Abstract
An important focus for innovation in photodynamic therapy (PDT) is theoretical investigations. They employ mostly methods based on Time-Dependent Density Functional Theory (TD-DFT) to study the photochemical properties of photosensitizers. In the current article we review the existing state-of-the-art TD-DFT methods (and beyond) which are employed to study the properties of porphyrinoid-based systems. The review is organized in such a way that each paragraph is devoted to a separate aspect of the PDT mechanism, e.g., correct prediction of the absorption spectra, determination of the singlet–triplet intersystem crossing, and interaction with molecular oxygen. Aspects of the calculation schemes are discussed, such as the choice of the most suitable functional and inclusion of a solvent. Finally, quantitative structure–activity relationship (QSAR) methods used to explore the photochemistry of porphyrinoid-based systems are discussed.
1. Introduction
Photodynamic therapy (PDT) is a possible treatment for various diseases [1,2,3], such as different kinds of cancer [4,5,6]; bacterial [7,8], fungal [9], and viral infections (including Covid-19) [10,11]; cutaneous manifestations [12]; dental caries [13]; and rheumatoid arthritis [14]. The mechanism of the PDT is schematically displayed in Scheme 1. It consists of the irradiation of infected/malignant cells in the presence of a light-absorbing compound (called photosensitizer). The photosensitizer is excited from its ground state (usually the singlet state S0) to the excited state (usually the first excited singlet state S1), which then undergoes intersystem crossing (and a series of de-excitations), generating an excited triplet state (T1). T1 is then deactivated to S0 following one of two processes. In the typical scenario, known also as type II photoreaction, the photosensitizer transfers its energy to the O2 molecule yielding singlet oxygen 1O2. In the less common process, known as type I photoreaction, the photosensitizer in the T1 state reacts with a molecule present nearby (e.g., from the cell membrane) and converts it into a highly reactive species (e.g., NO, O2−, ROO, or RO). Both processes can occur simultaneously and the ratio between them depends on the type of photosensitizer and the physico-chemical conditions in the place of action, mostly the concentration of oxygen. No matter which type of photoreaction occurs, the emerging reactive species are able to damage the targeted cells.
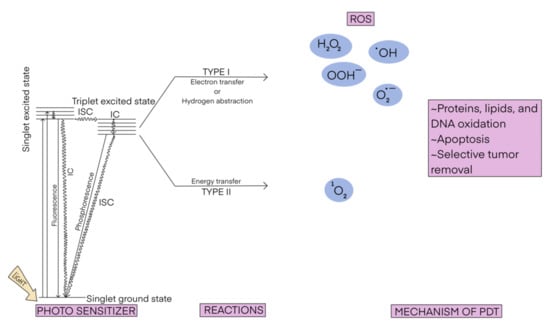
Scheme 1.
The scheme of photodynamic therapy.
In PDT, typically one uses visible light, which is able to penetrate tissues in the so-called therapeutic window to centimeters of depth [15], thereby reaching shallow tumors only. In order to overcome this limitation and target deeper tumors, other light sources have been tested [16]. Among them, electromagnetic radiation such as near infrared light [16,17,18], Cherenkov radiation [19,20,21], and X-rays [22,23,24,25] have been used. Self-illuminated systems based on chemiluminescent and/or bioluminescent mechanisms [16,26,27,28] are also under study.
Common photosensitizers originate usually from the following classes of molecules: different forms of porphyrins, including benzoporphyrins, hexaphyrins, and sapphyrins; porphyrin precursors—aminolevulinic acid (ALA); chlorins; bacteriochlorins; phthalocyanines; naphtalocyanines; corroles; indocyanine dyes; BODIPY dyes; Ru (II) complexes; Ir(III) complexes; Au (III) complexes; metal–organic frameworks (MOFs); covalent organic frameworks (COFs); hydrogen-bonded organic frameworks (HOFs); polymers; carbon-based nanomaterials; silicon-based nanomaterials; pure metals; transition metal carbides (TMCs); TiO2; mono- and bimetal oxides; and bimetal sulfides [4,29,30,31,32,33]. Among them, porphyrinoids have been in use the longest, are the most studied, and are the most widely known. Most of photosensitizers accepted for clinical use come from this group [1]. Additionally, their structure allows various modifications, such as metalation or substitution of the porphyrin ring, to improve their photophysical properties or to construct biohybrid materials [34]. In this context, porphyrinoids may also have dual effects as therapeutic and diagnostic agents, e.g., in PET imaging with 64Cu or in MRI with Gd as a central atom [31]. Their photodynamic actions are usually exerted via type II photoreactions, i.e., they transfer the excitation energy from the triplet excited state to the oxygen molecule.
The mechanism of PDT enforces the following key requirements of effective photosensitizers: (1) the ability to absorb light to the first excited singlet state in the so-called therapeutic window (600–800 nm); (2) a high intersystem spin-crossing probability, enabling effective population of the triplet excited state; (3) a T1–S0 energy gap larger than 0.98 eV, which is the amount of energy required to activate molecular oxygen.
Nowadays, new studies on photosensitizers, in particular, on their properties and activity, are based not only on experimental work, but are also complemented by theoretical calculations. They allow for determination of the geometries and electronic parameters of photosensitizers (bond lengths and orders, angles between atoms, charges accumulated on different parts of molecules, polarizabilities, etc.), and their spectroscopic properties (excited state energies, including the T1–S0 energy gap and estimation of excited state lifetimes). They employ mostly methods based on Time-Dependent Density Functional Theory (TD-DFT), but other, “higher” level theories are also gaining attention due to constantly increasing computational resources.
