
Ionic Liquids and Water: Hydrophobicity vs. Hydrophilicity


Abstract
:1. Introduction
2. Results and Discussion
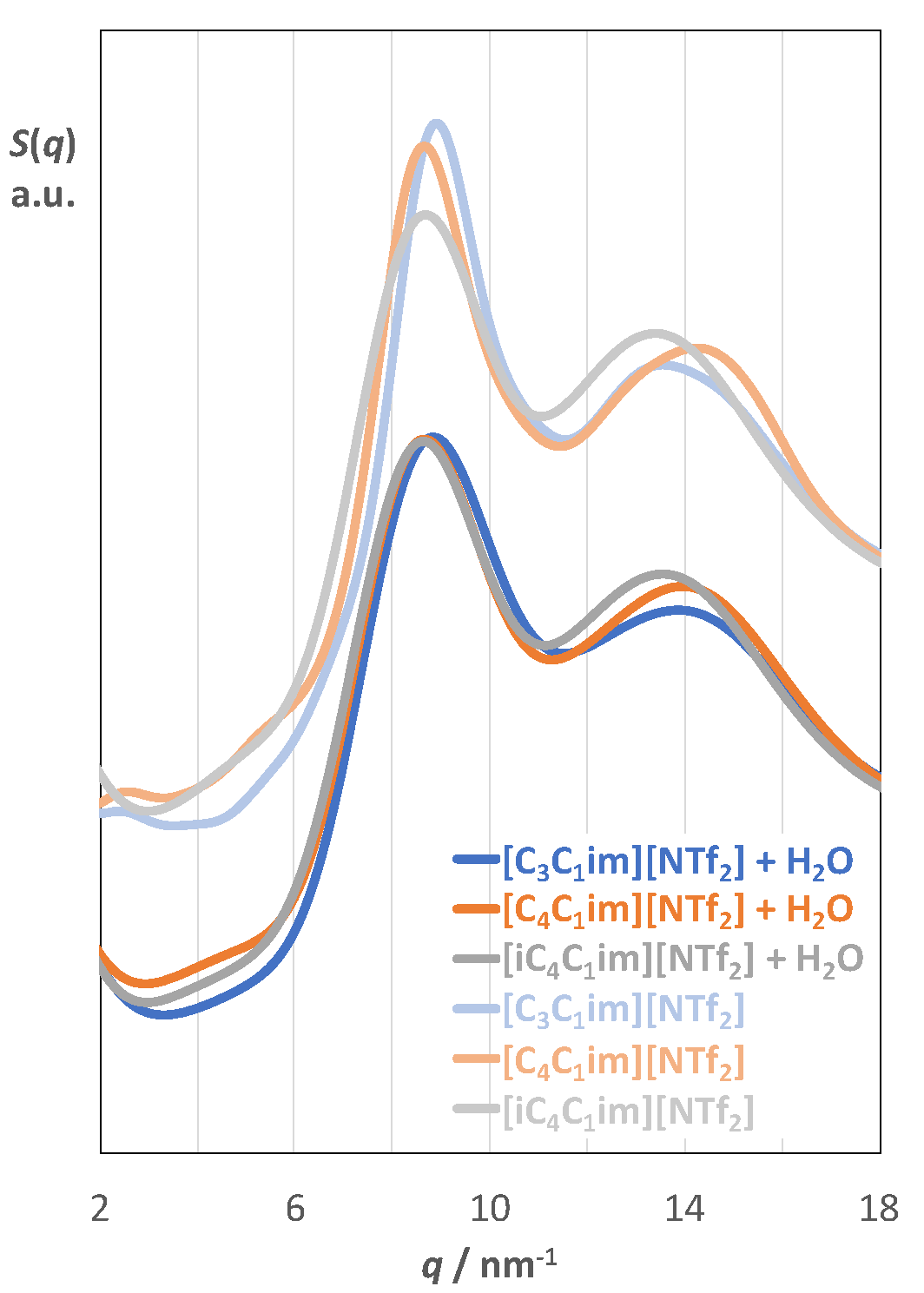
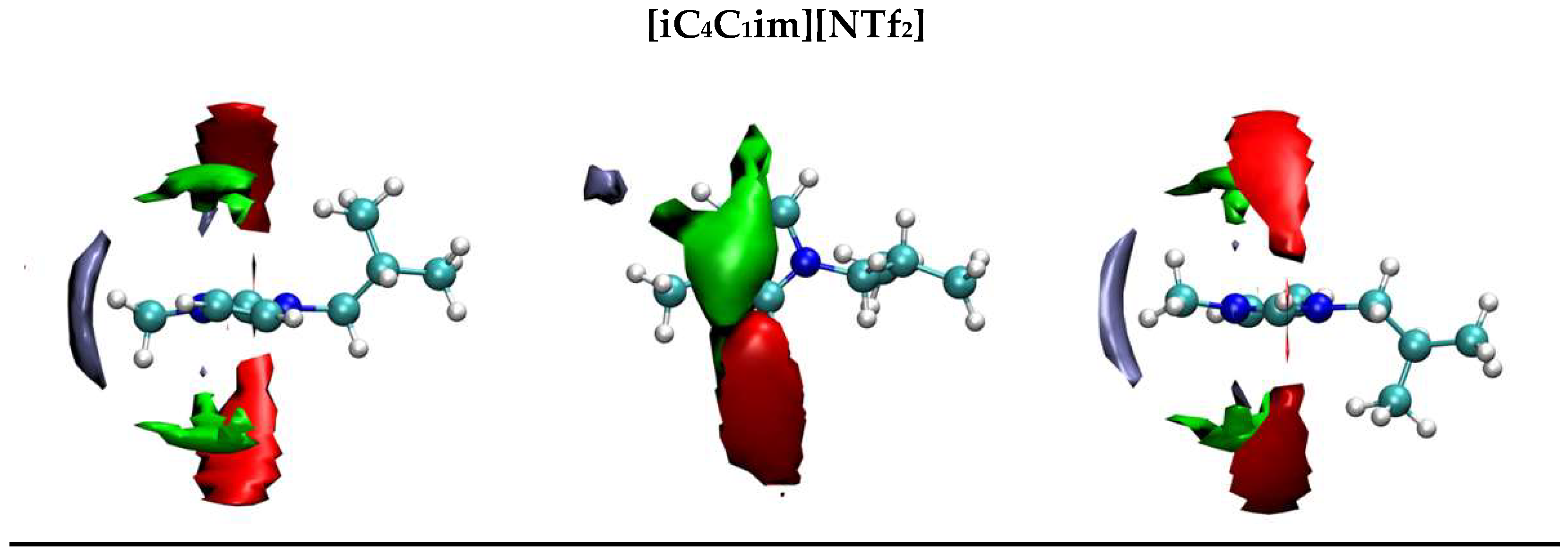
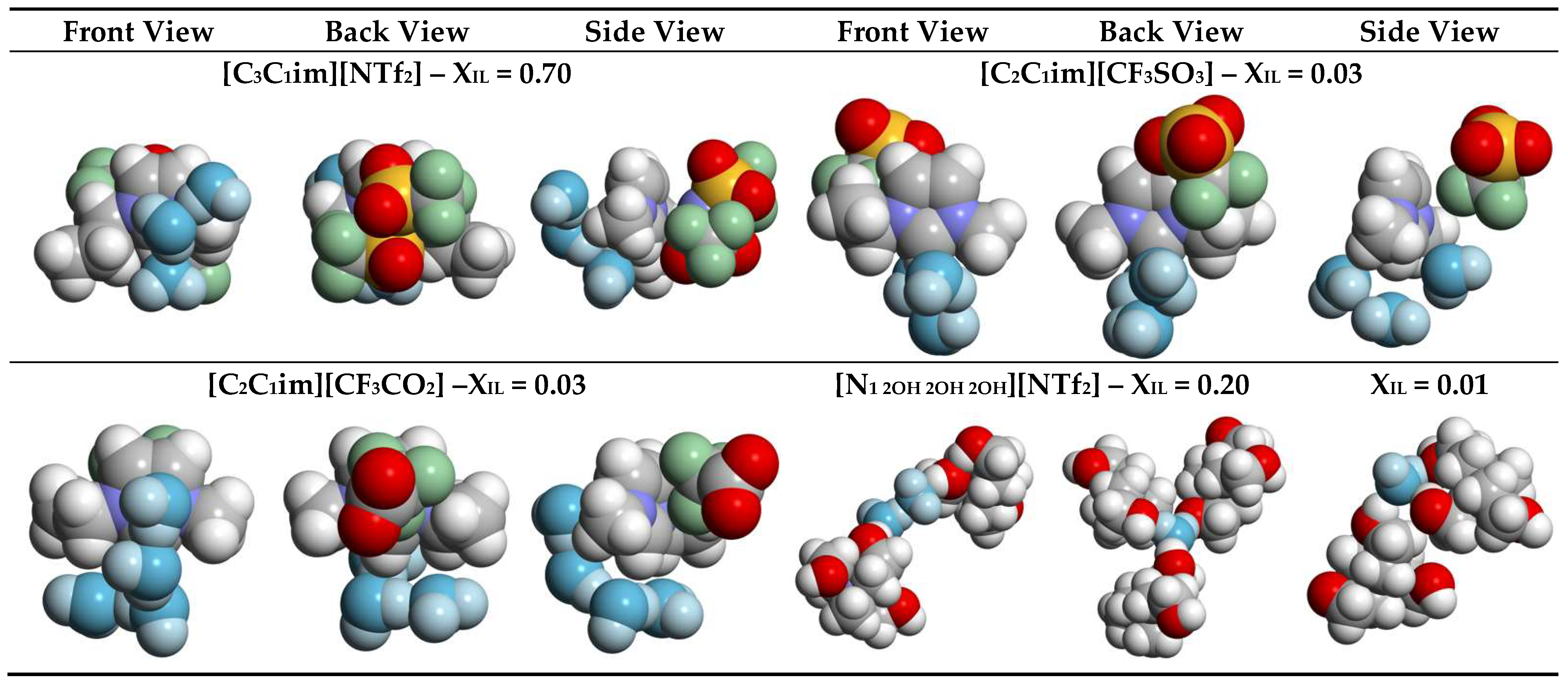
2.1. Aqueous Solutions of [C3C1im][NTf2], [C4C1im][NTf2], and [iC4C1im][NTf2]
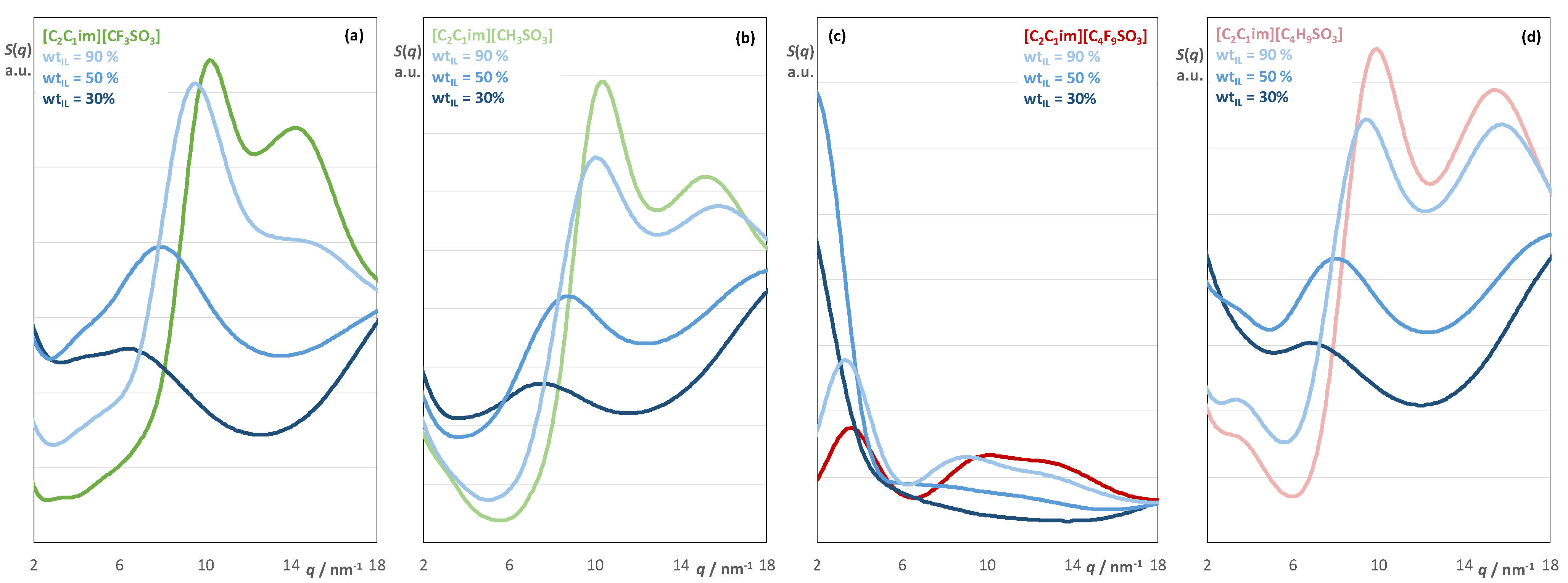
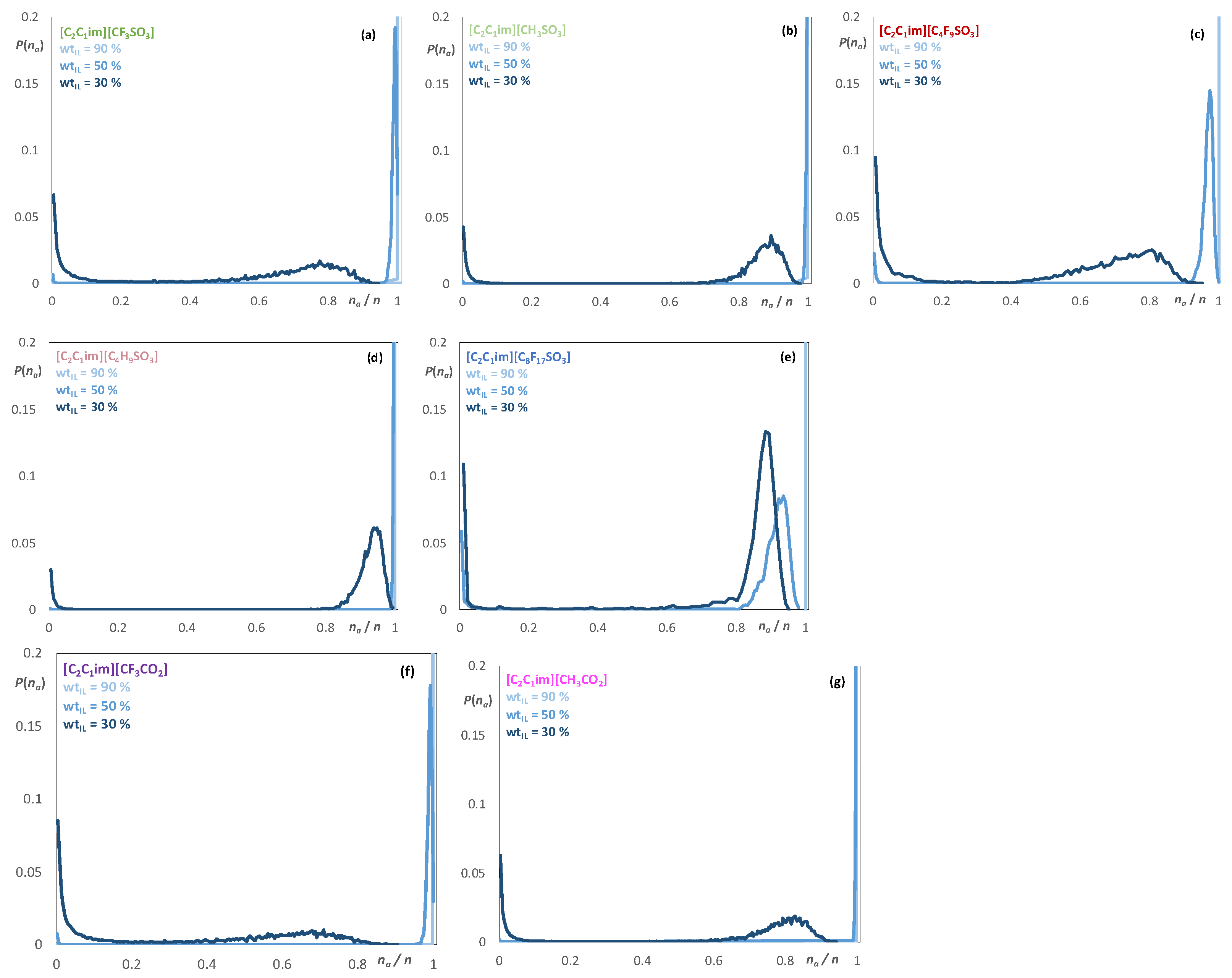
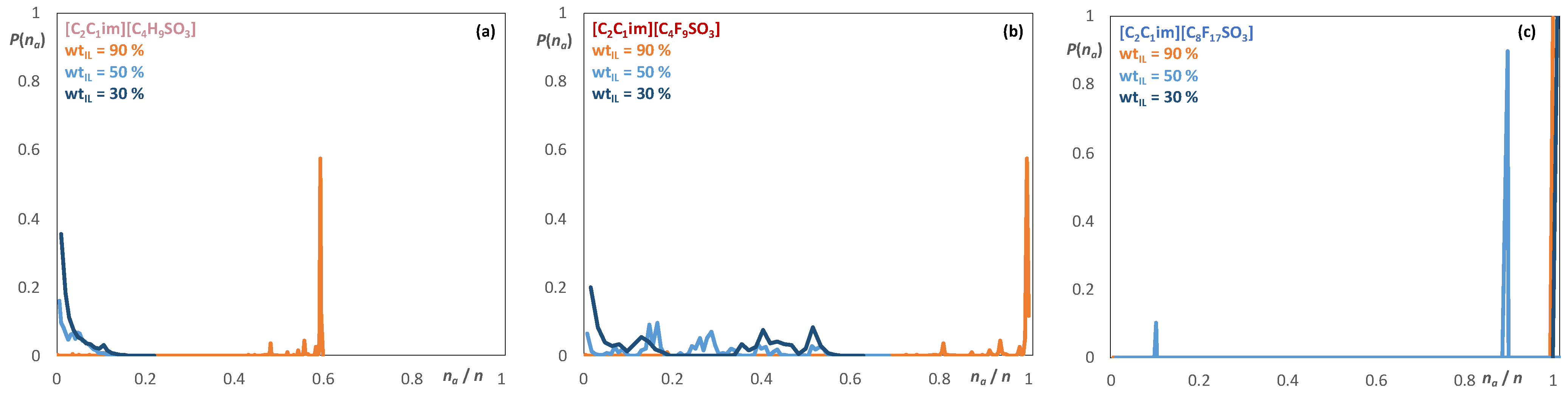
2.2. Aqueous Solutions of [C2C1im][CnF2n+1SO3], [C2C1im][CnH2n+1SO3], [C2C1im][CF3CO2], and [C2C1im][CH3CO2]
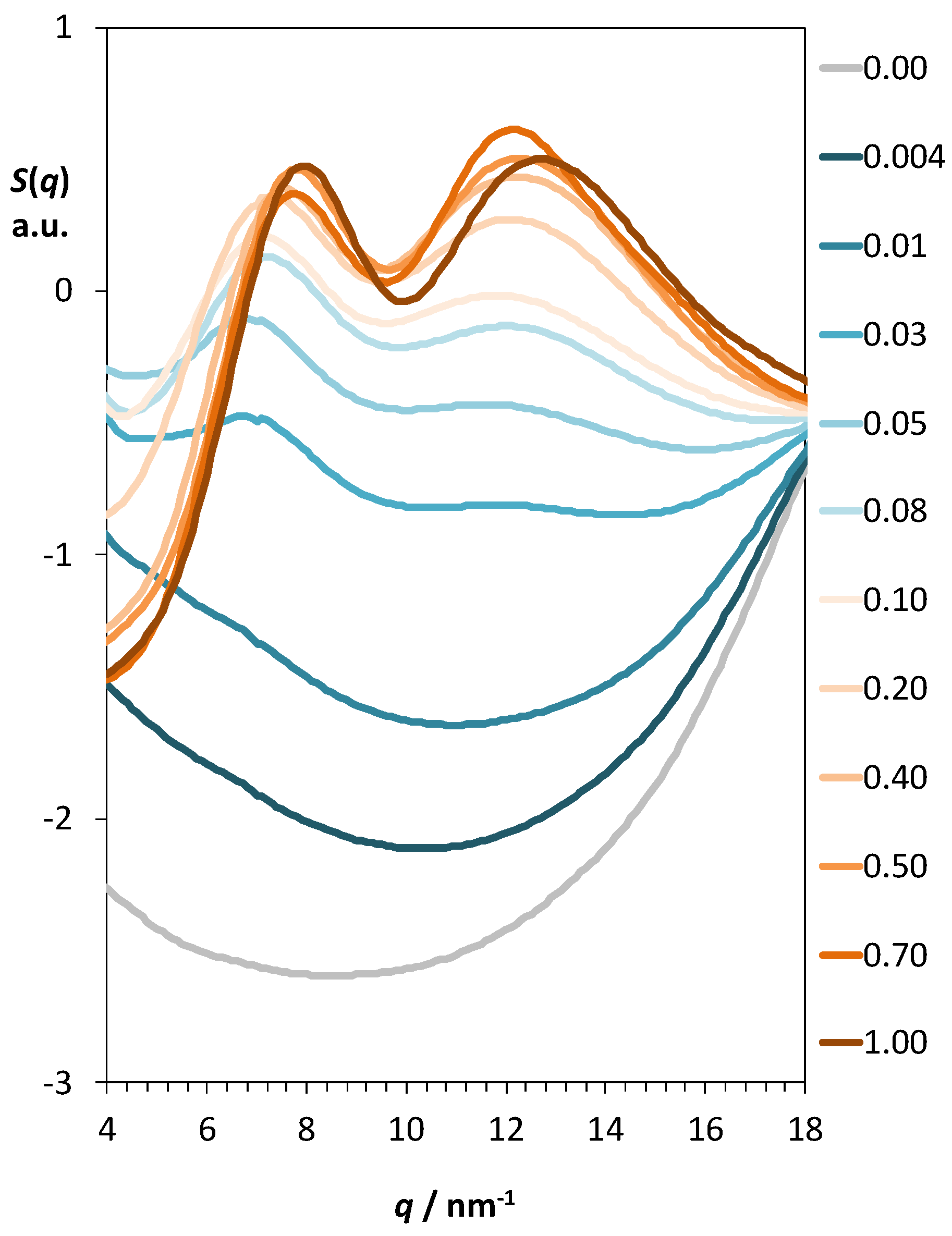
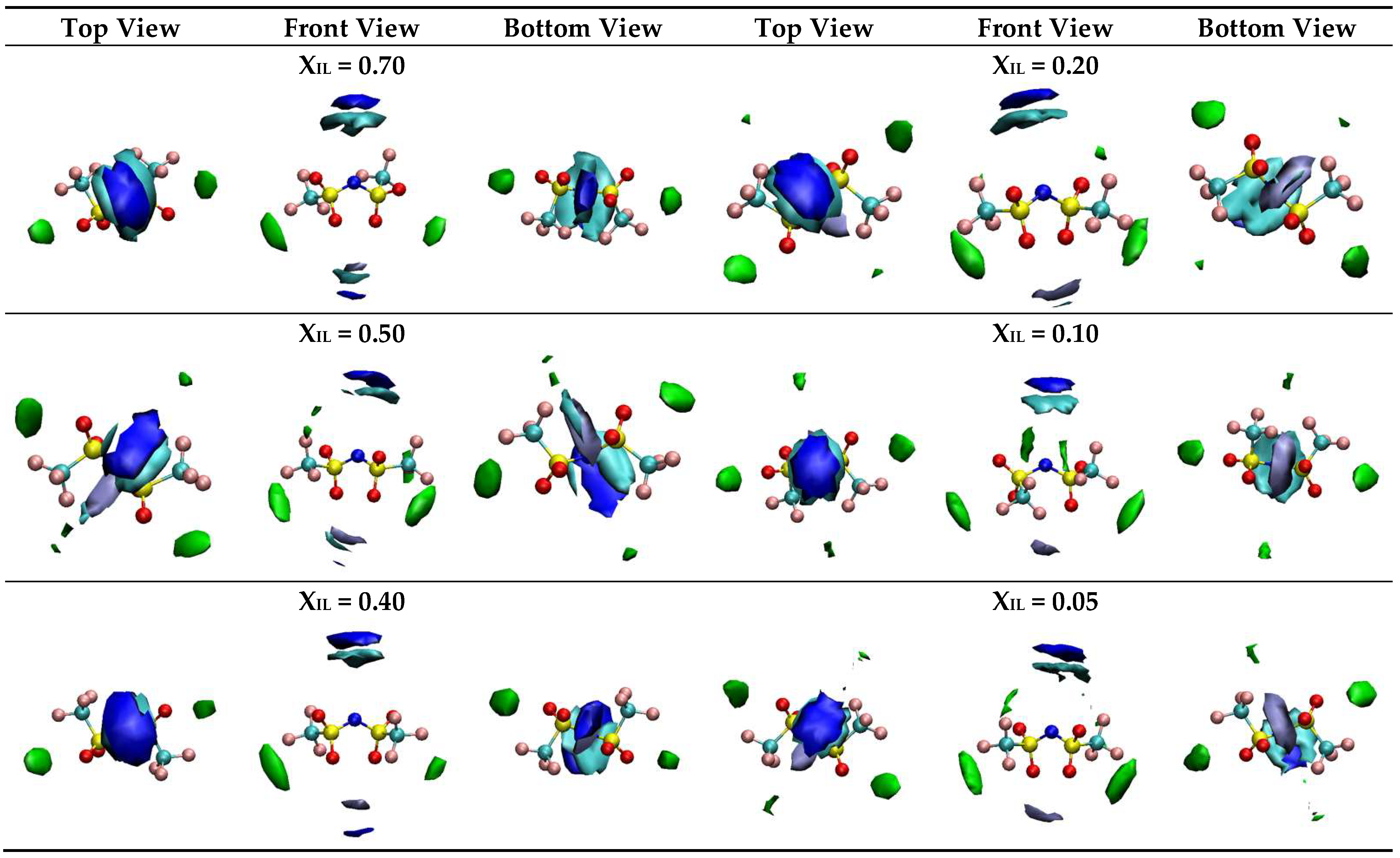
2.3. Aqueous Solutions of [N1 2OH 2OH 2OH][NTf2]
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997; ISBN 0-9678550-9-8. [Google Scholar]
- Shimizu, K.; Costa Gomes, M.F.; Pádua, A.A.H.; Rebelo, L.P.N.; Canongia Lopes, J.N. Three commentaries on the nano-segregated structure of ionic liquids. J. Mol. Struct. THEOCHEM 2010, 946, 70–76. [Google Scholar] [CrossRef]
- Rogers, R.D.; Seddon, K.R. Ionic Liquids—Solvents of the Future? Science 2003, 302, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.G.; Willauer, H.D.; Swatloski, R.P.; Visser, A.E.; Rogers, R.D. Room temperature ionic liquids as novel media for ‘clean’ liquid–liquid extraction. Chem. Commun. 1998, 16, 1765–1766. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green solvents for sustainable organic synthesis: State of the art. Green Chem. 2005, 7, 267–278. [Google Scholar] [CrossRef]
- Neves, C.M.S.S.; Ventura, S.P.M.; Freire, M.G.; Marrucho, I.M.; Coutinho, J.A.P. Evaluation of cation influence on the formation and extraction capability of ionic-liquid-based aqueous biphasic systems. J. Phys. Chem. B 2009, 113, 5194–5199. [Google Scholar] [CrossRef] [Green Version]
- Cláudio, A.F.M.; Freire, M.G.; Freire, C.S.; Silvestre, A.J.; Coutinho, J.A. Extraction of vanillin using ionic-liquid-based aqueous two-phase systems. Sep. Purif. Technol. 2010, 75, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.M.; Coutinho, J.A.; Fernandes, A.M.; Freire, M.G. Complete removal of textile dyes from aqueous media using ionic-liquid-based aqueous two-phase systems. Sep. Purif. Technol. 2014, 128, 58–66. [Google Scholar] [CrossRef]
- Marrucho, I.; Branco, L.; Rebelo, L. Ionic Liquids in Pharmaceutical Applications. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 527–546. [Google Scholar] [CrossRef]
