
Oxandrastins: Antibacterial Meroterpenes from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD14




Abstract
:1. Introduction
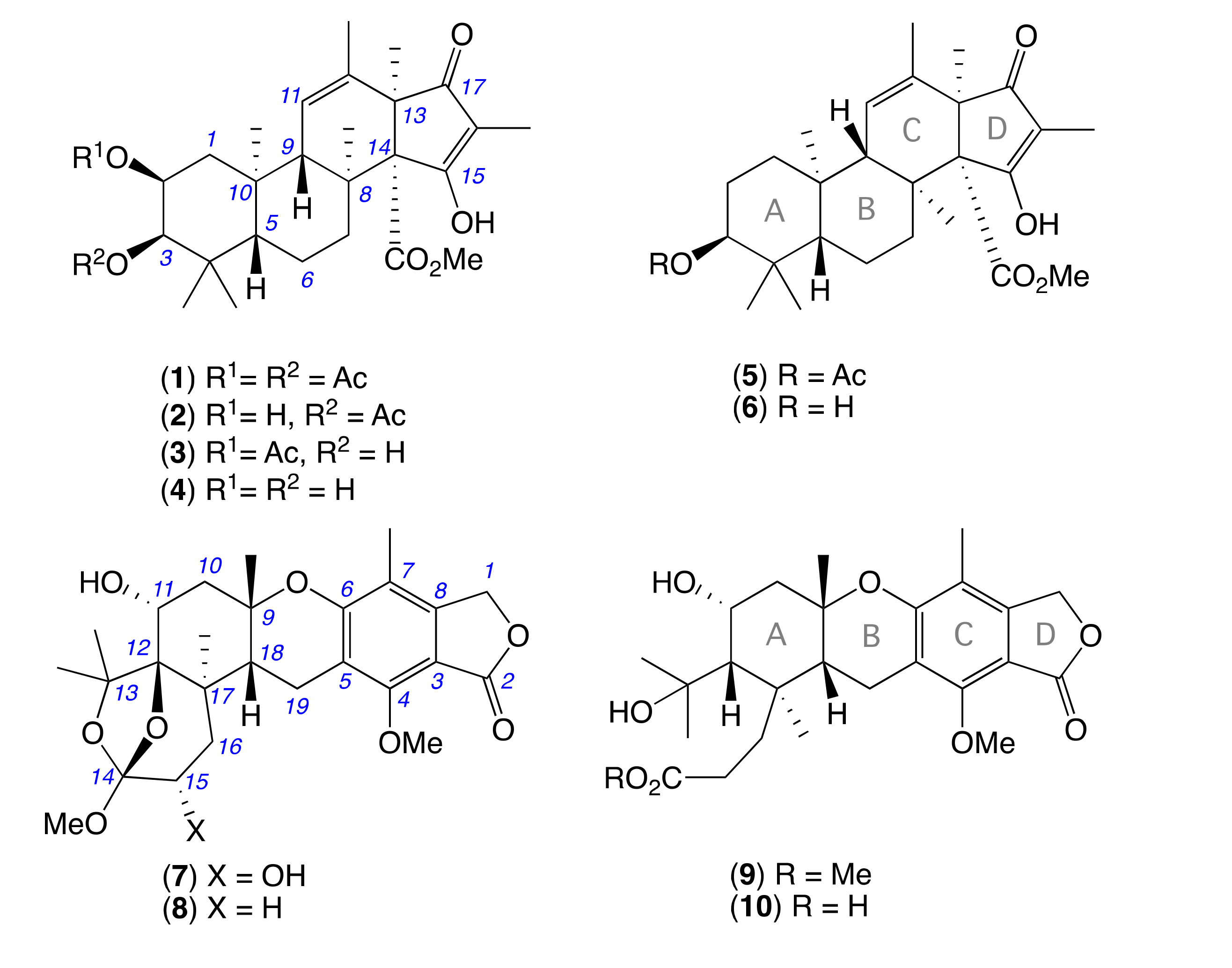
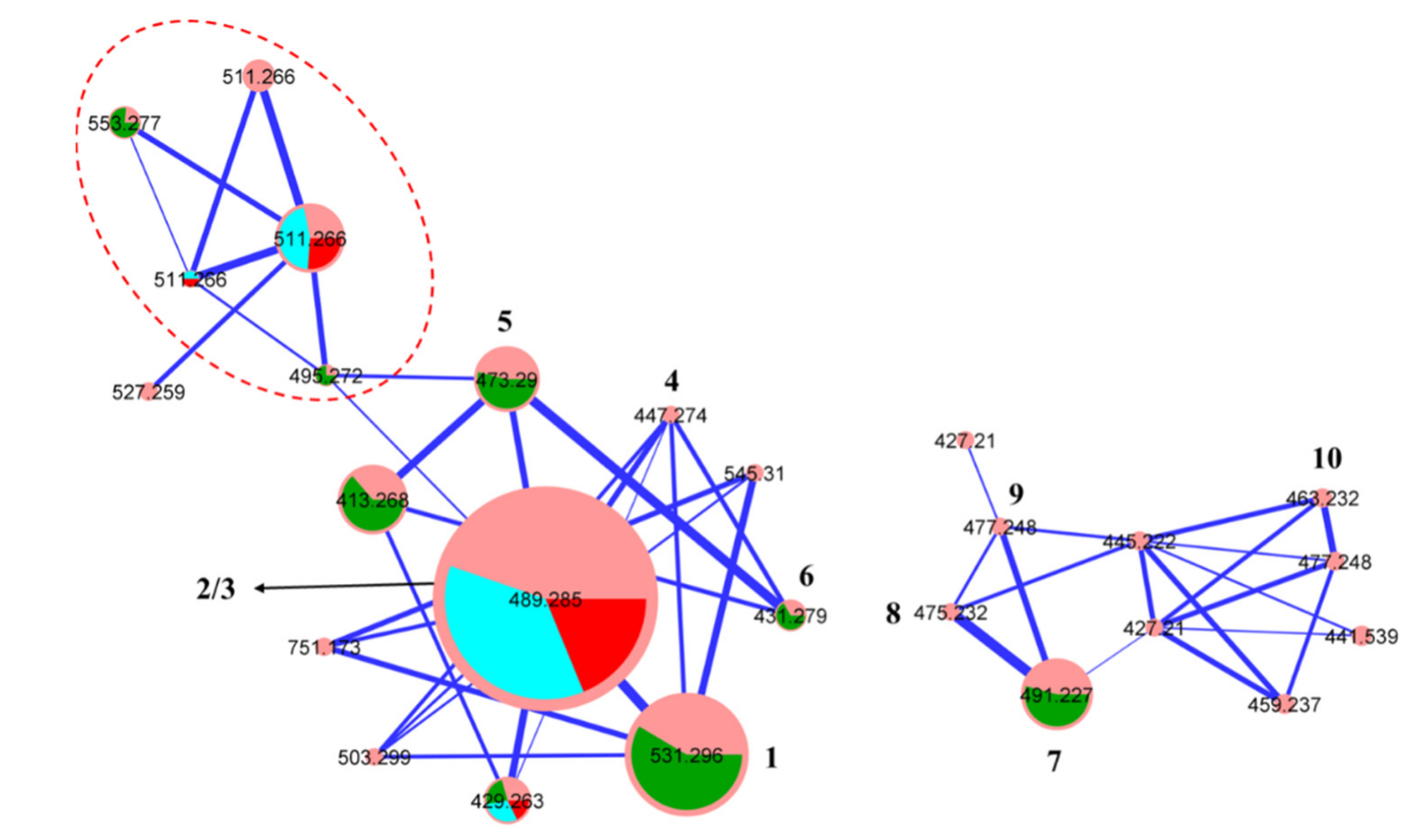
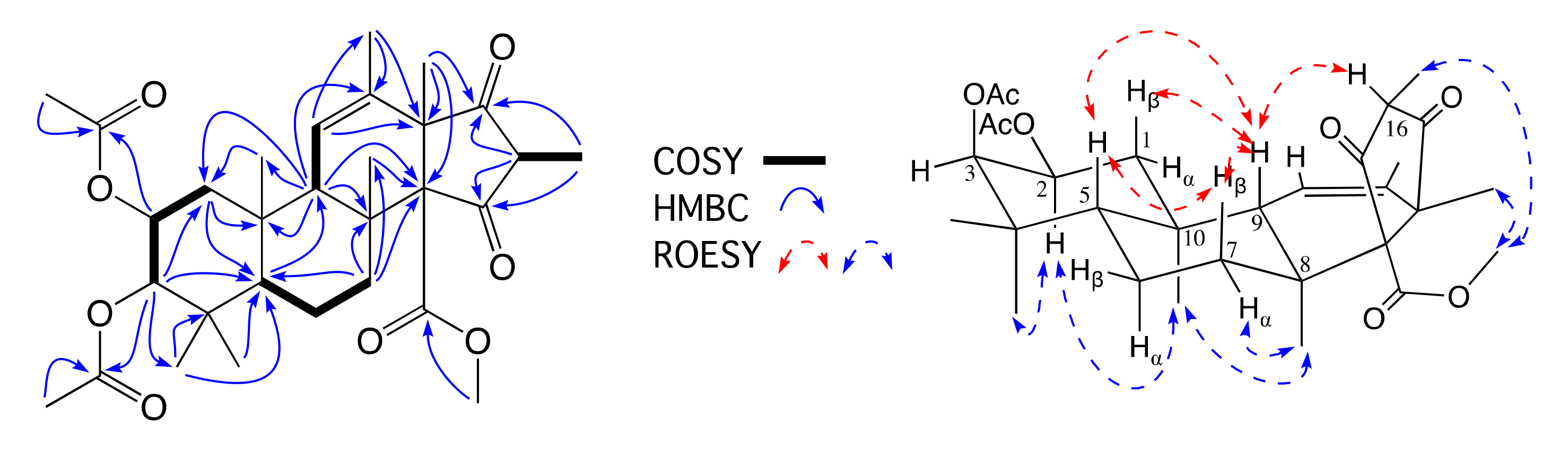
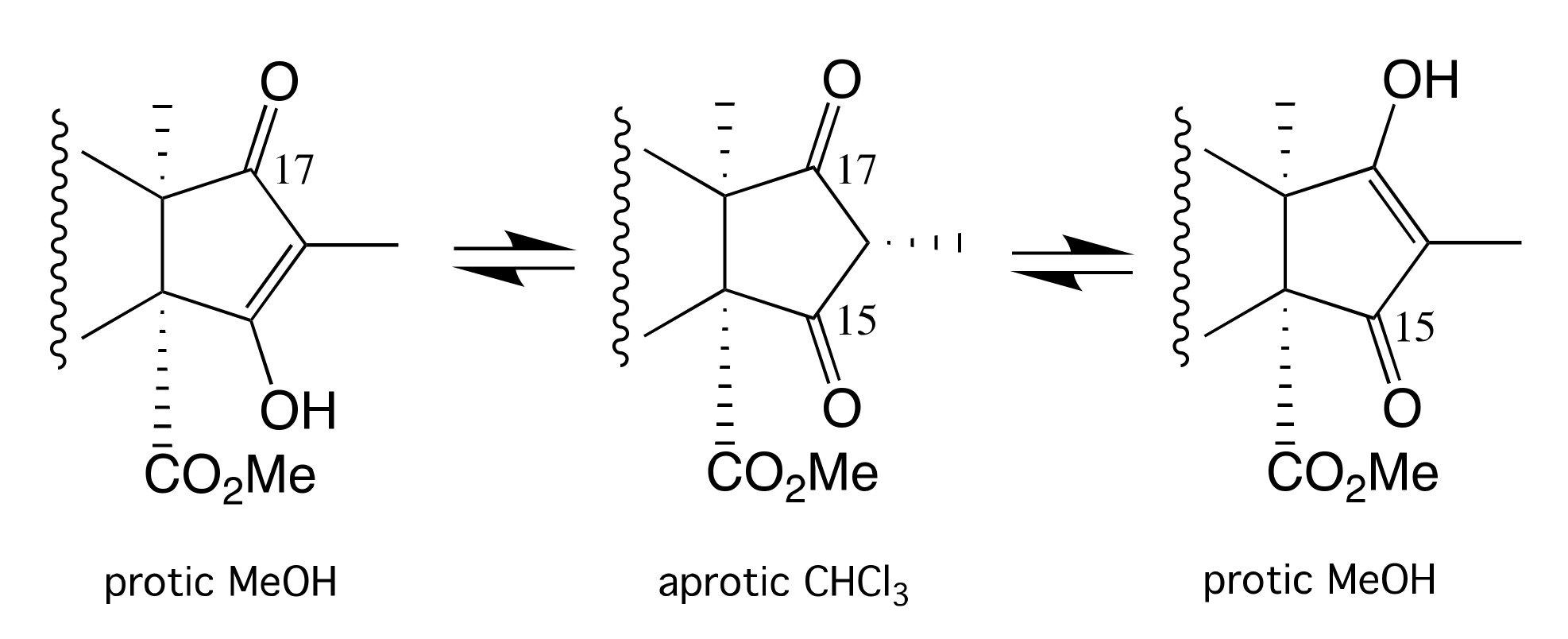
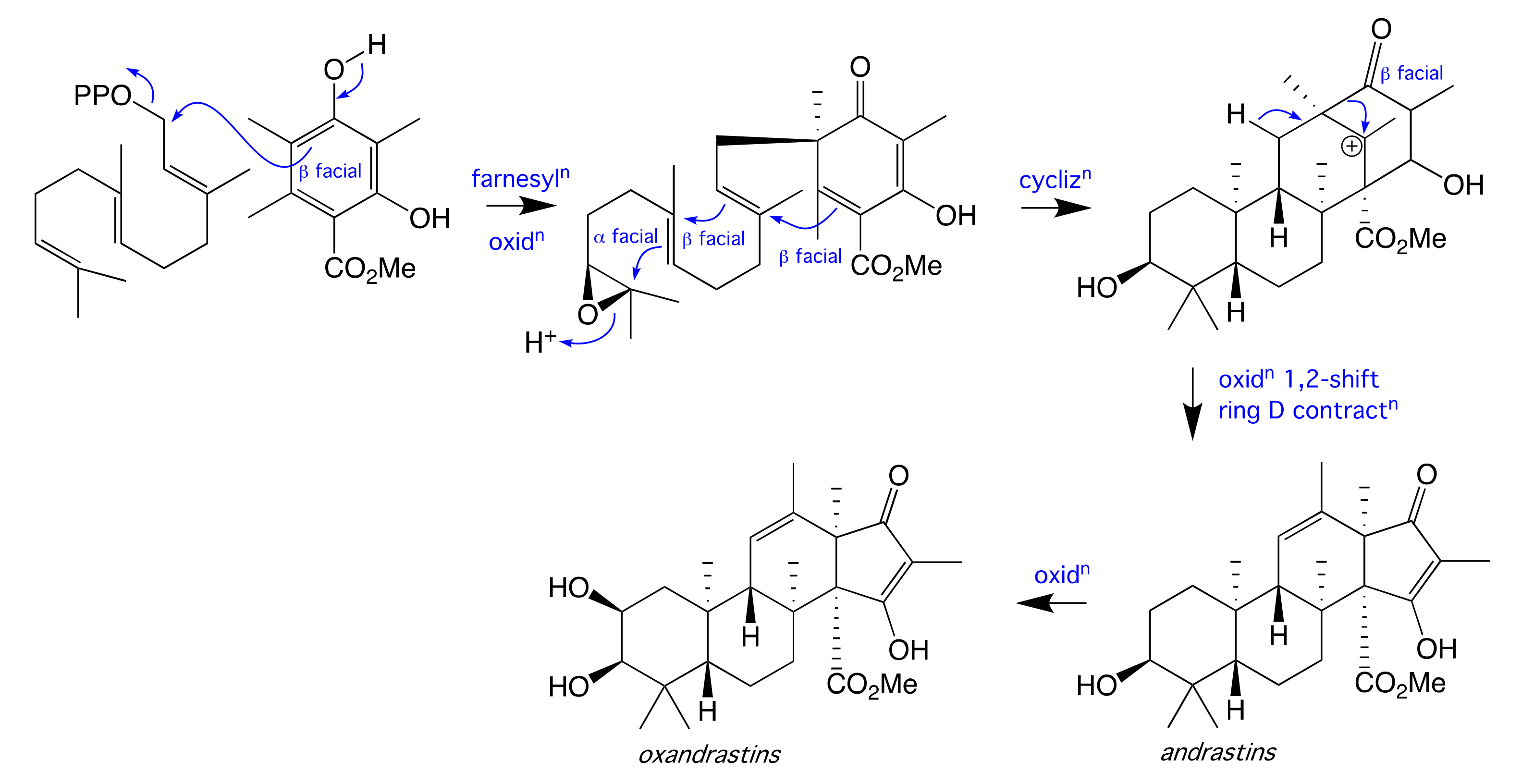
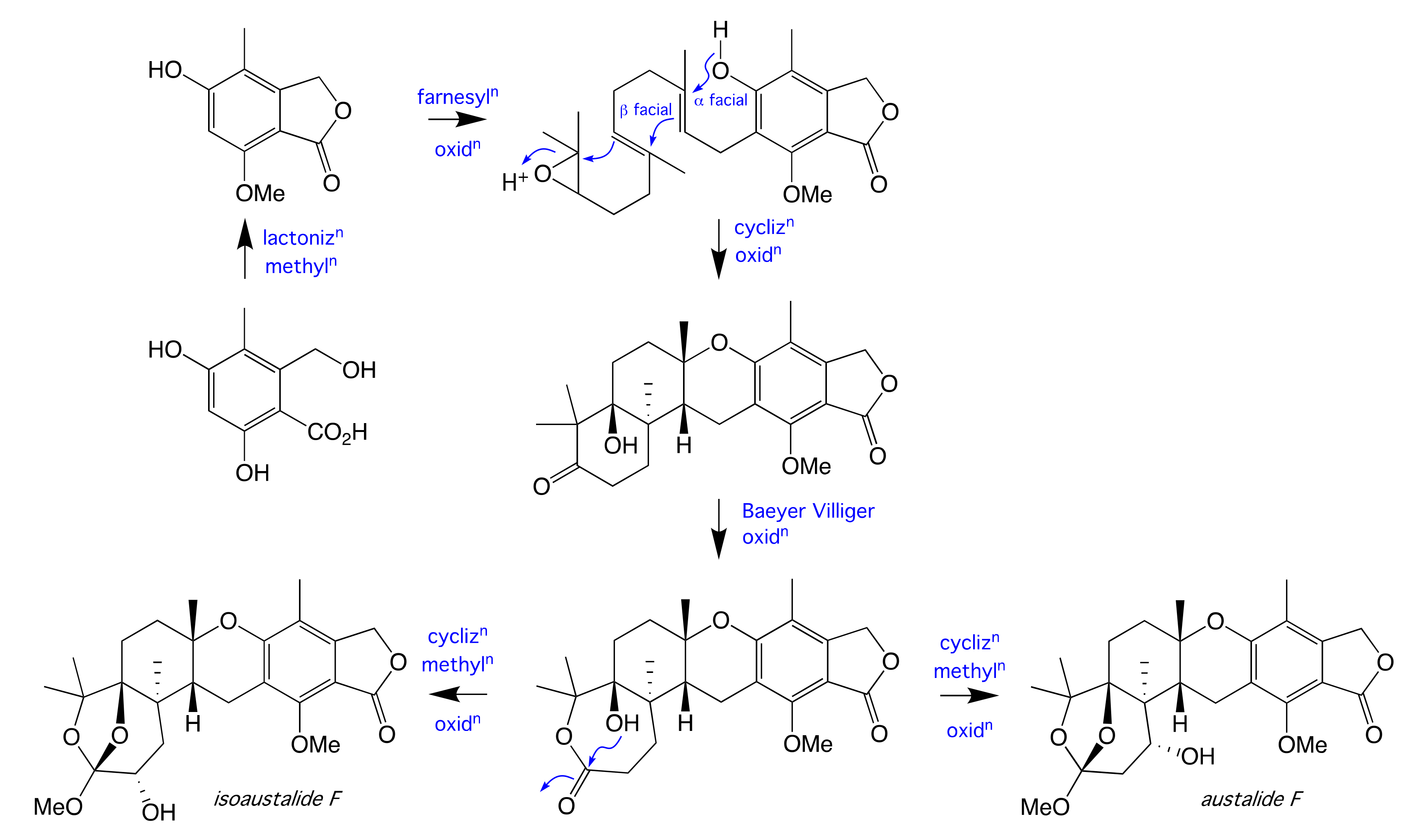
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Isolation
3.3. Fungal Taxonomy
3.4. Global Natural Product Social (GNPS) Molecular Networking
3.5. Extraction and Fractionation of an ISP-2 Agar Cultivation of CMB-MD14
3.6. Extraction and Fractionation of a Rice Cultivation of CMB-MD14
3.7. Metabolite Characterization
3.8. Hydrolysis of Oxandrastin B (2) to Oxandrastin D (4)
3.9. Antibiotic Assays
3.10. Cytotoxicity Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Quezada, M.; Shang, Z.; Kalansuriya, P.; Salim, A.A.; Lacey, E.; Capon, R.J. Waspergillamide A, a Nitro depsi-Tetrapeptide Diketopiperazine from an Australian Mud Dauber Wasp-Associated Aspergillus sp. (CMB-W031). J. Nat. Prod. 2017, 80, 1192–1195. [Google Scholar] [CrossRef]
- Kalansuriya, P.; Quezada, M.; Espósito, B.P.; Capon, R.J. Talarazines A-E: Noncytotoxic Iron(III) Chelators from an Australian Mud Dauber Wasp-Associated Fungus, Talaromyces sp. (CMB-W045). J. Nat. Prod. 2017, 80, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kalansuriya, P.; Khalil, Z.G.; Salim, A.A.; Capon, R.J. Talarophenol sulfate and talarophilones from the Australian mud dauber wasp-associated fungus, Talaromyces sp. CMB-W045. Tet. Lett. 2019, 60, 151157. [Google Scholar] [CrossRef]