Time-Dependent Density Functional Theory (TD-DFT) is an extension of Density Functional Theory (DFT) to the time-dependent domain. As proposed by Runge and Gross [35], the central theorem of TD-DFT states that there is a one-to-one correspondence between the external time-dependent potential and the electronic one-body density for any many-body system evolving from a fixed initial state ( Ψ(0)). The time-dependent Schrödinger equation can be solved, and so all observables of the many-electron system, beginning in the initial Ψ(0) state, may be extracted from the one-body time-dependent density. Such an approach is analogous to the Hohenberg–Kohn theorem for the ground-state DFT [36]. extensive descriptions of the TD-DFT methodology can be found in, e.g., [37,38,39]. In 1995, Mark Casida proposed a constructive linear-response formalism for TD-DFT (known as random-phase approximation, RPA), allowing to determine the solutions of the TD-DFT equations [40].
TD-DFT is formally an exact theory, but the implementations require the selection of the exchange correlation functionals. Consequently, the accuracies of the TD-DFT solutions suffer from the limitations of the functionals applied. For instance, in spectroscopic applications, the quality of a solution depends on the system and type of excitation considered. Typical functionals often fail to correctly describe Rydberg and charge-transfer states [41,42]. This can be overcome by the application of range-separated hybrid functionals (see Section 2.2.1 for further discussion). They incorporate a considerable amount of the exact Hartree–Fock exchange at large electron–electron distances and so reflect the correct asymptotic exchange potential.
Despite the limitations outlined above, TD-DFT theory has been used successfully many times to compute properties of various chemical, biological, and physical systems. The most common use is in spectroscopy, where TD-DFT yields response properties and excitations of atoms, molecules, and solids. It also provides the baseline for simulations of real-time dynamics in non-perturbative fields. The success of the TD-DFT approach is due to its relatively high accuracy achieved with relatively low computational cost and ongoing efforts to push its limits. As such, one should mention capturing potential energy surfaces near a conical intersection or addressing the Coulomb blockade phenomenon in calculations of molecular transport [39].
In the current article, we investigate which of the widely available methods seems best suited to studying the physico-chemical properties of porphyrin-based photosensitizers, which are the most common approaches, and which of the descriptors can be predicted with modern theoretical tools.
2. Results
The discussion of the literature is divided according to the requirements imposed on the most effective photosensitizers. Each paragraph in a subsection is devoted to a stage of the mechanism of the PDT, e.g., the correct prediction of the photosensitizer structure and its absorption spectra, the determination of singlet–triplet intersystem crossing, and interaction with molecular oxygen. The practical aspects of the calculation schemes are also discussed. These include the choice of the most suitable functional and inclusion of a solvent. Finally, quantitative structure–activity relationship (QSAR) methods to explore the photochemistry of porphyrinoid-based systems are discussed.
2.1. Prediction of the Geometric Parameters of Porphyrinoid-Based Photosensitizers
Theoretical calculations of porphyrinoid-based systems are usually done with Density Functional Theory (DFT)—a choice justified by the size of these systems and earlier general benchmark studies [43,44]. The most common is the use of the B3LYP functional [45,46,47,48,49], which is applied to predict geometries of the ground and excited states of the sensitizers [50,51,52], but other functionals, such as M06 [53], are gaining popularity as well [54].
In order to assess the applicability of various computational schemes, the DFT geometries are compared both with the X-ray diffraction (XRD) structures and with those predicted by higher level calculations, because the experimentally derived data can be influenced by the fact that porphyrinoids easily aggregate due to π-stacking.
Typically, B3LYP bond lengths differ by not more than 0.01–0.02 Å as compared to XRD results (B3LYP/6-31G* data for meso-tetraphenyl substituted porphyrin (TPP) and its hetero-atom analogs [55] or substituted bacteriochlorins [56]). Much larger discrepancies (up to ca. 20 degrees) have been found for valence angles [56]. The choice of the basis set has only a minor effect on the computed geometries [57]. A study on methyl pheophorbide-a revealed, however, that M06-2X/6-311+G(d,p) [53] and LSDA/6-311+G(d,p) [45,47,58] are better than B3LYP at reproducing bond lengths and valence angles, respectively [59].
There are more discrepancies when metal ions are present in the porphyrinoid structure. For instance, a comparison of the B3LYP/SVP geometries of a series of acene-modified zinc porphyrins with those calculated with BH&HLYP/SVP [48,49,60] and ωB97X-D/SVP [61] revealed that the changes in parameters could be up to 0.4 Å, but eventually the electronic parameters were similar and not very geometry-dependent for the neutral, oxidized, and reduced ground state species [50]. Rydberg and Olsen claimed that for iron-containing porphyrins, B3LYP yields the worst results, and that TPSSH [62], PBE0 [63,64], and TPSS [65,66] functionals perform the best (absolute bond distance deviations of 0.015−0.016 Å) [67]. One should be aware, however, that iron-porphyrins are not popular in PDT studies.
Excited state geometries are rarely computed, because they involve long computational times, and comparisons with other theoretical or experimental data are challenging. More is known about the applicability of TD-DFT for obtaining geometries of model systems. Jun Wang and Bo Durbeej studied the applicability of TD-DFT for predicting the excited state geometry of medium-sized organic molecules, including heterocycles [68]. They found that that the studied hybrid functionals (B3LYP, PBE0, M06-2X, CAM-B3LYP [69], and ωB97XD) can reproduce the approximate coupled-cluster singles’ and doubles’ (CC2) excited-state bond lengths with an overall root mean square deviation (RMSD) of 0.011 Å. The BP96 functional [49,70] yielded much worse results. Out of the studied hybrids, B3LYP and PBE0 performed the best. These results were corroborated by the recent findings by Robin Grotjahn and Martin Kaupp, who reported that local-hybrid functionals reproduce better C-N, C-C, C-halogen, and element-H bond lengths in the excited state than other semi-local and range-separated hybrids (the calculations were done for a set of small molecules, and the data were compared with coupled cluster results) [71]. Bond angles are system-dependent, and it was not possible to draw general conclusions for the studied functionals.
