
Dipyridylmethane Ethers as Ligands for Luminescent Ir Complexes


Abstract
:1. Introduction
2. Results and Discussion
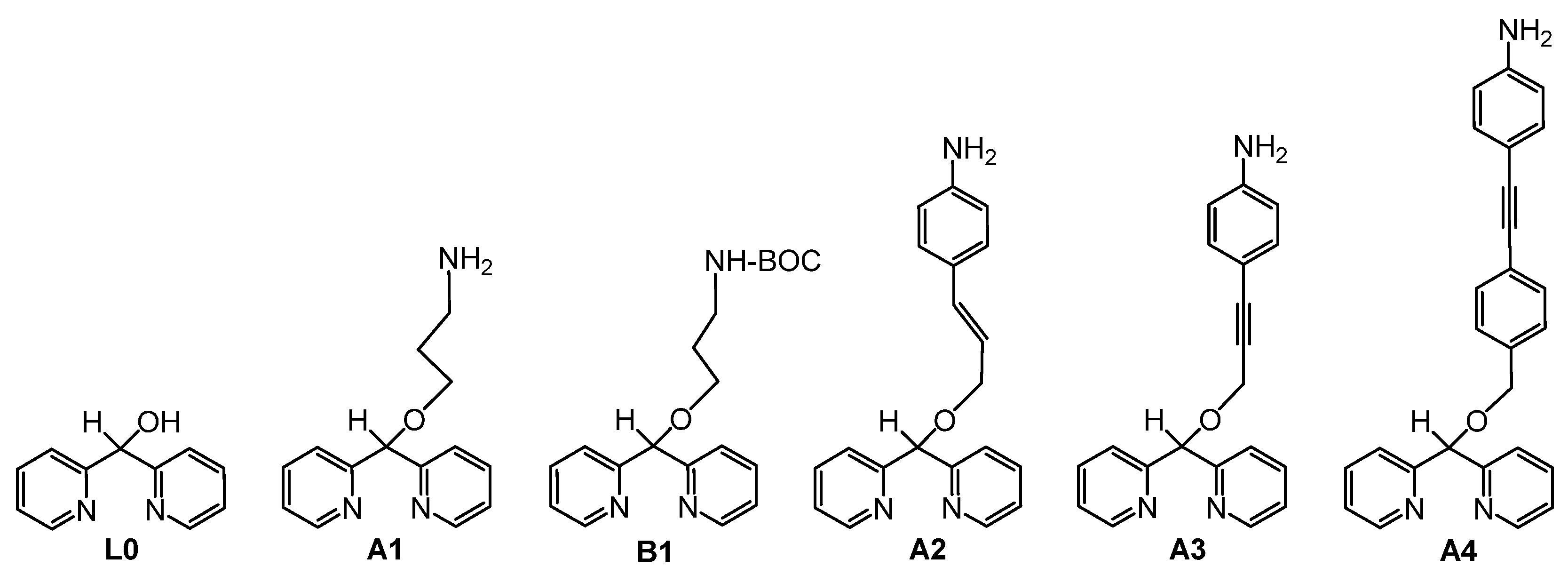
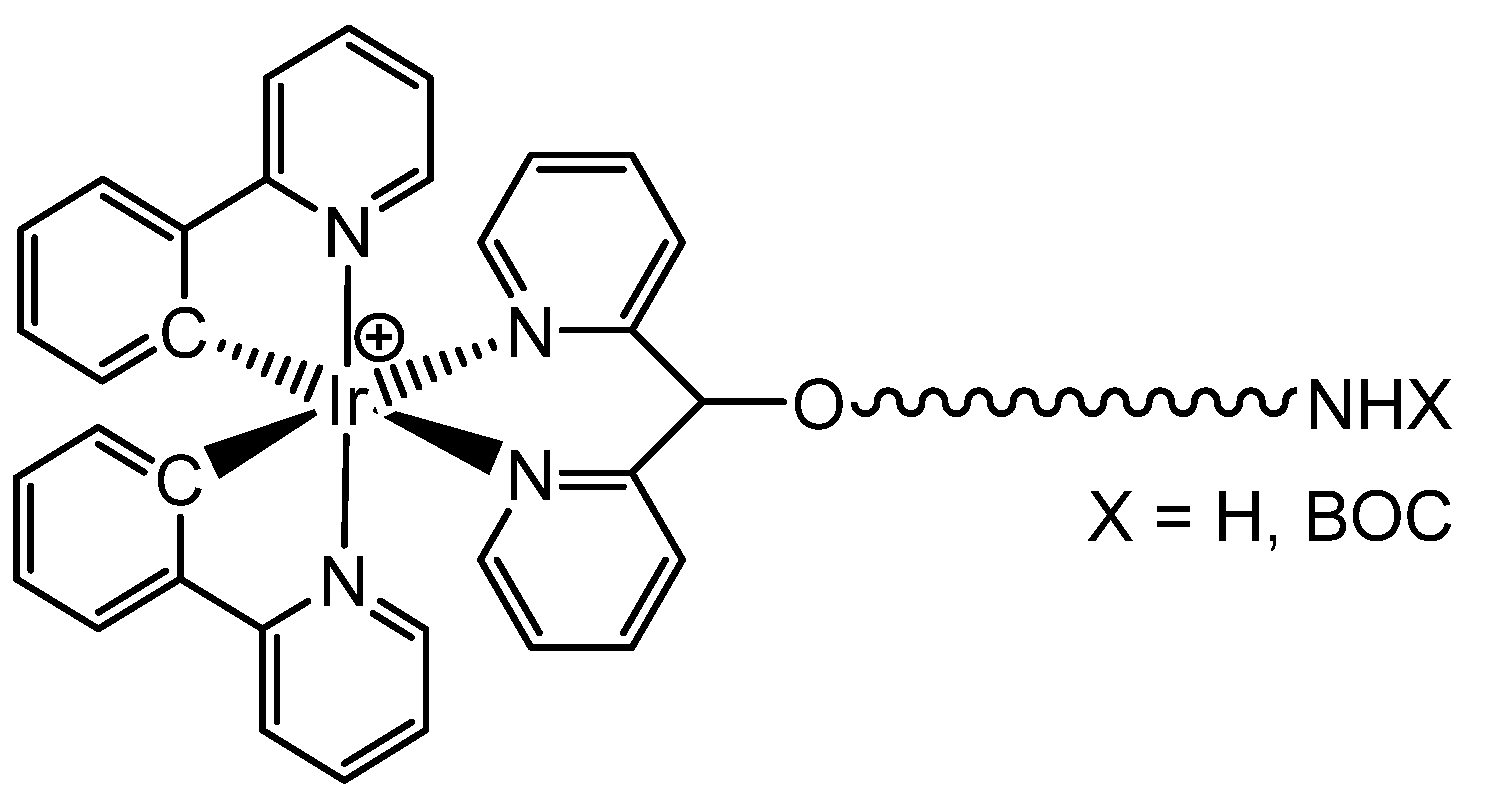
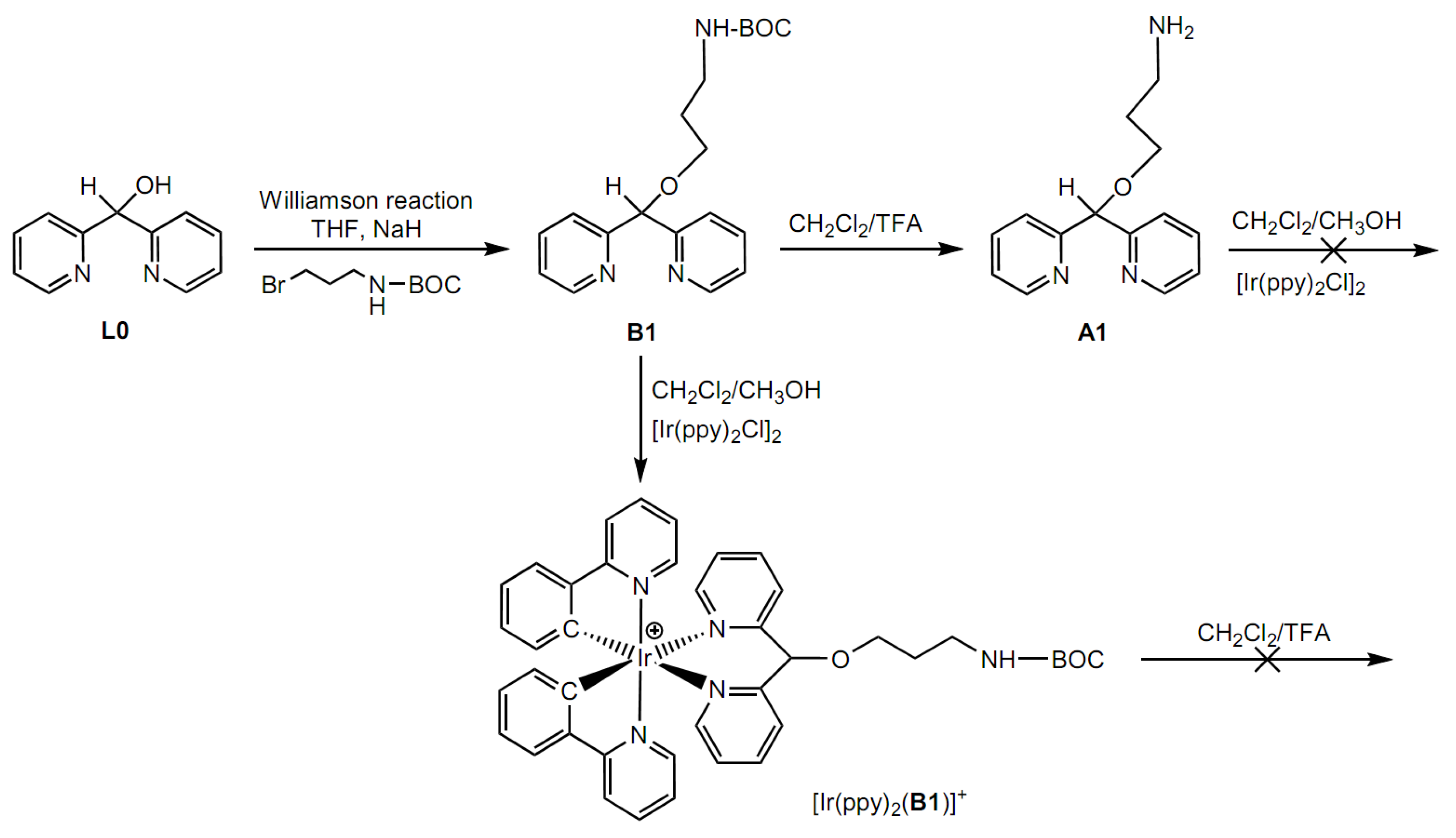
2.1. Syntheses
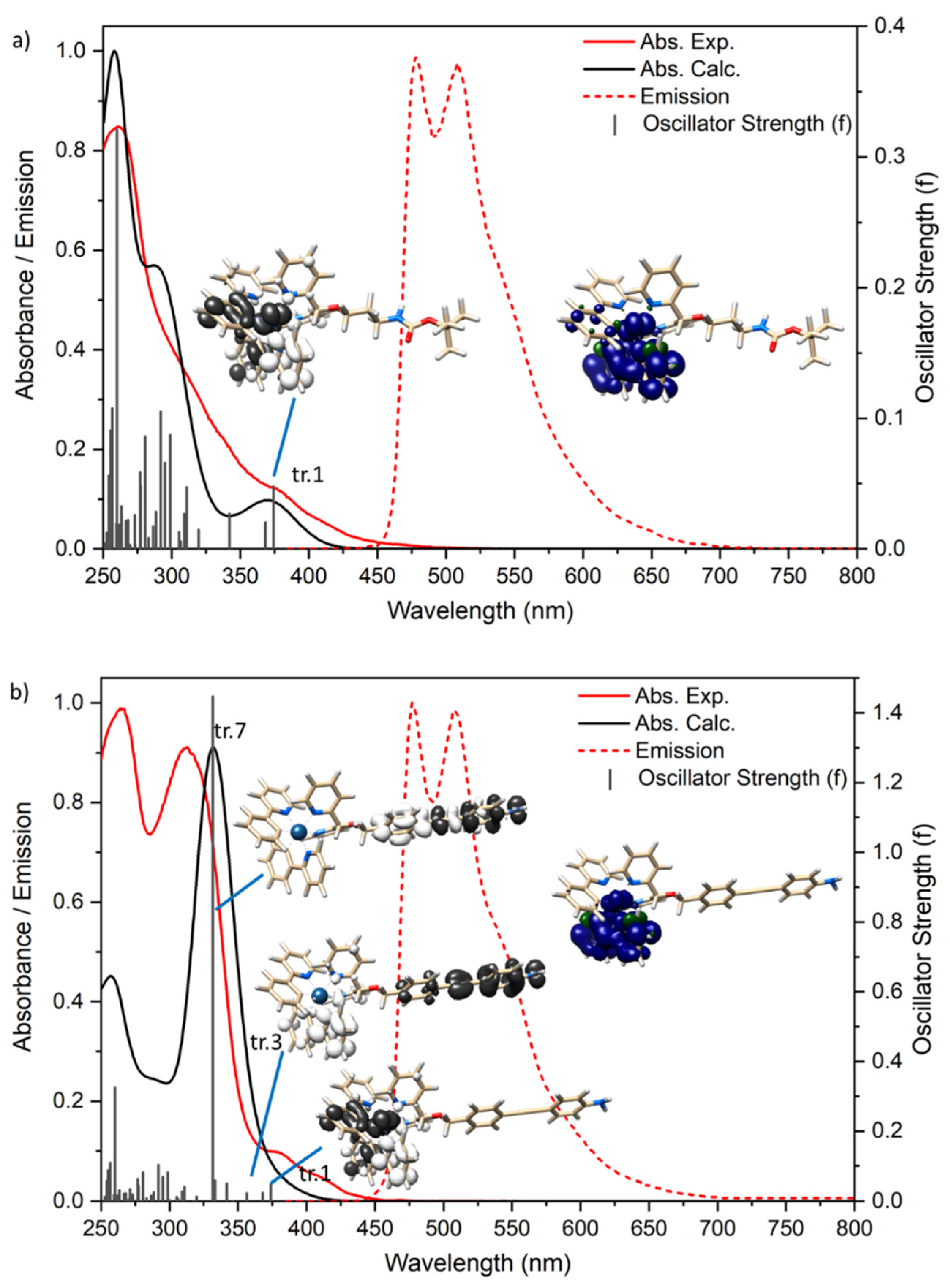
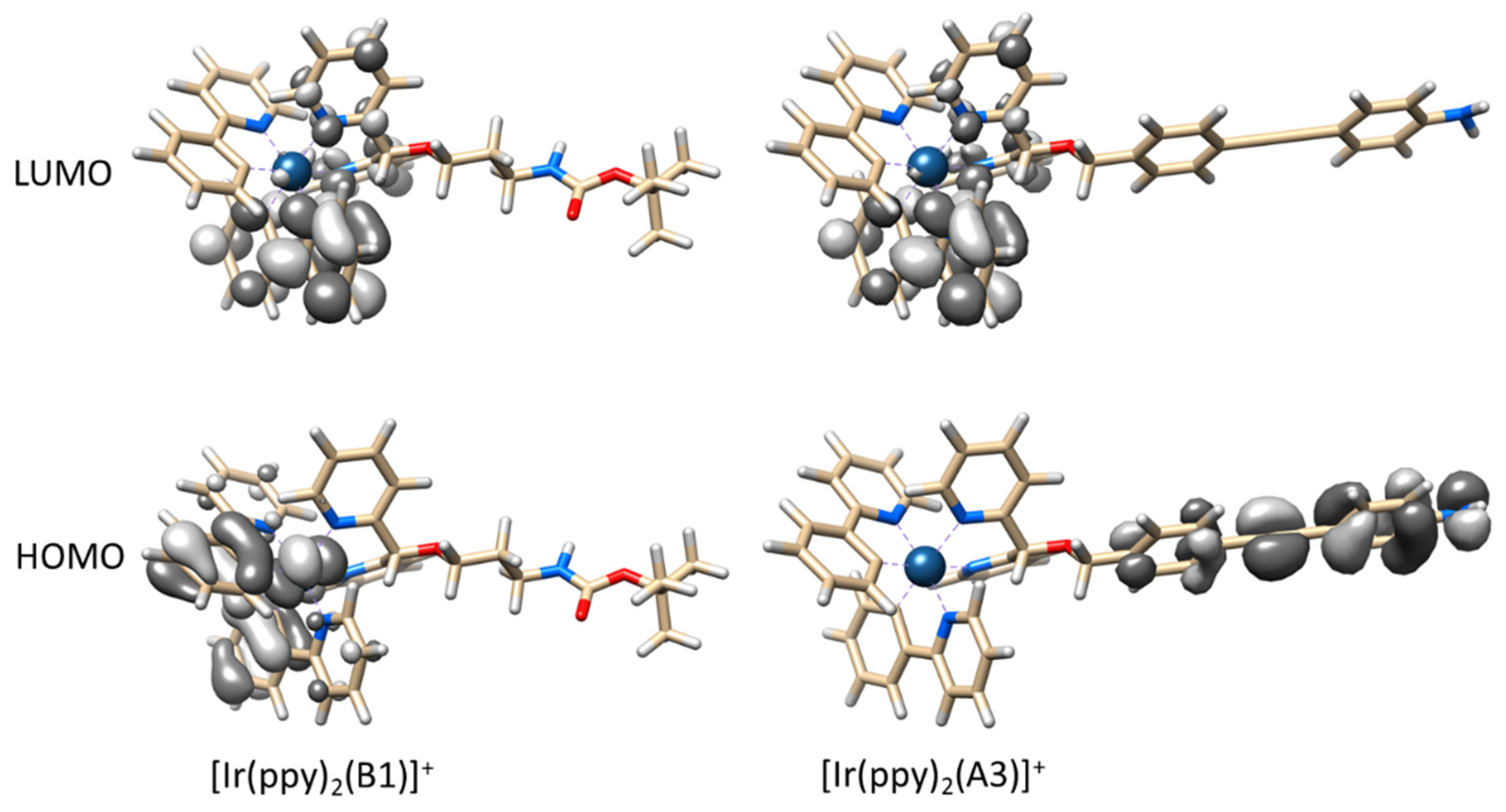
2.2. Absorption Spectra and Luminescence
2.3. Electrochemistry
3. Materials and Methods
3.1. Synthesis of Ligands
- B1: tert-butyl 3-(dipyridin-2-ylmethoxy)propylcarbamate
- A1: 3-(dipyridin-2-ylmethoxy)propan-1-amine
- A2: 4-(3-(di(pyridin-2-yl)methoxy)prop-1-en-1-yl)aniline
- A3: 4-(3-(benzhydryloxy)prop-1-ynyl)aniline
- A4: (4-((4-((dipyridin-2-ylmethoxy)methyl) phenyl)-ethynyl)aniline
3.2. Synthesis of Iridium Complexes
- [Ir(ppy)2(B1)]+
- [Ir(ppy)2(A4)]+
3.3. Characterization
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bazargan, M.; Mirzaei, M.; Franconetti, A.; Frontera, A. On the Preferences of Five-Membered Chelate Rings in Coordination Chemistry: Insights from the Cambridge Structural Database and Theoretical Calculations. Dalton Trans. 2019, 48, 5476–5490. [Google Scholar] [CrossRef] [PubMed]
- Kaes, C.; Katz, A.; Hosseini, M.W. Bipyridine: The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2‘-Bipyridine Units. Chem. Rev. 2000, 100, 3553–3590. [Google Scholar] [CrossRef]
- Yang, T.; Tang, N.; Wan, Q.; Yin, S.-F.; Qiu, R. Recent Progress on Synthesis of N,N′-Chelate Organoboron Derivatives. Molecules 2021, 26, 1401. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, L.; Braun, C.; Nieger, M.; Bräse, S. The Coordination- and Photochemistry of Copper(I) Complexes: Variation of N^N Ligands from Imidazole to Tetrazole. Dalton Trans. 2018, 47, 608–621. [Google Scholar] [CrossRef]