- Lopes, J.N.C.; Deschamps, J.; Pádua, A.A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Lopes, J.N.C.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids Composed of Triflate or Bistriflylimide Anions. J. Phys. Chem. B 2004, 108, 16893–16898. [Google Scholar] [CrossRef]
- Tomé, L.I.N.; Jorge, M.; Gomes, J.R.B.; Coutinho, J.A.P. Molecular Dynamics Simulation Studies of the Interactions between Ionic Liquids and Amino Acids in Aqueous Solution. J. Phys. Chem. B 2012, 116, 1831–1842. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canongia Lopes, J.N.; Pádua, A.A.H. Nanostructural organization in ionic liquids. J. Phys. Chem. B 2006, 110, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Li, N.; Zheng, L.; Yu, L.; Inoue, T. Surface adsorption and micelle formation of surface active ionic liquids in aqueous solution. Langmuir 2007, 23, 4178–4182. [Google Scholar] [CrossRef]
- Ribeiro, M.C.C. High Viscosity of Imidazolium Ionic Liquids with the Hydrogen Sulfate Anion: A Raman Spectroscopy Study. J. Phys. Chem. B 2012, 116, 7281–7290. [Google Scholar] [CrossRef]
- Blesic, M.; Lop, J.N.C.; Padua, A.; Shimizu, K.; Gomes, M.C.; Rebelo, L.P. Phase Equilibria in Ionic Liquid−Aromatic Compound Mixtures, Including Benzene Fluorination Effects. J. Phys. Chem. B 2009, 113, 7631–7636. [Google Scholar] [CrossRef]
- Dias, N.; Shimizu, K.; Morgado, P.; Filipe, E.J.M.; Lopes, J.N.C.; Chávez, F.V. Charge Templates in Aromatic Plus Ionic Liquid Systems Revisited: NMR Experiments and Molecular Dynamics Simulations. J. Phys. Chem. B 2014, 118, 5772–5780. [Google Scholar] [CrossRef]
- Shimizu, K.; Costa Gomes, M.F.; Pádua, A.A.H.; Rebelo, L.P.N.; Canongia Lopes, J.N. On the role of the dipole and quadrupole moments of aromatic compounds in the solvation by ionic liquids. J. Phys. Chem. B 2009, 113, 9894–9900. [Google Scholar] [CrossRef]
- Pereiro, A.B.; Rodriguez, A.; Blesic, M.; Shimizu, K.; Lopes, J.N.C.; Rebelo, L.P.N. Mixtures of Pyridine and Nicotine with Pyridinium-Based Ionic Liquids. J. Chem. Eng. Data 2011, 56, 4356–4363. [Google Scholar] [CrossRef]
- Freire, M.; Neves, C.S.; Carvalho, P.J.; Gardas, R.; Fernandes, A.M.; Marrucho, I.; Santos, L.; Coutinho, J. Mutual Solubilities of Water and Hydrophobic Ionic Liquids. J. Phys. Chem. B 2007, 111, 13082–13089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire, M.G.; Neves, C.M.S.S.; Shimizu, K.; Bernardes, C.E.S.; Marrucho, I.M.; Coutinho, J.A.P.; Lopes, J.N.C.; Rebelo, L.P.N. Mutual Solubility of Water and Structural/Positional Isomers of N-Alkylpyridinium-Based Ionic Liquids. J. Phys. Chem. B 2010, 114, 15925–15934. [Google Scholar] [CrossRef] [PubMed]
