
Quantum Mechanical-Based Stability Evaluation of Crystal Structures for HIV-Targeted Drug Cabotegravir


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
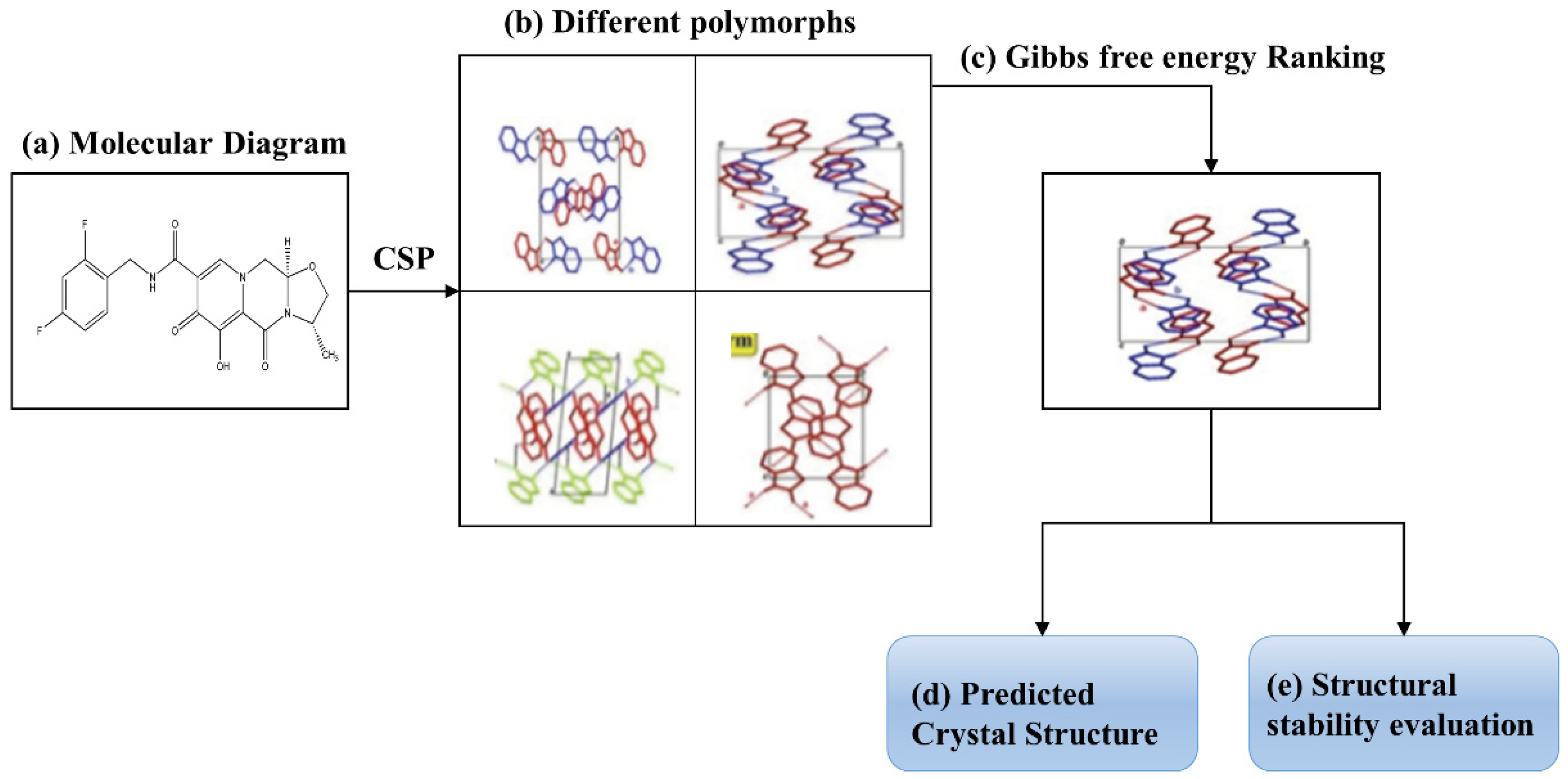
2. Results and Discussion
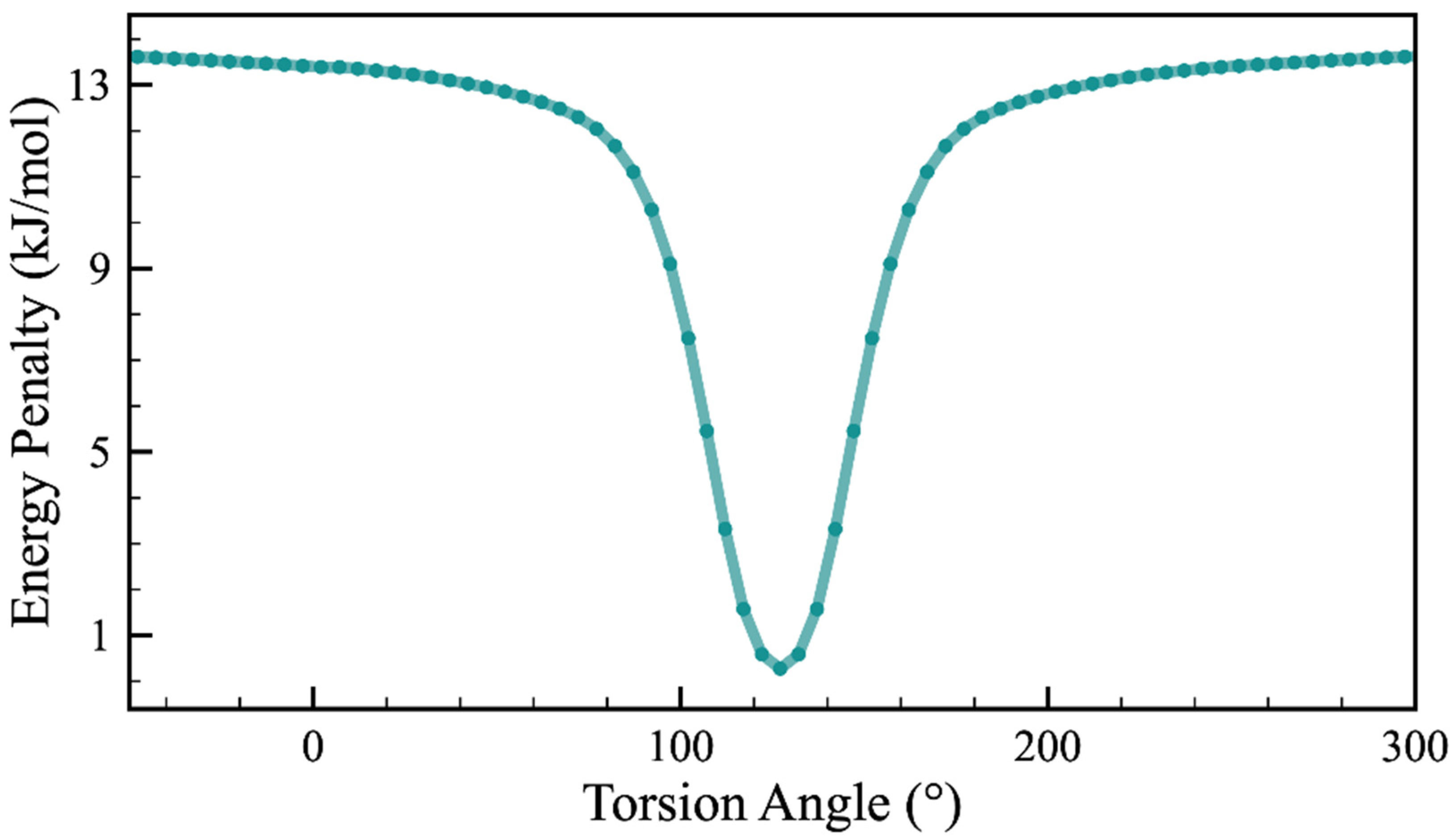
2.1. Conformational Search
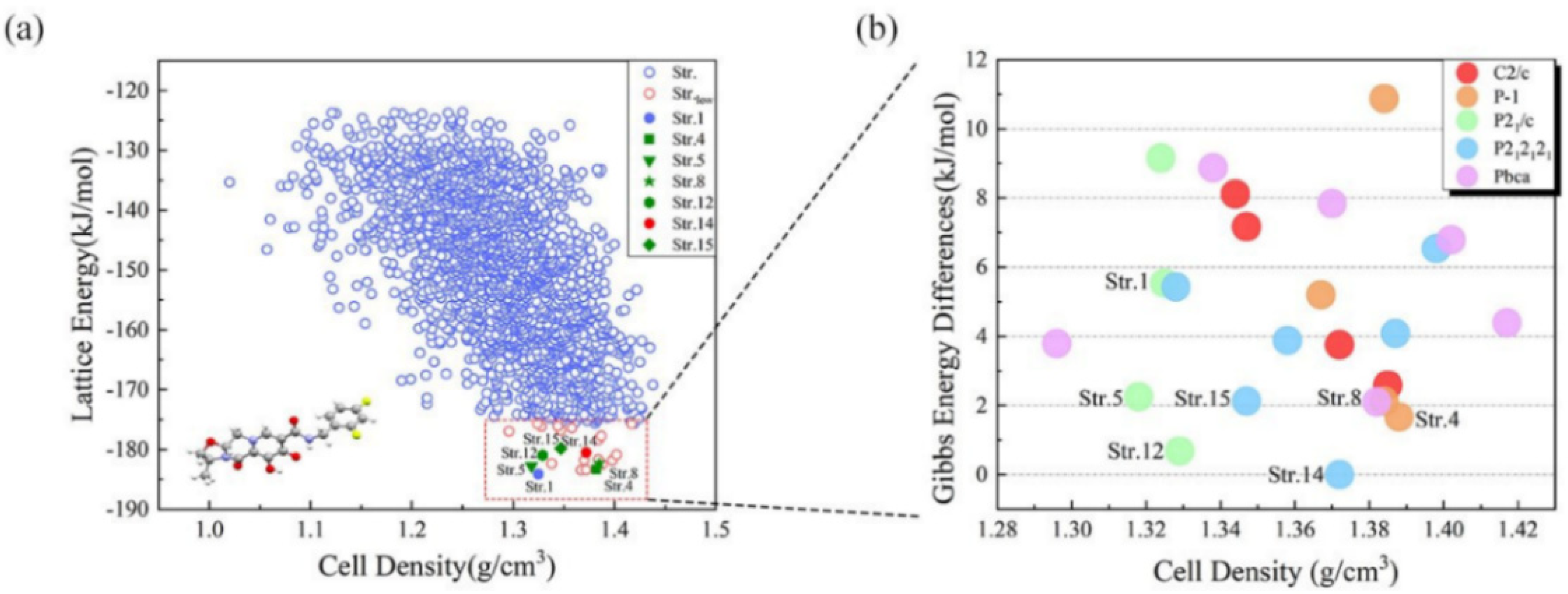
2.2. Gibbs Free Energy Guided CSP
3. Methods
3.1. Potential Energy Surface (PES) Scan
3.2. MOLPAK Global Search of Structures
3.3. Calculation of Gibbs Free Energy Parallel Execution Program
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pavlos, R.; Phillips, E.J. Individualization of antiretroviral therapy. Pharmgenom. Pers Med. 2011, 5, 1–17. [Google Scholar]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.; et al. Prevention of HIV-1 Infection with Early Antiretroviral Therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Ford, S.L.; Gould, E.; Chen, S.; Margolis, D.; Spreen, W.; Crauwels, H.; Piscitelli, S. Lack of Pharmacokinetic Interaction between Rilpivirine and Integrase Inhibitors Dolutegravir and GSK1265744. Antimicrob. Agents Chemother. 2013, 57, 5472–5477. [Google Scholar] [CrossRef] [Green Version]
- Andrews, C.D.; Spreen, W.R.; Mohri, H.; Moss, L.; Ford, S.; Gettie, A.; Russell-Lodrigue, K.; Bohm, R.P.; Cheng-Mayer, C.; Hong, Z.; et al. Long-Acting Integrase Inhibitor Protects Macaques from Intrarectal Simian/Human Immunodeficiency Virus. Science 2014, 343, 1151–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmon, S.L.; Mohri, H.; Spreen, W.; Markowitz, M. GSK1265744 Demonstrates Robust In Vitro Activity Against Various Clades of HIV-1. JAIDS J. Acquir. Immune Defic. Syndr. 2015, 68, e39–e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, T.D.; Sobieszczyk, M.E.; Markowitz, M. Cabotegravir in the treatment and prevention of Human Immunodeficiency Virus-1. Expert Opin. Investig. Drugs 2018, 27, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liang, J.K.; Chen, X.L.; Chen, J.R. Ab Initio Structure Determination of a New Compound, β-SrGaBO4, from Powder X-Ray Diffraction Data. J. Solid State Chem. 2002, 165, 119–124. [Google Scholar] [CrossRef]