- Constable; Housecroft The Early Years of 2,2′-Bipyridine—A Ligand in Its Own Lifetime. Molecules 2019, 24, 3951. [CrossRef] [PubMed] [Green Version]
- Xie, L.-M.; Bai, F.-Q.; Li, W.; Zhang, Z.-X.; Zhang, H.-X. Theoretical Research on the Effect of Regulated π-Conjugation on the Photophysical Properties of Ir(iii) Complexes. Phys. Chem. Chem. Phys. 2015, 17, 10014–10021. [Google Scholar] [CrossRef]
- Sakamoto, R.; Wu, K.-H.; Matsuoka, R.; Maeda, H.; Nishihara, H. π-Conjugated Bis(Terpyridine)Metal Complex Molecular Wires. Chem. Soc. Rev. 2015, 44, 7698–7714. [Google Scholar] [CrossRef]
- Schubert, C.; Margraf, J.T.; Clark, T.; Guldi, D.M. Molecular Wires—Impact of π-Conjugation and Implementation of Molecular Bottlenecks. Chem. Soc. Rev. 2015, 44, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, J.; Wang, Y.; Zhou, D.; Wang, P. Redox Couple Related Influences of π-Conjugation Extension in Organic Dye-Sensitized Mesoscopic Solar Cells. Chem. Sci. 2011, 2, 1401. [Google Scholar] [CrossRef]
- Volpi, G.; Garino, C.; Breuza, E.; Gobetto, R.; Nervi, C. Dipyridylketone as a Versatile Ligand Precursor for New Cationic Heteroleptic Cyclometalated Iridium Complexes. Dalton Trans. 2011, 41, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Volpi, G.; Garino, C.; Nervi, C. Exploring Synthetic Pathways to Cationic Heteroleptic Cyclometalated Iridium Complexes Derived from Dipyridylketone. Dalton Trans. 2012, 41, 7098–7108. [Google Scholar] [CrossRef]
- Wang, J.; Dyers, L.; Mason, R.; Amoyaw, P.; Bu, X.R. Highly Efficient and Direct Heterocyclization of Dipyridyl Ketone to N,N-Bidentate Ligands. J. Org. Chem. 2005, 70, 2353–2356. [Google Scholar] [CrossRef]