- Kurnia, K.A.; Sintra, T.E.; Neves, C.M.S.S.; Shimizu, K.; Canongia Lopes, J.N.; Gonçalves, F.; Ventura, S.P.M.; Freire, M.G.; Santos, L.M.N.B.F.; Coutinho, J.A.P. The effect of the cation alkyl chain branching on mutual solubilities with water and toxicities. Phys. Chem. Chem. Phys. 2014, 16, 19952–19963. [Google Scholar] [CrossRef]
- Neves, C.M.S.S.; Kurnia, K.A.; Shimizu, K.; Marrucho, I.M.; Rebelo, L.P.N.; Coutinho, J.A.P.; Freire, M.G.; Lopes, J.N.C. The impact of ionic liquid fluorinated moieties on their thermophysical properties and aqueous phase behaviour. Phys. Chem. Chem. Phys. 2014, 16, 21340–21348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, J.C.; Carvalho, S.F.; Welton, T.; Lopes, J.N.C.; Rebelo, L.P.N.; Shimizu, K.; Araújo, J.M.M.; Pereiro, A.B. Design of task-specific fluorinated ionic liquids: Nanosegregation versus hydrogen-bonding ability in aqueous solutions. Chem. Commun. 2018, 54, 3524–3527. [Google Scholar] [CrossRef]
- Ruivo, D.; Canongia Lopes, J.N.; Deive, F.J.; Esperança, J.M.S.S.; Rebelo, L.P.N.; Rodríguez, A.; Shimizu, K. Molecular dynamics studies on the structure and interactions of ionic liquids containing amino-acid anions. Phys. Chem. Chem. Phys. 2018, 20, 23864–23872. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.A.; Shimizu, K.; Canongia Lopes, J.N. Bio-inspired hydrophilic bistriflimide-based ionic liquids: Molecular dynam-ics modeling and simulations. J. Mol. Liq. 2020, 301, 112402. [Google Scholar] [CrossRef]
- Costa, A.J.L.; Soromenho, M.R.C.; Shimizu, K.; Marrucho, I.M.; Esperança, J.M.S.S.; Canongia Lopes, J.N.; Rebelo, L.P.N. Liquid–liquid equilibrium of cholinium-derived bistriflimide ionic liquids with water and octanol. J. Phys. Chem. B 2012, 116, 9186–9195. [Google Scholar] [CrossRef]
- Mão de Ferro, A.; Reis, P.M.; Soromenho, M.R.C.; Bernardes, C.E.S.; Shimizu, K.; Freitas, A.A.; Esperança, J.M.S.S.; Canongia Lopes, J.N.; Rebelo, L.P.N. Designing the ammonium cation to achieve a higher hydrophilicity of bistriflimide-based ionic liq-uids. Phys. Chem. Chem. Phys. 2018, 20, 19307–19313. [Google Scholar] [CrossRef]
- Tariq, M.; Shimizu, K.; Esperança, J.M.S.S.; Canongia Lopes, J.N.; Rebelo, L.P.N. Viscosity minima in binary mixtures of ionic liquids + molecular solvents. Phys. Chem. Chem. Phys. 2015, 17, 13480–13494. [Google Scholar] [CrossRef]