Winter and co-workers determined the first excited state geometries of tetraphenylporphyrin (TPP) and Zn-TPP with B3LYP/TZVP [72]. The appropriateness of the method was then judged based on the comparison of the energy of the first excitation of the resulting structure with experimental results, and/or results calculated with higher-order methods: algebraic diagrammatic construction through second order (ADC(2)), CC2, scaled opposite-spin CC2 (SOS-CC2), and spin-component scaled CC2 (SCS-CC2) within the TZVP basis set. B3LYP was proven to yield reasonable results.
2.2. Prediction of the Absorption Spectra
The experimental absorption spectra of porphyrinoids are divided into two characteristic spectral regions: an intense B band (called also Soret band) located near 350–400 nm (3–3.5 eV) and a weak Q band consisting of four bands in the region 500–800 nm (1.5–2.5 eV). Their origins are usually explained by Gouterman’s four-orbital model [73]. The bands are due to transitions within the two highest occupied molecular orbitals (HOMO, HOMO-1) and the two lowest unoccupied molecular orbitals (LUMO, LUMO-1)—see Scheme 2. The Q and B transitions are x-/y-polarized; therefore, they are denoted as Qx and Qy, and Bx and By, respectively.
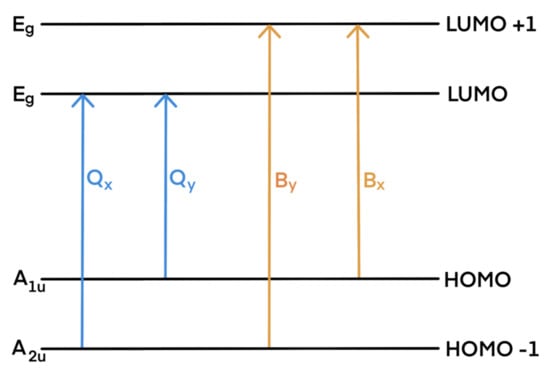
Scheme 2.
Gouterman’s model of porphyrinoids’ main absorption bands (note that substitution or other modifications in the porphyrin skeleton may result in modification of orbital levels, causing changes in the band assignment).
Theoretical calculations of the excited states of porphyrinoids are done with Time-Dependent Density Functional Theory or with higher level ab initio methods.
Literature concerning the applicability of the TD-DFT method for predicting adsorption spectra is very rich, and a number of excellent reviews exist—see, e.g., [74,75,76,77,78,79,80,81]. Those reviews indicated that there is no universal functional able to correctly describe all classes of dyes. In case of porphyrinoids, which are at focus of our study, it has been shown that there are two parameters which influence the results the most. These are (i) the choice of functional and (ii) the inclusion of a solvent. Therefore, in the following, those two aspects are presented. The choice of the basis set does not influence the results [82,83].
2.2.1. The Choice of Functional
A TD-DFT functional for UV-VIS spectra simulation can be benchmarked not only against experimental data (ultraviolet-visible light (UV-VIS) or magnetic circular dichroism (MCD) spectroscopy) [84,85], but also using high-level ab initio calculations. The latter option is of particular use when new, to-be-synthetized compounds are proposed for PDT.
Among the plethora of functionals, the most often used to simulate the spectra of porphyrins are: B3LYP, CAM-B3LYP, and PBE0 [51,83,86,87,88,89]. Those choices are well justified in light of the extended study presented recently by Batra and co-workers [83]. They found that hybrid functionals reproduce experimental porphyrinoid spectra with considerably smaller error rates than local general gradient functionals—see Table 1 for a comparison of the mean errors, the mean absolute errors, and the absolute maximum errors determined by comparing computed spectra of various porphyrinoids with different functionals to the experimental values.

Table 1.
The mean errors (ME), the mean absolute errors (MAE), and absolute maximum errors (MAXE) determined by comparing theoretical (RPA approach, def2-SVP basis set) and experimental spectra of various porphyrinoids (porphyrin, octaethylporphyrin, Mg octaethylporphyrin, Zn octaethylporphyrin, tetraphenylporphyrin, Mg tetraphenylporphyrin, Zn tetraphenylporphyrin, tetrakis(o-aminophenyl)porphyrin, Zn tetrakis(4-carboxyphenyl)porphyrin, Zn [5,15-dipiridyl- 10,20-bis(pentafluorophenyl)] porphyrin, Zn [5,15-di(pyridylacetyl)-0,20-diphenyl] porphyrin, octabromotetraphenyl porphyrin) determined with different functionals—data taken from [83].
The authors recommend energy scaling procedure for general hybrid functionals, whereas no scaling is required for range-separated hybrid ones. This is in agreement with the previous studies on the applicability of different functionals (including long-range separated and meta hybrid ones) to reproducing the absorption spectra of unsubstituted porphyrin [90] and Tookad® (Pd-bacteriopheophorbide) [82].
Comparisons between the theoretical and experimental spectra are often performed. Based on numerous reports, it is usually advised to use CAM-B3LYP to predict adsorption spectra [86]; see Table 2. The discrepancies between the energies of the theoretical and experimental Q and B bands are usually ca. 0.2 and 0.4 eV, respectively (calculations for porphyrins at CAM-B3LYP/6-31G and CAM-B3LYP/6-31G*—theoretical excitation energies overestimate the experimental ones) [51]. Analogous errors regarding the use of B3LYP for computing the UV–VIS spectra of free-base porphyrins were found to amount to 0.3 and 0.2 eV, respectively [51]; see Table 3. Additionally, B3LYP is sometimes reported to significantly underestimate the oscillator strength [91]. PBE0/6-31+G(d) results in a discrepancy of 0.4 eV for both bands [87]. M05-2X results in 0.3 and 0.5 eV errors for Q and B bands, respectively [92].
Table 2.
Performances of the CAM-B3LYP functional when reproducing experimental Q and B bands of selected porphyrinoids.
Table 3.
Performances of the B3LYP functional when reproducing experimental Q and B bands of selected porphyrinoids.
It is sometimes claimed that B3LYP and PBE0 reverse the energy ordering between the B and N (51A and 61A) transitions [96].