- Volpi, G.; Garino, C.; Conterosito, E.; Barolo, C.; Gobetto, R.; Viscardi, G. Facile Synthesis of Novel Blue Light and Large Stoke Shift Emitting Tetradentate Polyazines Based on Imidazo[1,5-a]Pyridine. Dyes Pigment. 2016, 128, 96–100. [Google Scholar] [CrossRef]
- Volpi, G.; Lace, B.; Garino, C.; Priola, E.; Artuso, E.; Cerreia Vioglio, P.; Barolo, C.; Fin, A.; Genre, A.; Prandi, C. New Substituted Imidazo[1,5-a]Pyridine and Imidazo[5,1-a]Isoquinoline Derivatives and Their Application in Fluorescence Cell Imaging. Dyes Pigment. 2018, 157, 298–304. [Google Scholar] [CrossRef]
- Volpi, G.; Rabezzana, R. Imidazo[1,5-a]Pyridine Derivatives: Useful, Luminescent and Versatile Scaffolds for Different Applications. New J. Chem. 2021, 5737–5743. [Google Scholar] [CrossRef]
- Volpi, G.; Garino, C.; Priola, E.; Magistris, C.; Chierotti, M.R.; Barolo, C. Halogenated Imidazo[1,5-a]Pyridines: Chemical Structure and Optical Properties of a Promising Luminescent Scaffold. Dyes Pigment. 2019, 171, 107713. [Google Scholar] [CrossRef]
- Volpi, G.; Galliano, S.; Buscaino, R.; Viscardi, G.; Barolo, C. Fluorescent Trifluoromethylated Imidazo[1,5-a]Pyridines and Their Application in Luminescent down-Shifting Conversion. J. Lumines. 2022, 242, 118529. [Google Scholar] [CrossRef]
- Volpi, G.; Garino, C.; Fresta, E.; Casamassa, E.; Giordano, M.; Barolo, C.; Viscardi, G. Strategies to Increase the Quantum Yield: Luminescent Methoxylated Imidazo[1,5-a]Pyridines. Dyes Pigment. 2021, 192, 109455. [Google Scholar] [CrossRef]
- Ardizzoia, G.A.; Ghiotti, D.; Therrien, B.; Brenna, S. Homoleptic Complexes of Divalent Metals Bearing N,O-Bidentate Imidazo[1,5-a]Pyridine Ligands: Synthesis, X-Ray Characterization and Catalytic Activity in the Heck Reaction. Inorg. Chim. Acta 2018, 471, 384–390. [Google Scholar] [CrossRef]