- Rammohan, A.; Sarjeant, A.A.; Kaduk, J.A. Crystal structure of dicesium hydrogen citrate from laboratory single-crystal and powder X-ray diffraction data and DFT comparison. Acta Crystallogr. Sect. E Crystallogr. Commun. 2017, 73, 231–234. [Google Scholar] [CrossRef]
- Lesage, A.; Bardet, A.M.; Emsley, L. Through-Bond Carbon−Carbon Connectivities in Disordered Solids by NMR. J. Am. Chem. Soc. 1999, 121, 10987–10993. [Google Scholar] [CrossRef]
- Brown, S.; Spiess, H. Advanced Solid-State NMR Methods for the Elucidation of Structure and Dynamics of Molecular, Macromolecular, and Supramolecular Systems. Chem. Rev. 2001, 101, 4125–4156. [Google Scholar] [CrossRef]
- Baias, M.; Widdifield, C.M.; Dumez, J.-N.; Thompson, H.P.G.; Cooper, T.G.; Salager, E.; Bassil, S.; Stein, R.S.; Lesage, A.; Day, G.M.; et al. Powder crystallography of pharmaceutical materials by combined crystal structure prediction and solid-state 1H NMR spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 8069–8080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beran, G.J.O.; Hartman, J.D.; Heit, Y.N. Predicting Molecular Crystal Properties from First Principles: Finite-Temperature Thermochemistry to NMR Crystallography. Accounts Chem. Res. 2016, 49, 2501–2508. [Google Scholar] [CrossRef]
- Shtukenberg, A.G.; Hu, C.T.; Zhu, Q.; Schmidt, M.U.; Xu, W.; Tan, M.; Kahr, B. The Third Ambient Aspirin Polymorph. Cryst. Growth Des. 2017, 17, 3562–3566. [Google Scholar] [CrossRef]
- Price, L.; Braun, D.M.; Reutzel-Edens, S. Can computed crystal energy landscapes help understand pharmaceutical solids? Chem. Commun. 2016, 52, 7065–7077. [Google Scholar] [CrossRef]
- Day, G.M. Current approaches to predicting molecular organic crystal structures. Crystallogr. Rev. 2011, 17, 3–52. [Google Scholar] [CrossRef]