The success of CAM-B3LYP (and other range-separated functionals) is due to the fact that it includes a long-range correction of the exchange potential, which incorporates an increasing fraction of Hartree–Fock (HF) exchange as the interelectronic separation increases.
2.2.2. Inclusion of Solvent
In theoretical calculations, solvent can be modelled by inclusion of the solvent molecules into the theoretical model (the so-called explicit solvation scheme) or by immersing the studied molecule inside a cavity in a continuum of a given dielectric constant (the so-called implicit solvation scheme). Most of the DFT calculations for PDT use the second approach. The most popular is the polarized continuum model (PCM), which has several variants, such as dielectric D-PCM and the conductor-like polarized continuum model (C-PCM), also called COSMO [97,98]—see, e.g., [87,99,100].
A comparison of the PCM and C-PCM results for Zn-TPP and its analogues revealed that both approaches yield similar results, which agrees with the experimental values. A slight decrease in computational time for C-PCM as compared with PCM was found [101].
Further, inclusion of a solvent within TD-DFT can be accomplished either via linear response (LR) method or external iteration (EI) (known also as the state-specific method, SS). Whereas in the first case, the excitation energies are determined without computing the exact excited state electronic density, in the latter, a different Schrödinger equation is solved for each excited state [102,103,104]. These approaches were tested by M. Dulski and coworkers [89], who computed the geometries and photochemical properties of two derivatives of meso-tetraphenylporphyrin (meso-tetrakis (3-methoxy-4-hydroxyphenyl)-porphyrin and meso-tetrakis (3,5-dimethoxy-4-hydroxyphenyl)-porphyrin) in acetonitrile. Both calculation schemes yielded spectra in agreement with experimental data; however, a small red shift of the spectrum resulted from the LR approach as compared with the EI one. LR calculations give higher oscillator strengths for both the B and Q bands. The value of the T1–S0 energy gap remains about the same regardless of the calculation scheme. A comparison of theoretical fluorescence emission spectra with experimental spectra indicated that the LR method gives better results than the EI approach. The LR method produces high values of dipole force; the values produced by EI approximations are significantly lower. This suggests that the effect of the solvent on molecules in excited states has a greater impact on calculations done using the LR method.
Similar results were also obtained by Fukuda et al. [105]. Their results indicate that the LR approach reproduced the experimentally observed trends of the solvatochromic shifts of porphyrin in dichloromethane and benzene (separately), as opposed to EI.
Petit et al. [87] compared results obtained with explicit and implicit solvation schemes (C-PCM) for a set of porphyrinoids (porphyrin, chlorin, bacteriochlorin, pheophytin a, porphyrazin, and texaphyrin). The explicit solvation (i.e., inclusion of two or four water molecules for one sensitizer molecule) did not affect the geometry of the studied porphyrinoids, but Qx and B bands were shifted upwards, minimizing the discrepancies between theoretical and experimental data. The C-PCM strongly stabilizes orbitals and makes transition moments higher. As a result, the computed excitations are stronger.
Fukuda et al. also compared results for porphyrin in CCl2H2 and benzene with explicit and implicit solvation [105]. The calculations reproduced the bathochromic shifts of the B band states.
Peng-Dong Fan and collaborators studied the inclusion of solvent by quantum mechanics/molecular mechanics (QM/MM) calculations of ZnP (being the quantum region) immersed in a 30 Å cubic box with 869 water molecules (being the molecular mechanics region) [106]. The ZnP moiety exhibited small out-of-plane distortion after QM/MM optimization (QM: DFT-B3LYP/VTZ basis set on Zn and 6-31*G for the rest of atoms; MM: SPC/E water model). The values of the vertical excitation energies (VEE) of ZnP in the gas phase and in solution were comparable for both methods. The difference between VEE in gas and in solvent was smaller than 0.02 eV for all states corresponding to the Q band (21A and 31A states) or B band (41A and 51A states) for both the TD-DFT (B3LYP functional, Ahlrich’s VTZ basis set on Zn, and 6-31G* basis set for the rest of the atoms) and the coupled cluster with singles and doubles, with the equation of motion approach (EOMCCSD) methods. The discrepancy was larger for the configuration interaction (CI), but it did not exceed 0.05 eV. When higher excitations were computed, the difference between VEE in solution and in the gas phase was maintained for the wave function methods, but for the TD-DFT it amounted to ca. 0.29 eV. Furthermore, TD-DFT reversed the ordering of the 61A and 71A states compared to the B band. The effect was ascribed to the fact that the 61A and 71A states are of charge transfer character.
Regarding the reproduction of the lowest T1 excitation energies of porphyrin, chlorin, and porphyrazine, it was found that B3LYP/6-31G(d,p) calculations within the PCM approach (in benzene and dichloromethane) did not differ considerably in T1 excitation energies determined in the gas phase, and well reproduced experimental ones [107].
In our opinion, solvent inclusion should be recommended for the calculations of the properties of the photosensitizers. The requirement of photodynamic therapy is to inject photosensitizers into tissues where the environment differs from the in vitro experimental conditions. Protein and/or lipid matrices inside tissues can affect the geometries of the molecules via interactions with charged groups, electron rich aromatic systems, or H-donors in proximity of the photosensitizer species. Consequently, a molecule’s geometry can be distorted from that encountered in the gas phase. Examples include the formation of salt bridges, the emergence of non-bonding solute–solvent interactions, and changes in the photosensitizer’s protonation state. Theoretical studies have revealed that these environmental factors can considerably affect the spectral properties of molecules (in particular, when charge transfer states are considered) and should not be neglected—see e.g., [108,109,110,111,112,113,114,115]. Therefore, the inclusion of solvent effects into calculations is forecast to be one of hot topics in the years to come.