- Ardizzoia, G.A.; Brenna, S.; Durini, S.; Therrien, B. Synthesis and Characterization of Luminescent Zinc(II) Complexes with a N,N-Bidentate 1-Pyridylimidazo[1,5-a]Pyridine Ligand. Polyhedron 2015, 90, 214–220. [Google Scholar] [CrossRef]
- Ardizzoia, G.A.; Brenna, S.; Durini, S.; Therrien, B.; Veronelli, M. Synthesis, Structure, and Photophysical Properties of Blue-Emitting Zinc(II) Complexes with 3-Aryl-Substituted 1-Pyridylimidazo[1,5-a]Pyridine Ligands: Blue-Emitting Zinc(II) Complexes. Eur. J. Inorg. Chem. 2014, 2014, 4310–4319. [Google Scholar] [CrossRef]
- Volpi, G.; Priola, E.; Garino, C.; Daolio, A.; Rabezzana, R.; Benzi, P.; Giordana, A.; Diana, E.; Gobetto, R. Blue Fluorescent Zinc(II) Complexes Based on Tunable Imidazo[1,5-a]Pyridines. Inorg. Chim. Acta 2020, 509, 119662. [Google Scholar] [CrossRef]
- Weber, M.D.; Garino, C.; Volpi, G.; Casamassa, E.; Milanesio, M.; Barolo, C.; Costa, R.D. Origin of a Counterintuitive Yellow Light-Emitting Electrochemical Cell Based on a Blue-Emitting Heteroleptic Copper(i) Complex. Dalton Trans. 2016, 45, 8984–8993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Rodríguez, A.M.; Kvapilova, H.; Sykora, J.; Towrie, M.; Nervi, C.; Volpi, G.; Zalis, S.; Vlcek, A. Photophysics of Singlet and Triplet Intraligand Excited States in [ReCl(CO)3(1-(2-Pyridyl)-Imidazo[1,5-α]Pyridine)] Complexes. J. Am. Chem. Soc. 2014, 136, 5963–5973. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Volpi, G.; Bonomo, M.; Mariani, P.; Garino, C.; Viscardi, G. Methoxy-Substituted Copper Complexes as Possible Redox Mediators in Dye-Sensitized Solar Cells. New J. Chem. 2021, 45, 15303–15311. [Google Scholar] [CrossRef]
