W.; Abas, F.; Lam, K.W.; Shaari, K.; Lajis, N.H. 2-Benzoyl-6-benzylidenecyclohexanone analogs as potent dual inhibitors of acetylcholinesterase and butyrylcholinesterase. Bioorganic Med. Chem. 2016, 24, 3742–3751. [Google Scholar] [CrossRef]
- Leong, S.W.; Abas, F.; Lam, K.W.; Yusoff, K. In vitro and in silico evaluations of diarylpentanoid series as α-glucosidase inhibitor. Bioorganic Med. Chem. Lett. 2018, 28, 302–309. [Google Scholar] [CrossRef]
- Singh, M.; Raghav, N. Synthesis, docking, and in vitro studies of some substituted bischalcones on acid and alkaline phosphatases. Med. Chem. Res. 2014, 23, 1781–1788. [Google Scholar] [CrossRef]
- Aditama, R.; Eryanti, Y.; Mujahidin, D.; Syah, Y.M.; Hertadi, R. Determination of activities of human carbonic anhydrase ii inhibitors from curcumin analogs. Trop. J. Pharm. Res. 2017, 16, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Nalli, M.; Ortar, G.; Schiano Moriello, A.; Di Marzo, V.; De Petrocellis, L. Effects of curcumin and curcumin analogues on TRP channels. Fitoterapia 2017, 122, 126–131. [Google Scholar] [CrossRef]
- Revalde, J.L.; Li, Y.; Hawkins, B.C.; Rosengren, R.J.; Paxton, J.W. Heterocyclic cyclohexanone monocarbonyl analogs of curcumin can inhibit the activity of ATP-binding cassette transporters in cancer multidrug resistance. Biochem. Pharmacol. 2015, 93, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.; Almeida, J.; Loureiro, J.B.; Ramos, H.; Palmeira, A.; Pinto, M.M.; Saraiva, L.; Cidade, H. A Diarylpentanoid with Potential Activation of the p53 Pathway: Combination of in silico Screening Studies, Synthesis, and Biological Activity Evaluation. ChemMedChem 2021, 16, 2969–2981. [Google Scholar] [CrossRef] [PubMed]
- Leão, M.; Soares, J.; Gomes, S.; Raimundo, L.; Ramos, H.; Bessa, C.; Queiroz, G.; Domingos, S.; Pinto, M.; Inga, A.; et al. Enhanced cytotoxicity of prenylated chalcone against tumour cells via disruption of the p53-MDM2 interaction. Life Sci. 2015, 142, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Lima, R.T.; Palmeira, A.; Seca, H.; Soares, J.; Gomes, S.; Raimundo, L.; Maciel, C.; Pinto, M.; Sousa, E.; et al. Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arab. J. Chem. 2019, 12, 4150–4161. [Google Scholar] [CrossRef]
- Brandão, P.; Loureiro, J.B.; Carvalho, S.; Hamadou, M.H.; Cravo, S.; Moreira, J.; Pereira, D.; Palmeira, A.; Pinto, M.; Saraiva, L.; et al. Targeting the MDM2-p53 protein-protein interaction with prenylchalcones: Synthesis of a small library and evaluation of potential antitumor activity. Eur. J. Med. Chem. 2018, 156, 711–721. [Google Scholar] [CrossRef]
- Raimundo, L.; Espadinha, M.; Soares, J.; Loureiro, J.B.; Alves, M.G.; Santos, M.M.M.; Saraiva, L. Improving anticancer activity towards colon cancer cells with a new p53-activating agent. Br. J. Pharmacol. 2018, 175, 3947–3962. [Google Scholar] [CrossRef]
- Masawang, K.; Pedro, M.; Cidade, H.; Reis, R.M.; Neves, M.P.; Corrêa, A.G.; Sudprasert, W.; Bousbaa, H.; Pinto, M.M. Evaluation of 2’,4’-dihydroxy-3,4,5-trimethoxychalcone as antimitotic agent that induces mitotic catastrophe in MCF-7 breast cancer cells. Toxicol. Lett. 2014, 229, 393–401. [Google Scholar] [CrossRef]
- Pinto, P.; Machado, C.M.; Moreira, J.; Almeida, J.D.P.; Silva, P.M.A.; Henriques, A.C.; Soares, J.X.; Salvador, J.A.R.; Afonso, C.; Pinto, M.; et al. Chalcone derivatives targeting mitosis: Synthesis, evaluation of antitumor activity and lipophilicity. Eur. J. Med. Chem. 2019, 184, 111752. [Google Scholar] [CrossRef]
- Fonseca, J.; Marques, S.; Silva, P.; Brandão, P.; Cidade, H.; Pinto, M.; Bousbaa, H. Prenylated Chalcone 2 Acts as an Antimitotic Agent and Enhances the Chemosensitivity of Tumor Cells to Paclitaxel. Molecules 2016, 21, 982. [Google Scholar] [CrossRef] [Green Version]
- Logarinho, E.; Bousbaa, H. Kinetochore-microtubule interactions “in check” by Bub1, Bub3 and BubR1: The dual task of attaching and signalling. Cell Cycle 2008, 7, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Logarinho, E.; Resende, T.; Torres, C.; Bousbaa, H. The human spindle assembly checkpoint protein Bub3 is required for the establishment of efficient kinetochore-microtubule attachments. Mol. Biol. Cell 2008, 19, 1798–1813. [Google Scholar] [CrossRef] [Green Version]