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Neobulgarones Revisited: Anti and Syn Bianthrones from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD22. J. Nat. Prod. 2021, 84, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Sunazuka, T.; Tanaka, H.; Iwai, Y.; Takayanagi, H.; Omura, S. Andrastins A-C, new protein farnesyltransferase inhibitors produced by Penicillium sp. FO-3929. II. Structure elucidation and biosynthesis. J. Antibiot. 1996, 49, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Awakawa, T.; Abe, I. Reconstituted biosynthesis of fungal meroterpenoid andrastin A. Tetrahedron 2013, 69, 8199–8204. [Google Scholar] [CrossRef]
- Wang, X.; Filho, J.G.S.; Hoover, A.R.; King, J.B.; Ellis, T.K.; Powell, D.R.; Cichewicz, R.H. Chemical epigenetics alters the secondary metabolite composition of guttate excreted by an atlantic-forest-soil-derived Penicillium citreonigrum. J. Nat. Prod. 2010, 73, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalsgaard, P.W.; Petersen, B.O.; Duus, J.Ø.; Zidorn, C.; Frisvad, J.C.; Christophersen, C.; Larsen, T.O. Atlantinone A, a Meroterpenoid Produced by Penicillium ribeum and Several Cheese Associated Penicillium Species. Metabolites 2012, 2, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Kosemura, S. Meroterpenoids from Penicillium citreo-viride B. IFO 4692 and 6200 hybrid. Tetrahedron 2003, 59, 5055–5072. [Google Scholar] [CrossRef]