- Costa, A.J.L.; Soromenho, M.R.C.; Shimizu, K.; Esperança, J.M.S.S.; Canongia Lopes, J.N.; Rebelo, L.P.N. Unusual LCST-type behaviour found in binary mixtures of choline-based ionic liquids with ethers. RSC Adv. 2013, 3, 10262–10271. [Google Scholar] [CrossRef]
- Deive, F.J.; Rodríguez, A.; Pereiro, A.B.; Shimizu, K.; Forte, P.A.S.; Romão, C.C.; Canongia Lopes, J.N.; Esperança, J.M.S.S.; Rebelo, L.P.N. Phase equilibria of haloalkanes dissolved in ethylsulfate- or ethylsulfonate-based ionic liquids. J. Phys. Chem. B 2010, 114, 7329–7337. [Google Scholar] [CrossRef] [PubMed]
- Morgado, P.; Shimizu, K.; Esperança, J.M.S.S.; Reis, P.M.; Rebelo, L.P.N.; Canongia Lopes, J.N.; Filipe, E.J.M. Using 129Xe NMR to probe the structure of ionic liquids. J. Phys. Chem. Lett. 2013, 4, 2758–2762. [Google Scholar] [CrossRef]
- Pison, L.; Shimizu, K.; Tamas, G.; Lopes, J.N.C.; Quitevis, E.L.; Gomes, M.F.C. Solubility of n-butane and 2-methylpropane (isobutane) in 1-alkyl-3-methylimidazolium-based ionic liquids with linear and branched alkyl side-chains. Phys. Chem. Chem. Phys. 2015, 17, 30328–30342. [Google Scholar] [CrossRef] [PubMed]
- Cláudio, A.F.M.; Neves, M.C.; Shimizu, K.; Canongia Lopes, J.N.; Freire, M.G.; Coutinho, J.A.P. The magic of aqueous solu-tions of ionic liquids: Ionic liquids as a powerful class of catanionic hydrotropes. Green Chem. 2015, 17, 3948–3963. [Google Scholar] [CrossRef] [PubMed]
- Sintra, T.E.; Shimizu, K.; Ventura, S.P.M.; Shimizu, S.; Canongia Lopes, J.N.; Coutinho, J.A.P. Enhanced dissolution of ibu-profen using ionic liquids as catanionic hydrotropes. Phys. Chem. Chem. Phys. 2018, 20, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Freitas, A.A.; Atkin, R.; Warr, G.G.; FitzGerald, P.A.; Doi, H.; Saito, S.; Ueno, K.; Umebayashi, Y.; Watanabe, M.; et al. Structural and aggregate analyses of (Li salt + glyme) mixtures: The complex nature of solvate ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 22321–22335. [Google Scholar] [CrossRef]
- Murphy, T.; Callear, S.K.; Yepuri, N.; Shimizu, K.; Watanabe, M.; Canongia Lopes, J.N.; Darwish, T.; Warr, G.G.; Atkin, R. Bulk nanostructure of the prototypical ‘good’ and ‘poor’ solvate ionic liquids [Li(G4)][TFSI] and [Li(G4)][NO3]. Phys. Chem. Chem. Phys. 2016, 18, 17224–17236. [Google Scholar] [CrossRef] [Green Version]
- Thum, A.; Heuer, A.; Shimizu, K.; Lopes, J.N.C. Solvate ionic liquids based on lithium bis(trifluoromethanesulfonyl)imide–glyme systems: Coordination in MD simulations with scaled charges. Phys. Chem. Chem. Phys. 2020, 22, 525–535. [Google Scholar] [CrossRef]