- Nyman, J.; Reutzel-Edens, S.M. Crystal structure prediction is changing from basic science to applied technology. Faraday Discuss. 2018, 211, 459–476. [Google Scholar] [CrossRef]
- Luo, H.; Hao, X.; Gong, Y.; Zhou, J.; He, X.; Li, J. Rational Crystal Polymorph Design of Olanzapine. Cryst. Growth Des. 2019, 19, 2388–2395. [Google Scholar] [CrossRef]
- Bhardwaj, R.M.; Price, L.S.; Price, S.L.; Reutzel-Edens, S.M.; Miller, G.J.; Oswald, I.D.H.; Johnston, B.F.; Florence, A.J. Exploring the Experimental and Computed Crystal Energy Landscape of Olanzapine. Cryst. Growth Des. 2013, 13, 1602–1617. [Google Scholar] [CrossRef]
- Tang, J.; Han, Y.; Ali, I.; Luo, H.; Nowak, A.; Li, J. Stability and phase transition investigation of olanzapine polymorphs. Chem. Phys. Lett. 2021, 767, 138384. [Google Scholar] [CrossRef]
- Vasileiadis, M.; Pantelides, C.C.; Adjiman, C. Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule. Chem. Eng. Sci. 2015, 121, 60–76. [Google Scholar] [CrossRef] [Green Version]
- Vasileiadis, M.; Kazantsev, A.V.; Karamertzanis, P.G.; Adjiman, C.S.; Pantelides, C.C. The polymorphs of ROY: Application of a systematic crystal structure prediction technique. Acta Crystallogr. Sect. B Struct. Sci. 2012, 68, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.; Leusen, F.J.J.; Neumann, M.A.; van de Streek, J. Progress in Crystal Structure Prediction. Chem. A Eur. J. 2011, 17, 10736–10744. [Google Scholar] [CrossRef] [PubMed]
- Pantelides, C.C.; Adjiman, C.S.; Kazantsev, A.V. General Computational Algorithms for Ab Initio Crystal Structure Prediction for Organic Molecules. In Topics in Current Chemistry; Springer: Singapore, 2014; Volume 345, pp. 25–58. [Google Scholar]
- Neumann, M.A.; van de Streek, J.; Fabbiani, F.P.A.; Hidber, P.; Grassmann, O. Combined crystal structure prediction and high-pressure crystallization in rational pharmaceutical polymorph screening. Nat. Commun. 2015, 6, 7793. [Google Scholar] [CrossRef] [Green Version]