2.2.3. Prediction of Multiphoton Adsorption Spectra
It is often claimed that one of the major limitations of porphyrins use in PDT is the need to activate them by blue or green light, which penetrates poorly into tissues. This can be overcome by use of two-photon absorption (TPA)—a nonlinear optical process involving the simultaneous absorption of two near-infrared photons. Such an approach allows for high spatial resolution of the PDT event and enables one to target tumors at greater depths. The theoretical studies of two-photon absorption are scarce, but gaining importance and predictive power [116]. The studies are focused on the prediction of the adsorption spectra by calculating two-photon absorption cross-sections or two-photon probabilities, which require quadratic response theory [117,118,119]. The probabilities of the two-photon absorption can be computed either by sum-over-state (SOS) expression or from the single residue of the quadratic response (SRQR) in TD-DFT [120,121].
Day et al. showed the sensitivity of TPA cross-section predictions to the accuracy of the functionals used to predict the most relevant state energies. They demonstrated that the overestimation by 0.5 eV (in respect to the experimental data) of the transition energy for the relevant TPA state in [5,15-bis(3,5-bitert-butylphenyl)-10,20-bis(trihexylsilylethynyl)porphyrinato]zinc by B3LYP led to a 5-fold overprediction of the TPA cross-section for this molecule, whereas for 5,5′-(ethyne)bis[[10,20-bis(3,5-bitert- butylphenyl)-porphyrinato]zinc], underestimation of the transition energy by 0.5 eV with CAM-B3LYP and by 0.8 eV with B3LYP lead to underestimations of the TPA cross-section by factors of 2 and 4, respectively [122].
Greco and co-workers computed two-photon absorptivities using the B3LYP/6-31G(d) equilibrium geometries and found that they were very sensitive to the ground-state geometry [123].
Further, it was found that the B3LYP/6-31G* and CAM-B3LYP/6-31G*-computed two-photon cross-sections of porphyrin derivatives with asymmetric substitution at the meso-positions were overestimated in comparison with the experimental data [124]. It was shown that all excitation wavelengths were lowered when CAM-B3LYP was used (in contrast to B3LYP results). CAM-B3LYP cross-section values were much smaller than those determined with B3LYP.
Only recently was the possibility of three-photon absorption of porphyrinoids explored experimentally and theoretically [125,126,127]. The results based on the SOS formulas indicate fair agreement between the computational and experimental results for a range of substituted porphyrins. The predicted cross-sections were overestimated by a factor of 2, but all trends were reproduced correctly [125].
2.3. Determination of the Singlet–Triplet Intersystem Crossing
The transfer of the excitation energy from the photosensitizer to, e.g., molecular oxygen requires the excited photosensitizer to change its spin state from singlet to triplet. This is done via intersystem crossing. The efficiency of the intersystem spin crossing (ISC) depends on the amplitudes of the spin-orbit matrix elements for the S1→T1 radiationless transitions, which can be computed with the response theory [128].
The methodology usually employed for spin-orbit coupling, within both variational and perturbative approaches, is presented in, e.g., [129,130].
The rate constant () for the ISC between the excited Si state and the jth triplet state (Tj) can be estimated as [131]:
where is the matrix element of the spin-orbit coupling interaction operator (expressed here in cm−1). For macrocyclic molecules such as porphyrinoids, the main contribution to the Franck–Condon factor arises from vibrational states with energies of ca. 1400 cm−1, corresponding to Huang–Rhys factors y of ca. 0.3. The density of the final states can then be estimated from the semiempirical expression for the density of states:
The reported calculations employ mostly the B3LYP functional with cc-pVDZ or 6-31G(d) basis sets for substituted TPP and ZnTPP, free and metalated meso-substituted tetrabenzotriazaporphyrin, expanded bacteriochlorins, and temocene (the porphycene analogue of Foscan®) [56,95,132,133]. The computed spin-orbit couplings are in line with the trend of intersystem crossing yields determined experimentally.
Alberto and co-workers reported spin-orbit couplings determined for Foscan® and temocene at the B3LYP/6-31G(d) level, showing correlations with the experimental singlet oxygen quantum yields [95]. By contrast, based on the values spin-orbit matrix elements computed for free tetrabenzotriazaporphyrin and its Mg and Zn complexes, the same group explained that these photosensitizers would not be able to act as singlet oxygen activators [133].
De Simone et al. computed the spin-orbit couplings for a set of tetraphenyl porphyrins and their zinc(II) complexes; halogen atoms (F, Cl, Br, I) were introduced into their macrocycle skeletons [132]. It was shown that the computed values of the spin-orbit couplings increased with the halogen atom mass, showing a heavy atom effect. In such a way, an efficient intersystem crossing for different decay routes was ensured. The authors demonstrated that the presence of halogen atoms influenced the composition of relevant orbitals, especially in the triplet state, for which the contribution of the halogen atom lone pairs increased with the atomic mass of the halogens. Based on spin-orbit couplings and S0–T1 energy gap values, different deactivation channels were considered, out of which the S1−T1 ISC process was the most probable.
These observations prove that the adopted scheme of calculations, although not frequently used so far, can be used to predict the photochemical properties of the new systems.
2.4. Interaction with Molecular Oxygen
Although this step is of crucial importance for PDT, theoretical studies on it are very limited. There are two approaches to tackle the problem of the interaction between the photosensitizer and molecular oxygen.
The first one comprises the comparison of the photosensitizer’s S0–T1 energy gap with the energy needed to excite oxygen from 3O2 to 1O2. The energy needed for the formation of singlet oxygen is 0.98 eV, but experimental studies determined that at least 1.13 eV is needed in solution [134,135,136]. In calculations, one is better to refer to the energy required to excite the triplet molecular oxygen determined at the same level of theory, rather than that used for porphyrinoid characterization (e.g., for the B3LYP/6-31G* it is 0.91 eV [56]).
The theoretical studies of various porphyrinoids done in Russo’s group indicate that the PBE0PBE and B3LYP functionals are the most suitable for reproducing the singlet–triplet energy gaps, and both have an average error of 0.1 eV [137]. B3LYP with both 6-31G and 6-31G(d,p) gives S0–T1 energy gaps in agreement with experimental results for porphyrin, chlorin, porphyrazine, and zinc porphyrins bearing meso-4-methylthiophenium groups [107,138].