- Rieder, C.L. The structure of the cold-stable kinetochore fiber in metaphase PtK1 cells. Chromosoma 1981, 84, 145–158. [Google Scholar] [CrossRef]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural Basis of cis- and trans-Combretastatin Binding to Tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Antúnez-Mojica, M.; Rodríguez-Salarichs, J.; Redondo-Horcajo, M.; León, A.; Barasoain, I.; Canales, Á.; Cañada, F.J.; Jiménez-Barbero, J.; Alvarez, L.; Díaz, J.F. Structural and Biochemical Characterization of the Interaction of Tubulin with Potent Natural Analogues of Podophyllotoxin. J. Nat. Prod. 2016, 79, 2113–2121. [Google Scholar] [CrossRef]
- Fu, D.J.; Liu, S.M.; Li, F.H.; Yang, J.J.; Li, J. Antiproliferative benzothiazoles incorporating a trimethoxyphenyl scaffold as novel colchicine site tubulin polymerisation inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1050–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, D.J.; Li, P.; Wu, B.W.; Cui, X.X.; Zhao, C.B.; Zhang, S.Y. Molecular diversity of trimethoxyphenyl-1,2,3-triazole hybrids as novel colchicine site tubulin polymerization inhibitors. Eur. J. Med. Chem. 2019, 165, 309–322. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 2021, 13, 1011. [Google Scholar] [CrossRef]
- Paulraj, F.; Abas, F.; Lajis, N.H.; Othman, I.; Naidu, R. Molecular pathways modulated by curcumin analogue, diarylpentanoids in cancer. Biomolecules 2019, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potapova, T.; Gorbsky, G. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; Mcphail, A.T. Plant Antitumor Agents.VI.The Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.C.; Silva, P.M.A.; Sarmento, B.; Bousbaa, H. Antagonizing the spindle assembly checkpoint silencing enhances paclitaxel and Navitoclax-mediated apoptosis with distinct mechanistic. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Kohyama, A.; Fukuda, M.; Sugiyama, S.; Yamakoshi, H.; Kanoh, N.; Ishioka, C.; Shibata, H.; Iwabuchi, Y. Reversibility of the thia-Michael reaction of cytotoxic C5-curcuminoid and structure-activity relationship of bis-thiol-adducts thereof. Org. Biomol. Chem. 2016, 14, 10683–10687. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Xia, Y.; Zou, P.; Shen, M.; Hu, J.; Ying, S.; Pan, J.; Liu, Z.; Dai, X.; Zhuge, W.; et al. Curcumin analog L48H37 induces apoptosis through ROS-mediated endoplasmic reticulum stress and STAT3 pathways in human lung cancer cells. Mol. Carcinog. 2017, 56, 1765–1777. [Google Scholar] [CrossRef]
- Kudo, C.; Yamakoshi, H.; Sato, A.; Nanjo, H.; Ohori, H.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Synthesis of 86 species of 1,5-diaryl-3-oxo-1,4-pentadienes analogs of curcumin can yield a good lead in vivo. BMC Pharmacol. 2011, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided. Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GI50 (μM) 1 | |||
|---|---|---|---|
| A375-C5 | MCF-7 | NCI-H460 | |
| BP-M345 | 0.24 ± 0.01 | 0.45 ± 0.06 | 0.37 ± 0.00 |
| Doxorubicin | 0.030 ± 0.04 | 0.028 ± 0.01 | 0.028 ± 0.01 |
| HPAEpiC 1 | Selectivity Index 2 | |||
|---|---|---|---|---|
| GI50 (μM) | A375-C5 | MCF-7 | NCI-H460 | |
| BP-M345 | 1.07 ± 0.16 | 4.45 | 2.38 | 2.89 |
| Doxorubicin | 0.054 ± 0.012 | 1.81 | 1.91 | 1.95 |
| Ligand | Docking Score (kcal mol−1) |
|---|---|
| Colchicine | −10.1 |
| Combretastatin A4 | −7.8 |
| Podophyllotoxin | −8.4 |
| BP-M345 | −8.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novais, P.; Silva, P.M.A.; Moreira, J.; Palmeira, A.; Amorim, I.; Pinto, M.; Cidade, H.; Bousbaa, H. BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity. Molecules 2021, 26, 7139. https://doi.org/10.3390/molecules26237139
Novais P, Silva PMA, Moreira J, Palmeira A, Amorim I, Pinto M, Cidade H, Bousbaa H. BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity. Molecules. 2021; 26(23):7139. https://doi.org/10.3390/molecules26237139
Chicago/Turabian StyleNovais, Pedro, Patrícia M. A. Silva, Joana Moreira, Andreia Palmeira, Isabel Amorim, Madalena Pinto, Honorina Cidade, and Hassan Bousbaa. 2021. "BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity" Molecules 26, no. 23: 7139. https://doi.org/10.3390/molecules26237139
APA StyleNovais, P., Silva, P. M. A., Moreira, J., Palmeira, A., Amorim, I., Pinto, M., Cidade, H., & Bousbaa, H. (2021). BP-M345, a New Diarylpentanoid with Promising Antimitotic Activity. Molecules, 26(23), 7139. https://doi.org/10.3390/molecules26237139