- Zhu, S.; Lin, X.; Ran, P.; Xia, Q.; Yang, C.; Ma, J.; Fu, Y. A Novel Luminescence-Functionalized Metal-Organic Framework Nanoflowers Electrochemiluminesence Sensor via “on-off” System. Biosens. Bioelectron. 2017, 91, 436–440. [Google Scholar] [CrossRef]
- Miomandre, F.; Stancheva, S.; Audibert, J.-F.; Brosseau, A.; Pansu, R.B.; Lepeltier, M.; Mayer, C.R. Gold and Silver Nanoparticles Functionalized by Luminescent Iridium Complexes: Synthesis and Photophysical and Electrofluorochromic Properties. J. Phys. Chem. C 2013, 117, 12806–12814. [Google Scholar] [CrossRef]
- Liu, J.; Dong, S. Grafting of Diaminoalkane on Glassy Carbon Surface and Its Functionalization. Electrochem. Commun. 2000, 2, 707–712. [Google Scholar] [CrossRef]
- Kanazawa, A.; Daisaku, T.; Okajima, T.; Uchiyama, S.; Kawauchi, S.; Ohsaka, T. Characterization by Electrochemical and X-Ray Photoelectron Spectroscopic Measurements and Quantum Chemical Calculations of N-Containing Functional Groups Introduced onto Glassy Carbon Electrode Surfaces by Electrooxidation of a Carbamate Salt in Aqueous Solutions. Langmuir 2014, 30, 5297–5305. [Google Scholar] [CrossRef]
- Hayashida, E.; Takahashi, Y.; Nishi, H.; Uchiyama, S. Electrolytic Aminated Carbon Materials for the Electrocatalytic Redox Reactions of Inorganic and Organic Compounds. J. Environ. Sci. 2011, 23, S124–S127. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Applications of Functionalized Transition Metal Complexes in Photonic and Optoelectronic Devices. Coord. Chem. Rev. 1998, 177, 347–414. [Google Scholar] [CrossRef]