The second strategy aims at the theoretical description of the energy transfer processes. It requires explicit modeling of the coupled electronic and vibrational dynamics, which is not a trivial task. One of the first attempts was done by Liang Shen, who considered total energy changes accompanying possible reactions of porphyrin, chlorin, and porphyrazine (abbreviated below as PS) with molecular oxygen at the B3LYP/6-31+G(d,p) level [107]:
PS(T1) + 3O2 → PS(S0) + 1O2
PS(T1) + 3O2 → PS(S0)⋅+ + O2−
PS− + 3O2 → PS + O2−
It was postulated that the singlet oxygen can be generated according to reaction (1) in water and benzene by all studied porphyrinoids, whereas O2− can be obtained only in water according to reactions (2) and (3), whereas mechanism (3) can involve only porphyrazine as the photosensitizer.
More advanced calculations in which the dynamics of the PS to O2 energy transfer process are studied, are to the best of our knowledge, non-existent.
2.5. Quantitative Structure–Activity Relationship (QSAR) Studies
QSAR studies aiming at elucidation of relationships between structural and electronic properties of photosensitizers and their activity in PDT are scarce.
The oldest QSAR analyses link porphyrinoid activity with the octanol/water partition coefficient and basic structural parameters (length and shape of alkyl chains) [139,140]. Banfi and co-workers proposed predicting the photodynamic cytotoxicity of porphyrins (bearing variable numbers of aryl substituents in meso-positions) towards human colon adenocarcinoma cells [141]. Three molecular descriptors were selected by genetic algorithms as the most relevant to obtaining models with the highest predictive power. These were: (i) the autocorrelation descriptor GATS6v [142], which accounts for the correlation among atoms, weighted by the van der Waals volumes, with a distance of six bonds in the molecule; (ii) the topological descriptor PW3 [143], which is linked to molecular shape information; (iii) GETAWAY descriptor R4u+ [144], which accounts for the local structural aspects of the molecule.
Bettanin et al. also searched for the best descriptors of the photodynamic activity of porphyrin, phthalocyanine, and chlorin [94]. They proposed molecular volume, LUMO energy, oscillator strength, dipole moment, and free energy of solvation as the most relevant for the desired activity.
The experimental Qx band energy of metallobacteriochlorins (M-BChl) correlates with the HOMO–LUMO energy gap determined at the BP/def2-TZVP level [145]; therefore, the latter can be used to predict the adsorption edges of the unknown derivatives of M-BChl.
One can see that the photodynamic activity of the studied set of porphyrins is mostly determined by such structural aspects as their sizes and shapes, and descriptors of their solubility in water (octanol/water partition coefficient, free energy of solvation, or dipole moment).
3. Conclusions
- As can be seen, TD-DFT is one of the most widely applied methods in these studies, due to its good balance between accuracy and computational cost. The fair agreement between theoretical and experiment spectra found for various porphyrinoids indicates that an a priori computation of some important photophysical properties may guide the design of new drugs that can be proposed as potential photosensitizers for photodynamic therapy.
- Most studies advise the use of B3LYP or M06-2X functionals for geometry optimization and the CAM-B3LYP functional for predicting the UV–VIS spectra of porphyrinoids. The latter well handles charge-transfer states, which dominate the photochemistry of these macrocycles.
- The inclusion of a solvent usually induces only minor geometry changes in the porphyrinoids. There are no significant differences in outcome among the implicit solvent models that are employed for the calculations.
- The S0–T1 gap is generally well reproduced by the B3LYP or PBE0PBE functionals.
- The spin-orbit couplings, computed at the B3LYP level, quantifying the singlet-to-triplet intersystem crossing, scale well with such experimental data as intersystem crossing yields and singlet oxygen quantum yields.
- Regarding the interactions between the photosensitizer and molecular oxygen, the relevant calculations are still to be determined. The mechanism of the energy transfer between the photosensitizer and molecular oxygen is sought, as pathways involved in this process are essential to the design of effective PDT drugs. Studies similar to [146] are highly demanded.
- QSAR analyses indicate that the structural parameters characterizing porphyrinoids’ shapes, volumes, and solubility levels are the most important factors determining their photodynamic activity. This is in line with the common observation that an important parameter for anticancer drugs is their solubility in aqueous solutions.