- Özkaya, F.C.; Ebrahim, W.; Klopotowski, M.; Liu, Z.; Janiak, C.; Proksch, P. Isolation and X-ray structure analysis of citreohybridonol from marine-derived Penicillium atrovenetum. Nat. Prod. Res. 2017, 32, 840–843. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. Metabolites of Aspergillus ustus. Part 1. Application of the heteronuclear selective population inversion (SPI) n.m.r. technique to the structure elucidation of the austalides A–F, novel ortho ester meroterpenoids. J. Chem. Soc. Perkin Trans. 1 1985, 345–356. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Rooyen, P.H.V.; Vleggaar, R.; Rabie, C.J. Structures of the austalides A–E, five noval toxic metabolites from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1981, 1265–1267. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. Metabolites of Aspergillus ustus. Part 3. Structure elucidation of austalides G–L. J. Chem. Soc. Perkin Trans. 1 1985, 363–367. [Google Scholar] [CrossRef]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Leshchenko, E.V.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Zakharenko, A.M.; Kim, N.Y.; Afiyatullov, S.S. Meroterpenoids from the Alga-Derived Fungi Penicillium thomii Maire and Penicillium lividum Westling. J. Nat. Prod. 2014, 77, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.F.; Dalsgaard, P.W.; Smedsgaard, J.; Larsen, T.O. Andrastins A-D, Penicillium roqueforti Metabolites consistently produced in blue-mold-ripened cheese. J. Agric. Food. Chem. 2005, 53, 2908–2913. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, X.; Wang, W.; Zhu, T.; Gu, Q.; Li, D. Austalides S-U, New Meroterpenoids from the Sponge-Derived Fungus Aspergillus aureolatus HDN14-107. Mar. Drugs 2016, 14, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillen, J.L.M.; Horak, R.M.; Maharaj, V.J.; Marais, S.F.; Vleggaar, R. Absolute configuration and biosynthesis of the austalides, meroterpenoid metabolites of Aspergillus ustus: Mode of cyclisation of the farnesyl moiety. J. Chem. Soc. Chem. Commun. 1989, 7, 393–394. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Total Synthesis of (-)-Austalide B. A Generic Solution to Elaboration of the Pyran/p-Cresol/Butenolide Triad. J. Am. Chem. Soc. 1994, 116, 11323–11334. [Google Scholar] [CrossRef]
- Yang, Z.; Goldman, N.; Friday, A. Comparison of models for nucleotide substitution used in maximum-likelihood phylogenetic estimation. Mol. Biol. Evol. 1994, 11, 316–324. [Google Scholar] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Salim, A.A.; Khalil, Z.; Quezada, M.; Bernhardt, P.V.; Capon, R.J. Viridicatumtoxins: Expanding on a Rare Tetracycline Antibiotic Scaffold. J. Org. Chem. 2015, 80, 12501–12508. [Google Scholar] [CrossRef] [PubMed]