- Bruce, D.W.; Gao, Y.; Lopes, J.N.C.; Shimizu, K.; Slattery, J.M. Liquid-Crystalline Ionic Liquids as Ordered Reaction Media for the Diels-Alder Reaction. Chem. A Eur. J. 2016, 22, 16113–16123. [Google Scholar] [CrossRef]
- Bruce, D.W.; Cabry, C.P.; Canongia Lopes, J.N.; Costen, M.L.; D’Andrea, L.; Grillo, I.; Marshall, B.C.; McKendrick, K.G.; Minton, T.K.; Purcell, S.M.; et al. Nanosegregation and structuring in the bulk and at the surface of ionic-liquid mixtures. J. Phys. Chem. B 2017, 121, 6002–6020. [Google Scholar] [CrossRef] [Green Version]
- Cabry, C.P.; D’Andrea, L.; Shimizu, K.; Grillo, I.; Li, P.; Rogers, S.; Bruce, D.W.; Canongia Lopes, J.N.; Slattery, J.M. Exploring the bulk-phase structure of ionic liquid mixtures using small-angle neutron scattering. Faraday Discuss. 2018, 206, 265–289. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Lopes, J.N.C. Probing the structural features of the 1-alkyl-3-methylimidazolium hexafluorophosphate ionic liquid series using Molecular Dynamics simulations. J. Mol. Liq. 2015, 210, 257–263. [Google Scholar] [CrossRef]
- Shimizu, K.; Lopes, J.N.C. Comparing the structure of different ionic liquid series: Bistriflamide v. hexafluorophosphate; pure v. equimolar mixtures. Fluid Phase Equilibria 2016, 418, 181–191. [Google Scholar] [CrossRef]
- Danten, Y.; Cabaço, M.I.; Besnard, M. Interaction of water diluted in 1-butyl-3-methyl imidazolium ionic liquids by vibrational spectroscopy modeling. J. Mol. Liq. 2010, 153, 57–66. [Google Scholar] [CrossRef]
- Stange, P.; Fumino, K.; Ludwig, R. Ion Speciation of Protic Ionic Liquids in Water: Transition from Contact to Solvent-Separated Ion Pairs. Angew. Chem. Int. Ed. 2013, 52, 2990–2994. [Google Scholar] [CrossRef] [PubMed]
- Macchieraldo, R.; Esser, L.; Elfgen, R.; Voepel, P.; Zahn, S.; Smarsly, B.M.; Kirchner, B. Hydrophilic Ionic Liquid Mixtures of Weakly and Strongly Coordinating Anions with and without Water. ACS Omega 2018, 3, 8567–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, N.S.M.; Reis, P.M.; Shimizu, K.; Cortes, O.A.; Marrucho, I.M.; Araújo, J.M.M.; Esperança, J.M.S.S.; Canongia Lopes, J.N.; Pereiro, A.B.; Rebelo, L.P.N. A thermophysical and structural characterization of ionic liquids with alkyl and perfluoroalkyl side chains. RSC Adv. 2015, 5, 65337–65350. [Google Scholar] [CrossRef]