Author Contributions
Conceptualization, A.D.-M. and D.R.-Z.; investigation, A.D.-M. and D.R.-Z.; writing—original draft preparation, D.R.-Z.; writing—review and editing, A.D.-M. and D.R.-Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by statutory funds of the Jerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Ada Zbik for preparing figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Niculescu, A.-G.; Grumezescu, A. Photodynamic Therapy—An Up-to-Date Review. Appl. Sci. 2021, 11, 3626. [Google Scholar] [CrossRef]
- Dąbrowski, J.M.; Pucelik, B.; Regiel-Futyra, A.; Brindell, M.; Mazuryk, O.; Kyzioł, A.; Stochel, G.; Macyk, W.; Arnaut, L.G. Engineering of relevant photodynamic processes through structural modifications of metal-lotetrapyrrolic photosensitizers. Coord. Chem. Rev. 2016, 325, 67–101. [Google Scholar] [CrossRef]
- Dąbrowski, J.M.; Arnaut, L.G. Photodynamic therapy (PDT) of cancer: From local to systemic treatment. Photochem. Photobiol. Sci. 2015, 14, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Dou, D.; Yang, L. Porphyrin photosensitizers in photodynamic therapy and its applications. Oncotarget 2017, 8, 81591–81603. [Google Scholar] [CrossRef] [Green Version]
- Hu, T.; Wang, Z.; Shen, W.; Liang, R.; Yan, D.; Wei, M. Recent advances in innovative strategies for enhanced cancer photodynamic therapy. Theranostics 2021, 11, 3278–3300. [Google Scholar] [CrossRef]
- Wang, Z.; Peng, H.; Shi, W.; Gan, L.; Zhong, L.; He, J.; Xie, L.; Wu, P.; Zhao, Y.; Deng, Z. Application of photodynamic therapy in cancer: Challenges and advancements. BIOCELL 2021, 45, 489–500. [Google Scholar] [CrossRef]
- Amos-Tautua, B.M.; Songca, S.P.; Oluwafemi, O.S. Application of Porphyrins in Antibacterial Photodynamic Therapy. Molecules 2019, 24, 2456. [Google Scholar] [CrossRef] [Green Version]
- Galstyan, A. Turning Photons into Drugs: Phthalocyanine-Based Photosensitizers as Efficient Photoantimicrobials. Chem. Eur. J. 2021, 27, 1903–1920. [Google Scholar] [CrossRef] [PubMed]
- Roomaney, I.A.; Holmes, H.K.; Engel, M.E. Treatment of oral fungal infections using photodynamic therapy: Systematic review and meta-analysis. Clin. Exp. Dent. Res. 2021, 7, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Bhapkar, S.; Kumbhar, N.; Gacche, R.; Jagtap, S.; Jadhav, U. Photodynamic Therapy (PDT): An Alternative Ap-proach for Combating COVID-19. Biointerface Res. Appl. Chem. 2021, 11, 12808–12830. [Google Scholar]
- Sabino, C.P.; Ball, A.R.; Baptista, M.S.; Dai, T.; Hamblin, M.R.; Ribeiro, M.S.; Santos, A.L.; Sellera, F.P.; Tegos, G.P.; Wainwright, M. Light-based technologies for management of COVID-19 pandemic crisis. J. Photochem. Photobiol. B Biol. 2020, 212, 111999. [Google Scholar] [CrossRef]
- De Annunzio, S.R.; Costa, N.C.S.; Mezzina, R.D.; Graminha, M.A.S.; Fontana, C.R. Chlorin, Phthalocyanine, and Porphyrin Types Derivatives in Phototreatment of Cutaneous Manifestations: A Review. Int. J. Mol. Sci. 2019, 20, 3861. [Google Scholar] [CrossRef] [Green Version]
- Nagata, J.Y.; Hioka, N.; Kimura, E.; Batistela, V.R.; Terada, R.S.S.; Graciano, A.X.; Baesso, M.L.; Hayacibara, M.F. Antibacterial photodynamic therapy for dental caries: Evaluation of the photosensitizers used and light source properties. Photodiagnosis Photodyn. Ther. 2012, 9, 122–131. [Google Scholar] [CrossRef]
- Gallardo-Villagrán, M.; Leger, D.Y.; Liagre, B.; Therrien, B. Photosensitizers Used in the Photodynamic Therapy of Rheumatoid Arthritis. Int. J. Mol. Sci. 2019, 20, 3339. [Google Scholar] [CrossRef] [Green Version]
- Bashkatov, A.; Genina, E.; Kochubey, V.; Tuchin, V. Optical properties of human skin, subcutaneous and mucous tissues in the wavelength range from 400 to 2000 nm. J. Phys. D Appl. Phys. 2005, 38, 2543–2555. [Google Scholar] [CrossRef]
- Algorri, J.; Ochoa, M.; Roldán-Varona, P.; Rodríguez-Cobo, L.; López-Higuera, J. Light Technology for Efficient and Effective Photodynamic Therapy: A Critical Review. Cancers 2021, 13, 3484. [Google Scholar] [CrossRef]
- Swamy, P.C.A.; Sivaraman, G.; Priyanka, R.N.; Raja, S.O.; Ponnuvel, K.; Shanmugpriya, J.; Gulyani, A. Near Infrared (NIR) absorbing dyes as promising photosensitizer for photo dynamic therapy. Co-Ord. Chem. Rev. 2020, 411, 213233. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, R.; Kim, E.; Lee, S.; Park, Y.I. Near-Infrared Light-Triggered Photodynamic Therapy and Apoptosis Using Upconversion Nanoparticles With Dual Photosensitizers. Front. Bioeng. Biotechnol. 2020, 8, 275. [Google Scholar] [CrossRef]
- Hartl, B.A.; Hirschberg, H.; Marcu, L.; Cherry, S.R. Activating Photodynamic Therapy in vitro with Cerenkov Radiation Generated from Yttrium-90. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Boschi, F.; Spinelli, A.E. Nanoparticles for Cerenkov and Radioluminescent Light Enhancement for Imaging and Radiotherapy. Nanomaterials 2020, 10, 1771. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, A.E.; Boschi, F. Photodynamic Therapy Using Cerenkov and Radioluminescence Light. Front. Phys. 2021, 9, 9. [Google Scholar] [CrossRef]
- Clement, S.; Chen, W.; Deng, W.; Goldys, E.M. X-ray radiation-induced and targeted photodynamic therapy with folic acid-conjugated biodegradable nanoconstructs. Int. J. Nanomed. 2018, 13, 3553–3570. [Google Scholar] [CrossRef] [Green Version]
- Larue, L.; Ben Mihoub, A.; Youssef, Z.; Colombeau, L.; Acherar, S.; André, J.C.; Arnoux, P.; Baros, F.; Vermandel, M.; Frochot, C. Using X-rays in photodynamic therapy: An overview. Photochem. Photobiol. Sci. 2018, 17, 1612–1650. [Google Scholar] [CrossRef]