- Sandroni, M.; Volpi, G.; Fiedler, J.; Buscaino, R.; Viscardi, G.; Milone, L.; Gobetto, R.; Nervi, C. Iridium and Ruthenium Complexes Covalently Bonded to Carbon Surfaces by Means of Electrochemical Oxidation of Aromatic Amines. Catal. Today 2010, 158, 22–28. [Google Scholar] [CrossRef]
- Ezquerro, C.; Fresta, E.; Serrano, E.; Lalinde, E.; García-Martínez, J.; Berenguer, J.R.; Costa, R.D. White-Emitting Organometallo-Silica Nanoparticles for Sun-like Light-Emitting Diodes. Mater. Horiz. 2019, 6, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.; Dodge, H.M.; Farquhar, A.H.; Gardner, K.E.; Miller, A.J.M. Decarbonylative Ether Dissection by Iridium Pincer Complexes. Chem. Sci. 2020, 11, 12130–12138. [Google Scholar] [CrossRef]
- Haibach, M.C.; Lease, N.; Goldman, A.S. Catalytic Cleavage of Ether C–O Bonds by Pincer Iridium Complexes. Angew. Chem. Int. Ed. 2014, 53, 10160–10163. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, N.; Curchod, B.F.E.; Male, L.; Ma, D.; Fan, J.; Liu, Y.; Zhu, W.; Baranoff, E. Tuning the Oxidation Potential of 2-Phenylpyridine-Based Iridium Complexes to Improve the Performance of Bluish and White OLEDs. J. Mater. Chem. C 2016, 4, 3738–3746. [Google Scholar] [CrossRef]
- Tehfe, M.-A.; Lepeltier, M.; Dumur, F.; Gigmes, D.; Fouassier, J.-P.; Lalevée, J. Structural Effects in the Iridium Complex Series: Photoredox Catalysis and Photoinitiation of Polymerization Reactions under Visible Lights. Macromol. Chem. Phys. 2017, 218, 1700192. [Google Scholar] [CrossRef]
- Herr, J.M.; Rössiger, C.; Locke, H.; Wilhelm, M.; Becker, J.; Heimbrodt, W.; Schlettwein, D.; Göttlich, R. Synthesis, Optical and Theoretical Characterization of Heteroleptic Iridium(III) Imidazo[1,5-a]Pyridine and -Quinoline Complexes. Dyes Pigment. 2020, 180, 108512. [Google Scholar] [CrossRef]