- Fujii, K.; Kanzaki, R.; Takamuku, T.; Kameda, Y.; Kohara, S.; Kanakubo, M.; Shibayama, M.; Ishiguro, S.-I.; Umebayashi, Y. Experimental evidences for molecular origin of low-Q peak in neutron/x-ray scattering of 1-alkyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide ionic liquids. J. Chem. Phys. 2011, 135, 244502. [Google Scholar] [CrossRef]
- Pereiro, A.B.; Araújo, J.M.M.; Martinho, S.; Alves, F.; Nunes, S.; Matias, A.; Duarte, C.M.M.; Rebelo, L.P.N.; Marrucho, I.M. Fluorinated ionic liquids: Properties and applications. ACS Sustain. Chem. Eng. 2013, 1, 427–439. [Google Scholar] [CrossRef]
- Sánchez, P.B.; García, J.; Pádua, A.A. Structural effects on dynamic and energetic properties of mixtures of ionic liquids and water. J. Mol. Liq. 2017, 242, 204–212. [Google Scholar] [CrossRef]
- Thomas, M.; Brehm, M.; Holloczki, O.; Kelemen, Z.; Nyulaszi, L.; Pasinszki, T.; Kirchner, B. Simulating the vibrational spectra of ionic liquid systems: 1-Ethyl-3-methylimidazolium acetate and its mixtures. J. Chem. Phys. 2014, 141, 024510. [Google Scholar] [CrossRef]
- Hall, C.A.; Le, K.A.; Rudaz, C.; Radhi, A.; Lovell, C.S.; Damion, R.; Budtova, T.; Ries, M.E. Macroscopic and Microscopic Study of 1-Ethyl-3-methyl-imidazolium Acetate–Water Mixtures. J. Phys. Chem. B 2012, 116, 12810–12818. [Google Scholar] [CrossRef]
- Strate, A.; Niemann, T.; Ludwig, R. Controlling the kinetic and thermodynamic stability of cationic clusters by the addition of molecules or counterions. Phys. Chem. Chem. Phys. 2017, 19, 18854–18862. [Google Scholar] [CrossRef]
- Niemann, T.; Neumann, J.; Stange, P.; Gartner, S.; Young, T.G.A.; Paschek, D.; Warr, G.G.; Atkin, R.; Ludwig, R. The double-faced nature of hydrogen bonding in hydroxy-functionalized ionic liquids shown by neutron diffraction and molecular dynamics simulations. Angew. Chem. Int. Ed. 2019, 58, 12887–12892. [Google Scholar] [CrossRef]
- Smith, W.; Forester, T.R. The DL_POLY Package of Molecular Simulation Routines; V.2.2; The Council for The Central Laboratory of Research Councils; Daresbury Laboratory: Warrington, UK, 2006. [Google Scholar]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Van Drunen, R.; Van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M. Gromacs: A parallel computer for molecular dynamics simulations. In Proceedings of the 4th International Con-ference Physics Computing, Prague, Czech Republic, 24–28 August 1992; pp. 252–256. [Google Scholar]