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Chrysosporazines A-E: P-Glycoprotein Inhibitory Piperazines from an Australian Marine Fish Gastrointestinal Tract-Derived Fungus, Chrysosporium sp. CMB-F214. Org. Lett. 2019, 21, 8097–8100. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.G.; Salim, A.A.; Khalil, Z.G.; Elbanna, A.H.; Bernhardt, P.V.; Capon, R.J. Chrysosporazines F-M: P-Glycoprotein Inhibitory Phenylpropanoid Piperazines from an Australian Marine Fish Derived Fungus, Chrysosporium sp. CMB-F294. J. Nat. Prod. 2020, 83, 497–504. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH, Mult (J in Hz) | |||
|---|---|---|---|---|
| (1) | (2) | (3) | (4) | |
| 1 | α. 1.82, dd (12.3, 4.2) | α. 1.81, dd (12.3, 4.1) | α. 1.72, dd (12.0, 4.1) | α. 1.71, dd (12.0, 4.1) |
| β. 1.27, dd (12.3, 12.3) | β. 1.14, dd (12.3, 12.1) | β. 1.36, dd (12.1, 12.0) | β. 1.17, dd (12.1, 12.0) | |
| 2 | 5.25, ddd (12.3, 4.2, 2.4) | 4.07, ddd (12.1, 4.1, 2.8) | 5.17, ddd (12.1, 4.1, 2.7) | 3.94, ddd (11.9, 4.3, 3.0) |
| 3 | 4.96, d (2.4) | 4.91, d (2.8) | 3.44, d (2.7) | 3.32, d (3.0) A |
| 5 | 1.40, dd (12.1, 2.2) | 1.33, dd (12.1, 2.9) | 1.45, dd (12.1, 2.0) | 1.39, dd (11.7, 2.7) |
| 6 | α. 1.56, ddd (13.3, 12.9, 3.0) | α. 1.53, ddd (12.8, 12.4, 3.1) | α. 1.52, ddd (13.1, 12.8, 3.1) | α. 1.51, ddd (12.8, 12.4, 3.2) |
| β. 1.52, m | β. 1.49, m | β. 1.46, m | β. 1.48, m | |
| 7 | α. 2.13, br d (13.4) | α. 2.11, ddd (12.8, 3.2, 3.1) | α. 2.09, ddd (13.1, 3.1, 2.4) | α. 2.07, ddd (13.2, 3.0, 2.8) |
| β. 2.77, m | β. 2.75, ddd (12.8, 12.4, 5.0) | β. 2.76, ddd (13.1, 12.8, 4.4) | β. 2.74, m | |