- Gavezzotti, A. Molecular Aggregation: Structure Analysis and Molecular Simulation of Crystals and Liquids; OUP: Oxford, UK, 2006; Volume 19. [Google Scholar]
- Holden, J.R.; Du, Z.; Ammon, H.L. Prediction of possible crystal structures for C-, H-, N-, O-, and F-containing organic compounds. J. Comput. Chem. 1993, 14, 422–437. [Google Scholar] [CrossRef]
- Neumann, M.A. Tailor-Made Force Fields for Crystal-Structure Prediction. J. Phys. Chem. B 2008, 112, 9810–9829. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tilbury, C.; Kim, S.H.; Doherty, M.F. A design aid for crystal growth engineering. Prog. Mater. Sci. 2016, 82, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Salager, E.; Day, G.; Stein, R.S.; Pickard, C.J.; Elena-Herrmann, B.; Emsley, L. Powder Crystallography by Combined Crystal Structure Prediction and High-Resolution 1H Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 2564–2566. [Google Scholar] [CrossRef] [PubMed]
- Van Eijck, B.P.; Mooij, W.T.M.; Kroon, J. Ab initio crystal structure predictions for flexible hydrogen-bonded molecules. Part II. Accurate energy minimization. J. Comput. Chem. 2001, 22, 805–815. [Google Scholar] [CrossRef]
- Lewis, C.T.; Tocher, D.A.; Day, G.M.; Price, S.L. A computational and experimental search for polymorphs of parabanic acid—A salutary tale leading to the crystal structure of oxo-ureido-acetic acid methyl ester. CrystEngComm 2003, 5, 3–9. [Google Scholar] [CrossRef]
- Rice, B.; LeBlanc, L.M.; Otero-De-La-Roza, A.; Fuchter, M.J.; Johnson, E.R.; Nelson, J.; Jelfs, K.E. A computational exploration of the crystal energy and charge-carrier mobility landscapes of the chiral [6]helicene molecule. Nanoscale 2018, 10, 1865–1876. [Google Scholar] [CrossRef] [Green Version]
- Whittleton, S.R.; Otero-De-La-Roza, A.; Johnson, E.R. Exchange-Hole Dipole Dispersion Model for Accurate Energy Ranking in Molecular Crystal Structure Prediction. J. Chem. Theory Comput. 2017, 13, 441–450. [Google Scholar] [CrossRef]