- Berendsen, H.; Van der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementa-tion. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics sim-ulations with GROMACS. In International Conference on Exascale Applications and Software; Springer: Cham, Switzerland, 2015; Volume 8759, pp. 3–27. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Praprotnik, M.; Janežič, D.; Mavri, J. Temperature dependence of water vibrational spectrum: A molecular dynamics simulation study. J. Phys. Chem. A 2004, 108, 11056–11062. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Mão de Ferro, A.; Reis, P.M.; Freitas, A.A.; Canongia Lopes, J.N.; Rebelo, L.P.N.; Shimizu, K.; Esperança, J.M.S.S. Evidences for a null molar volume contribution by hydroxyl groups in ammonium bistriflimide-based ionic liquids. J. Chem. Eng. Data 2019, 64, 4932–4945. [Google Scholar] [CrossRef]
- Blesic, M.; Marques, M.H.; Plechkova, N.V.; Seddon, K.R.; Rebelo, L.P.N.; Lopes, A. Self-aggregation of ionic liquids: Micelle formation in aqueous solution. Green Chem. 2007, 9, 481–490. [Google Scholar] [CrossRef]
- Brown, P.; Butts, C.; Dyer, R.; Eastoe, J.; Grillo, I.; Guittard, F.; Rogers, S.; Heenan, R. Anionic surfactants and surfactant ion-ic liquids with quaternary ammonium counterions. Langmuir 2011, 27, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- El Seoud, O.; Keppeler, N.; Malek, N.; Galgano, P. Ionic Liquid-Based Surfactants: Recent Advances in Their Syntheses, Solution Properties, and Applications. Polymers 2021, 13, 1100. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Ionic Liquid | nIL | nwater | wtIL | XIL | lbox | Section | Ionic Liquid | nIL | nwater | wtIL | XIL | lbox |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Section 2.1 | [C3C1im][NTf2] | 320 | – | 1.00 | 1.00 | 5.19 | Section 2.2 | [C2C1im][C4H9SO3] | 600 | – | 1.00 | 1.00 | 6.07 |
| [C4C1im][NTf2] | 300 | – | 1.00 | 1.00 | 5.28 | [C2C1im][C4H9SO3] | 420 | 620 | 0.90 | 0.40 | 5.57 | ||
| [iC4C1im][NTf2] | 300 | – | 1.00 | 1.00 | 5.20 | [C2C1im][C4H9SO3] | 190 | 2600 | 0.50 | 0.07 | 5.26 | ||
| [C3C1im][NTf2] | 300 | 130 | 0.98 | 0.70 | 5.16 | [C2C1im][C4H9SO3] | 105 | 3300 | 0.30 | 0.03 | 5.14 | ||
| [C4C1im][NTf2] | 300 | 130 | 0.98 | 0.70 | 5.25 | [C2C1im][CF3CO2] | 1000 | – | 1.00 | 1.00 | 6.55 | ||
| [iC4C1im][NTf2] | 300 | 130 | 0.98 | 0.70 | 5.25 | [C2C1im][CF3CO2] | 450 | 620 | 0.90 | 0.42 | 5.27 | ||
| Section 2.2 | [C2C1im][CF3SO3] | 1000 | – | 1.00 | 1.00 | 6.69 | [C2C1im][CF3CO2] | 210 | 2600 | 0.50 | 0.07 | 5.13 | |
| [C2C1im][CF3SO3] | 390 | 620 | 0.90 | 0.39 | 5.16 | [C2C1im][CF3CO2] | 115 | 3300 | 0.30 | 0.03 | 5.05 | ||