| 9 | 1.88, br s | 1.85, br s | 1.86, br s | 1.82, br s |
| 11 | 5.40, br s | 5.44, br s | 5.39, br s | 5.44, br s |
| 2-COCH3 | 1.92, s | - | 2.03, s | - |
| 3-COCH3 | 2.07, s | 2.07, s | - | - |
| 4-CH3 (α) | 1.00, s | 0.959, s | 0.91, s | 0.86, s |
| 4-CH3 (β) | 0.90, s | 0.88, s | 0.99, s | 0.99, s |
| 8-CH3 | 1.31, s | 1.30, s | 1.29, s | 1.28, s |
| 10-CH3 | 1.03, s | 0.99, s | 1.00, s | 0.95, s |
| 12-CH3 | 1.80, s | 1.80, br s | 1.78, br s | 1.79, br s |
| 13-CH3 | 1.18, s | 1.18, s | 1.17, s | 1.17, s |
| 14-CO2CH3 | 3.57, s | 3.56, s | 3.56, s | 3.56, s |
| 16-CH3 | 1.60, s | 1.59, s | 1.57, s | 1.57, s |
| Position | δC, Type | |||||||
|---|---|---|---|---|---|---|---|---|
| (1) | (2) | (3) | (4) | |||||
| 1 | 39.6 | CH2 | 42.8 | CH2 | 38.8 | CH2 | 42.2 | CH2 |
| 2 | 69.4 | CH | 65.6 | CH | 71.9 | CH | 67.0 | CH |
| 3 | 78.5 | CH | 81.6 | CH | 77.2 | CH | 79.7 | CH |
| 4 | 39.3 | C | 39.4 | C | 40.0 | C | 39.6 | C |
| 5 | 50.0 | CH | 49.9 | CH | 48.4 | CH | 48.3 | CH |
| 6 | 18.6 | CH2 | 18.6 | CH2 | 18.7 | CH2 | 18.8 | CH2 |
| 7 | 34.1 | CH2 | 34.1 | CH2 | 34.2 | CH2 | 34.3 | CH2 |
| 8 | 43.5 | C | 43.5 | C | 43.5 | C | 43.6 | C |
| 9 | 54.5 | CH | 54.6 | CH | 54.6 | CH | 54.6 | CH |
| 10 | 39.5 | C | 39.4 | C | 39.4 | C | 39.2 | C |
| 11 | 125.6 | CH | 125.9 | CH | 126.0 | CH | 126.4 | CH |
| 12 | 137.2 | C | 136.9 | C | 136.7 | C | 136.3 a | C |
| 13 | 58.6 a | C | 58.4 a | C | 58.4 | C | 58.8 a | C |