- Price, S.L.; Price, L.S. Toward Computational Polymorph Prediction. In Polymorphism in the Pharmaceutical Industry; Wiley: Hoboken, NJ, USA, 2018; pp. 133–157. [Google Scholar]
- Hušák, M. Successful Strategy for High Degree of Freedom Crystal Structure Determination from Powder X-Ray Diffraction Data: A Case Study for Selexipag Form I with 38 DOF. Cryst. Growth Des. 2019, 19, 4625–4631. [Google Scholar] [CrossRef]
- Zilka, M. Ab initio random structure searching of organic molecular solids: Assessment and validation against experimental data. Phys. Chem. Chem. Phys. 2017, 19, 25949–25960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baias, M.; Dumez, J.-N.; Svensson, P.H.; Schantz, S.; Day, G.M.; Emsley, L. De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sode, O.; Voth, G.A.; Hirata, S. A solid–solid phase transition in carbon dioxide at high pressures and intermediate temperatures. Nat. Commun. 2013, 4, 2647. [Google Scholar] [CrossRef]
- Han, Y.; Liu, J.; Huang, L.; He, X.; Li, J. Predicting the phase diagram of solid carbon dioxide at high pressure from first principles. npj Quantum Mater. 2019, 4, 10. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Z.; Li, J. Neural Networks Accelerate the Ab Initio Prediction of Solid–Solid Phase Transitions at High Pressures. J. Phys. Chem. Lett. 2021, 12, 132–137. [Google Scholar] [CrossRef]
- Han, Y.; Wang, Z.; Wei, Z.; Liu, J.; Li, J. Machine learning builds full-QM precision protein force fields in seconds. Briefings Bioinform. 2021. [Google Scholar] [CrossRef]
- Han, Y.; Ali, I.; Wang, Z.; Cai, J.; Wu, S.; Tang, J.; Zhang, L.; Ren, J.; Xiao, R.; Lu, Q.; et al. Machine learning accelerates quantum mechanics predictions of molecular crystals. Phys. Rep. 2021, 934, 1–71. [Google Scholar] [CrossRef]
- Han, Y.; Liu, J.; Li, J. Molecular structure determination of solid carbon dioxide phase IV at high pressures and temperatures based on Møller-Plesset perturbation theory. Int. J. Quantum Chem. 2020, 120, 26397. [Google Scholar] [CrossRef]