| [C2C1im][CF3SO3] | 180 | 2600 | 0.50 | 0.06 | 5.08 | [C2C1im][CH3CO2] | 1000 | – | 1.00 | 1.00 | 6.39 | ||
| [C2C1im][CF3SO3] | 100 | 3300 | 0.30 | 0.03 | 5.57 | [C2C1im][CH3CO2] | 600 | 620 | 0.90 | 0.49 | 5.56 | ||
| [C2C1im][C4F9SO3] | 350 | – | 1.00 | 1.00 | 5.30 | [C2C1im][CH3CO2] | 280 | 2600 | 0.50 | 0.10 | 5.25 | ||
| [C2C1im][C4F9SO3] | 250 | 620 | 0.90 | 0.29 | 5.00 | [C2C1im][CH3CO2] | 150 | 3300 | 0.30 | 0.04 | 5.11 | ||
| [C2C1im][C4F9SO3] | 115 | 2600 | 0.50 | 0.05 | 5.01 | Section 2.3 | [N1 2OH 2OH 2OH][NTf2] | 350 | – | 1.00 | 1.00 | 5.44 | |
| [C2C1im][C4F9SO3] | 62 | 3300 | 0.30 | 0.02 | 4.98 | [N1 2OH 2OH 2OH][NTf2] | 340 | 146 | 0.98 | 0.70 | 5.40 | ||
| [C2C1im][C8F17SO3] | 250 | – | 1.00 | 1.00 | 5.32 | [N1 2OH 2OH 2OH][NTf2] | 320 | 320 | 0.96 | 0.50 | 5.40 | ||
| [C2C1im][C8F17SO3] | 180 | 620 | 0.90 | 0.23 | 5.00 | [N1 2OH 2OH 2OH][NTf2] | 320 | 480 | 0.94 | 0.40 | 5.44 | ||
| [C2C1im][C8F17SO3] | 78 | 2600 | 0.50 | 0.05 | 4.97 | [N1 2OH 2OH 2OH][NTf2] | 280 | 1120 | 0.86 | 0.20 | 5.46 | ||
| [C2C1im][C8F17SO3] | 43 | 3300 | 0.30 | 0.01 | 4.97 | [N1 2OH 2OH 2OH][NTf2] | 220 | 1980 | 0.73 | 0.10 | 5.44 | ||
| [C2C1im][CH3SO3] | 1000 | – | 1.00 | 1.00 | 6.62 | [N1 2OH 2OH 2OH][NTf2] | 160 | 3040 | 0.56 | 0.05 | 5.50 | ||
| [C2C1im][CH3SO3] | 490 | 620 | 0.90 | 0.44 | 5.42 | [N1 2OH 2OH 2OH][NTf2] | 115 | 3718 | 0.43 | 0.03 | 5.51 | ||
| [C2C1im][CH3SO3] | 230 | 2600 | 0.50 | 0.08 | 5.22 | [N1 2OH 2OH 2OH][NTf2] | 48 | 4752 | 0.20 | 0.01 | 5.45 | ||
| [C2C1im][CH3SO3] | 125 | 3300 | 0.30 | 0.04 | 5.11 | [N1 2OH 2OH 2OH][NTf2] | 20 | 4980 | 0.09 | 0.004 | 5.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, R.F.; Freitas, A.A.; Canongia Lopes, J.N.; Shimizu, K. Ionic Liquids and Water: Hydrophobicity vs. Hydrophilicity. Molecules 2021, 26, 7159. https://doi.org/10.3390/molecules26237159
Rodrigues RF, Freitas AA, Canongia Lopes JN, Shimizu K. Ionic Liquids and Water: Hydrophobicity vs. Hydrophilicity. Molecules. 2021; 26(23):7159. https://doi.org/10.3390/molecules26237159
Chicago/Turabian StyleRodrigues, Rita F., Adilson A. Freitas, José N. Canongia Lopes, and Karina Shimizu. 2021. "Ionic Liquids and Water: Hydrophobicity vs. Hydrophilicity" Molecules 26, no. 23: 7159. https://doi.org/10.3390/molecules26237159
APA StyleRodrigues, R. F., Freitas, A. A., Canongia Lopes, J. N., & Shimizu, K. (2021). Ionic Liquids and Water: Hydrophobicity vs. Hydrophilicity. Molecules, 26(23), 7159. https://doi.org/10.3390/molecules26237159