| 14 | 68.9 a | C | 68.9 a | C | 68.8 | C | 68.8 a | C |
| 15 | ND | C | ND | C | ND | C | ND | C |
| 16 | 114.8 | C | 114.6 | C | 114.8 | C | 114.9 a | C |
| 17 | ND | C | 201.8 a | C | 202.3 a | C | ND | C |
| 2-COCH3 | 172.3 | C | - | - | 172.7 | C | - | - |
| 2-COCH3 | 21.1 | CH3 | - | - | 21.3 | CH3 | - | - |
| 3-COCH3 | 172.6 | C | 173.1 | C | - | - | - | - |
| 3-COCH3 | 20.9 | CH3 | 21.2 | CH3 | - | - | - | - |
| 4-CH3 (α) | 21.7 | CH3 | 21.9 | CH3 | 22.0 | CH3 | 22.1 | CH3 |
| 4-CH3 (β) | 28.2 | CH3 | 28.3 | CH3 | 29.1 | CH3 | 29.2 | CH3 |
| 8-CH3 | 18.3 | CH3 | 18.4 | CH3 | 18.3 | CH3 | 18.4 | CH3 |
| 10-CH3 | 18.4 | CH3 | 18.5 | CH3 | 18.5 | CH3 | 18.7 | CH3 |
| 12-CH3 | 19.9 | CH3 | 19.9 | CH3 | 19.9 | CH3 | 19.9 | CH3 |
| 13-CH3 | 16.2 | CH3 | 16.2 | CH3 | 16.2 | CH3 | 16.2 | CH3 |
| 14-CO2CH3 | 172.0 | C | 172.0 | C | 172.1 | C | 172.1 | C |
| 14-CO2CH3 | 52.2 | CH3 | 52.2 | CH3 | 52.1 | CH3 | 52.1 | CH3 |
| 16-CH3 | 6.4 | CH3 | 6.4 | CH3 | 6.4 | CH3 | 6.4 | CH3 |
| Position | δH, Mult (J in Hz) | δC | Type |
|---|---|---|---|
| 1 | 5.21, br s | 69.9 | CH2 |
| 2 | - | 171.9 | C |
| 3 | - | 108.4 | C |
| 4 | - | 156.7 | C |
| 5 | - | 117.5 | C |
| 6 | - | 160.0 | C |
| 7 | - | 116.2 | C |
| 8 | - | 147.6 | C |
| 9 | - | 77.6 | C |
| 10 | a. 2.33, dd (15.7, 2.0) | 43.4 | CH2 |
| b. 2.16, dd (15.7, 4.3) | |||
| 11 | 4.16, dd (3.9, 2.0) | 69.7 | CH |
| 12 | - | 87.5 | C |
| 13 | - | 87.0 | C |
| 14 | - | 120.1 | C |