- Han, Y.; Liu, J.; Li, J. Ab initio investigation of Solid Carbon Monoxide Phase Diagram at Low Temperature. Mater. Today Commun. 2021, 28, 102571. [Google Scholar] [CrossRef]
- Sode, O.; Hirata, S. Second-order many-body perturbation study of solid hydrogen fluoride under pressure. Phys. Chem. Chem. Phys. 2012, 14, 7765–7779. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Thompson, H.P.; Day, G.M. Which conformations make stable crystal structures? Mapping crystalline molecular geometries to the conformational energy landscape. Chem. Sci. 2014, 5, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Price, S.L. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef] [Green Version]
- Karamertzanis, P.G.; Price, S. Energy Minimization of Crystal Structures Containing Flexible Molecules. J. Chem. Theory Comput. 2006, 2, 1184–1199. [Google Scholar] [CrossRef]
- Gaussian, R.A. Gaussian 09 Citation; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Luo, H.; Lu, Q.; Liu, Z.; Liu, J.; Zhang, J.; Wei, Z.; Li, J. Quantum Mechanical-Based Stability Evaluation of Crystal Structures for HIV-Targeted Drug Cabotegravir. Molecules 2021, 26, 7178. https://doi.org/10.3390/molecules26237178
Han Y, Luo H, Lu Q, Liu Z, Liu J, Zhang J, Wei Z, Li J. Quantum Mechanical-Based Stability Evaluation of Crystal Structures for HIV-Targeted Drug Cabotegravir. Molecules. 2021; 26(23):7178. https://doi.org/10.3390/molecules26237178
Chicago/Turabian StyleHan, Yanqiang, Hongyuan Luo, Qianqian Lu, Zeying Liu, Jinyun Liu, Jiarui Zhang, Zhiyun Wei, and Jinjin Li. 2021. "Quantum Mechanical-Based Stability Evaluation of Crystal Structures for HIV-Targeted Drug Cabotegravir" Molecules 26, no. 23: 7178. https://doi.org/10.3390/molecules26237178
APA StyleHan, Y., Luo, H., Lu, Q., Liu, Z., Liu, J., Zhang, J., Wei, Z., & Li, J. (2021). Quantum Mechanical-Based Stability Evaluation of Crystal Structures for HIV-Targeted Drug Cabotegravir. Molecules, 26(23), 7178. https://doi.org/10.3390/molecules26237178