- Volpi, G.; Garino, C.; Salassa, L.; Fiedler, J.; Hardcastle, K.I.; Gobetto, R.; Nervi, C. Cationic Heteroleptic Cyclometalated Iridium Complexes with 1-Pyridylimidazo[1,5-a]Pyridine Ligands: Exploitation of an Efficient Intersystem Crossing. Chem.-Eur. J. 2009, 15, 6415–6427. [Google Scholar] [CrossRef]
- Adenier, A.; Chehimi, M.M.; Gallardo, I.; Pinson, J.; Vilà, N. Electrochemical Oxidation of Aliphatic Amines and Their Attachment to Carbon and Metal Surfaces. Langmuir 2004, 20, 8243–8253. [Google Scholar] [CrossRef]
- Orwat, B.; Oh, M.J.; Zaranek, M.; Kubicki, M.; Januszewski, R.; Kownacki, I. Microwave-Accelerated C,N-Cyclometalation as a Route to Chloro-Bridged Iridium(III) Binuclear Precursors of Phosphorescent Materials: Optimization, Synthesis, and Studies of the Iridium(III) Dimer Behavior in Coordinating Solvents. Inorg. Chem. 2020, 59, 9163–9176. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An Efficient Implementation of Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New Developments in the Polarizable Continuum Model for Quantum Mechanical and Classical Calculations on Molecules in Solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A Library for Package-Independent Computational Chemistry Algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Browne, W.R.; O’Boyle, N.M.; McGarvey, J.J.; Vos, J.G. Elucidating Excited State Electronic Structure and Intercomponent Interactions in Multicomponent and Supramolecular Systems. Chem. Soc. Rev. 2005, 34, 641–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headgordon, M.; Grana, A.M.; Maurice, D.; White, C.A. Analysis of Electronic-Transitions as the Difference of Electron-Attachment and Detachment Densities. J. Phys. Chem. 1995, 99, 14261–14270. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput Chem 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Rotundo, L.; Garino, C.; Nencini, L.; Yoon, S.S.; Gobetto, R.; Nervi, C. Electrochemical CO2 Reduction at Glassy Carbon Electrodes Functionalized by Mn I and Re I Organometallic Complexes. ChemPhysChem 2017, 18, 3219–3229. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Gobetto, R.; Nervi, C. Recent Advances in Catalytic CO2 Reduction by Organometal Complexes Anchored on Modified Electrodes. New J. Chem. 2016, 40, 5656–5661. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λabs (nm) | λem (nm) | Φ | τ (ns) | Eox (V) | Ered (V) | |
|---|---|---|---|---|---|---|
| [Ir(ppy)2(B1)]+ | 380 314 sh 260 | 549 509 479 | 8% | 605 | E1/2 = 0.953 | Ep = −2.363 Ep = −2.595 |
| [Ir(ppy)2(A4)]+ | 380 313 265 | 548 509 478 | 7% | 2170 | Ep= 0.494 Ep= 0.891 Ep= 1.031 | Ep= −2.093 Ep= −2.369 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volpi, G.; Garino, C.; Gobetto, R.; Nervi, C. Dipyridylmethane Ethers as Ligands for Luminescent Ir Complexes. Molecules 2021, 26, 7161. https://doi.org/10.3390/molecules26237161
Volpi G, Garino C, Gobetto R, Nervi C. Dipyridylmethane Ethers as Ligands for Luminescent Ir Complexes. Molecules. 2021; 26(23):7161. https://doi.org/10.3390/molecules26237161
Chicago/Turabian StyleVolpi, Giorgio, Claudio Garino, Roberto Gobetto, and Carlo Nervi. 2021. "Dipyridylmethane Ethers as Ligands for Luminescent Ir Complexes" Molecules 26, no. 23: 7161. https://doi.org/10.3390/molecules26237161
APA StyleVolpi, G., Garino, C., Gobetto, R., & Nervi, C. (2021). Dipyridylmethane Ethers as Ligands for Luminescent Ir Complexes. Molecules, 26(23), 7161. https://doi.org/10.3390/molecules26237161