| 15 | 3.71, dd (10.5, 5.9) | 68.8 | CH |
| 16 | a. 2.18, dd (13.9, 5.9) | 40.5 | CH2 |
| b. 1.68, dd (13.9, 10.5) | |||
| 17 | - | 44.6 | C |
| 18 | 2.40, d (8.1) | 39.4 | CH |
| 19 | a. 3.01, d (18.6) | 19.2 | CH2 |
| b. 2.94, dd (18.6, 8.1) | |||
| 7-CH3 | 2.07, s | 10.8 | CH3 |
| 9-CH3 | 1.23, s | 28.3 | CH3 |
| 13-CH3(a) | 1.52, s | 29.3 | CH3 |
| 13-CH3(b) | 1.63, s | 26.7 | CH3 |
| 17-CH3 | 1.03, s | 18.8 | CH3 |
| 4-OCH3 | 4.06, s | 62.4 | CH3 |
| 14-OCH3 | 3.41, s | 49.3 A | CH3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbanna, A.H.; Khalil, Z.G.; Capon, R.J. Oxandrastins: Antibacterial Meroterpenes from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD14. Molecules 2021, 26, 7144. https://doi.org/10.3390/molecules26237144
Elbanna AH, Khalil ZG, Capon RJ. Oxandrastins: Antibacterial Meroterpenes from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD14. Molecules. 2021; 26(23):7144. https://doi.org/10.3390/molecules26237144
Chicago/Turabian StyleElbanna, Ahmed H., Zeinab G. Khalil, and Robert J. Capon. 2021. "Oxandrastins: Antibacterial Meroterpenes from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD14" Molecules 26, no. 23: 7144. https://doi.org/10.3390/molecules26237144
APA StyleElbanna, A. H., Khalil, Z. G., & Capon, R. J. (2021). Oxandrastins: Antibacterial Meroterpenes from an Australian Mud Dauber Wasp Nest-Associated Fungus, Penicillium sp. CMB-MD14. Molecules, 26(23), 7144. https://doi.org/10.3390/molecules26237144