- Wang, G.D.; Nguyen, H.T.; Chen, H.; Cox, P.B.; Wang, L.; Nagata, K.; Hao, Z.; Wang, A.; Li, Z.; Xie, J. X-ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics 2016, 6, 2295–2305. [Google Scholar] [CrossRef]
- Overchuk, M.; Cheng, M.H.Y.; Zheng, G. X-ray-Activatable Photodynamic Nanoconstructs. ACS Cent. Sci. 2020, 6, 613–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hao, Y.; Chen, S.; Xu, M. Photodynamic Therapy of Cancers with Internal Light Sources: Chemiluminescence, Bioluminescence, and Cerenkov Radiation. Front. Chem. 2020, 8, 770. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.P.d.; Núnez-Montenegro, A.; Magalhães, C.M.; Ferreira, P.J.O.; Duarte, D.; González-Berdullas, P.; Rodríguez-Borges, J.E.; Vale, N.; Joaquim, C.G.; Silva, E. Single-molecule chemiluminescent photosensitizer for a self-activating and tumor-selective photodynamic therapy of cancer. Eur. J. Med. Chem. 2019, 183, 111683. [Google Scholar] [CrossRef]
- Magalhães, C.M.; González-Berdullas, P.; Duarte, D.; Correia, A.; Rodríguez-Borges, J.E.; Vale, N. Target-Oriented Synthesis of Marine Coelenterazine Derivatives with Anticancer Activity by Applying the Heavy-Atom Effect. Biomedicines 2021, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Lan, M.; Zhao, S.; Liu, W.; Lee, C.S.; Zhang, W.; Wang, P. Photosensitizers for Photodynamic Therapy. Adv. Healthc. Mater. 2019, 8, 1900132. [Google Scholar] [CrossRef]
- Lovell, J.F.; Liu, T.W.B.; Chen, J.; Zheng, G. Activatable Photosensitizers for Imaging and Therapy. Chem. Rev. 2010, 110, 2839–2857. [Google Scholar] [CrossRef]
- Aggarwal, A.; Samaroo, D.; Jovanovic, I.R.; Singh, S.; Tuz, M.P.; Mackiewicz, M.R. Porphyrinoid-based photosensitizers for diagnostic and therapeutic applications: An update. J. Porphyr. Phthalocyanines 2019, 23, 729–765. [Google Scholar] [CrossRef]
- Pucelik, B.; Sułek, A.; Drozd, A.; Stochel, G.; Pereira, M.M.; Pinto, S.M.A.; Arnaut, L.G.; Dąbrowski, J.M. Enhanced Cellular Uptake and Photodynamic Effect with Amphiphilic Fluorinated Porphyrins: The Role of Sulfoester Groups and the Nature of Reactive Oxygen Species. Int. J. Mol. Sci. 2020, 21, 2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sułek, A.; Pucelik, B.; Kobielusz, M.; Barzowska, A.; Dąbrowski, J.M. Photodynamic Inactivation of Bacteria with Porphyrin Derivatives: Effect of Charge, Lipophilicity, ROS Generation, and Cellular Uptake on Their Biological Activity In Vitro. Int. J. Mol. Sci. 2020, 21, 8716. [Google Scholar] [CrossRef]
- Almeida-Marrero, V.; van de Winckel, E.; Anaya-Plaza, E.; Torres, T.; de la Escosura, A. Porphyrinoid biohybrid materials as an emerging toolbox for biomedical light management. Chem. Soc. Rev. 2018, 47, 7369–7400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runge, E.; Gross, E. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.K.U.; Kohn, W. Time-Dependent Density-Functional Theory. Adv. Quantum Chem. 1990, 21, 255–291. [Google Scholar]
- Marques, M.A.L.; Gross, E.K.U. Time-dependent density-functional theory. Annu. Rev. Phys. Chem. 2004, 55, 427–455. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.K.U.; Maitra, N.T. Introduction to TDDFT. In Fundamentals of Time-Dependent Density Functional Theory; Marques, M.A.L., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 53–99. [Google Scholar]
- Casida, M.E. Time-Dependent Density Functional Response Theory for Molecules. In Recent Advances in Density Functional Methods; Chong, D.P., Ed.; World Scientific: Singapore, 1995; pp. 155–192. [Google Scholar]
- Dreuw, A.A.; Head-Gordon, M. Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: The Zincbacteriochlorin−Bacteriochlorin and Bacteriochlorophyll−Spheroidene Complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. [Google Scholar] [CrossRef]
- Tozer, D.J.; Amos, R.; Handy, N.C.; Roos, B.O.; Serrano-Andrés, L. Does density functional theory contribute to the understanding of excited states of unsaturated organic compounds? Mol. Phys. 1999, 97, 859–868. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Brandenburg, J.G. Simplified DFT methods for consistent structures and energies of large systems. J. Phys. Condens. Matter 2018, 30, 213001. [Google Scholar] [CrossRef] [PubMed]
- Zandler, M.E.; D’Souza, F. The remarkable ability of B3LYP/3-21G(*) calculations to describe geometry, spectral and electrochemical properties of molecular and supramolecular porphyrin–fullerene conjugates. Comptes. Rendus. Chim. 2006, 9, 960–981. [Google Scholar] [CrossRef]
- Dirac, P.A.M. Quantum mechanics of many-electron systems. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1929, 123, 714–733. [Google Scholar] [CrossRef] [Green Version]
- Slater, J.C. A Simplification of the Hartree-Fock Method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Liu, Y.F.; Guan, J.; Hu, D.; Du, L.; Sun, H.; Gao, J.; Zhao, J.; Lan, Z. Computational Investigation of Acene-Modified Zinc-Porphyrin Based Sensitizers for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2015, 119, 8417–8430. [Google Scholar] [CrossRef]
- Fransson, T.; Saue, T.; Norman, P. Four-Component Damped Density Functional Response Theory Study of UV/Vis Absorption Spectra and Phosphorescence Parameters of Group 12 Metal-Substituted Porphyrins. J. Chem. Theory Comput. 2016, 12, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, L.; Harper, S.R.; Arnold, D.P. A systematic theoretical study of the electronic structures of porphyrin dimers: DFT and TD-DFT calculations on diporphyrins linked by ethane, ethene, ethyne, imine, and azo bridges. Phys. Chem. Chem. Phys. 2013, 15, 18951–18964. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Simone, B.C.D.; Mazzone, G.; Russo, N.; Sicilia, E.; Toscano, M. Metal Atom Effect on the Photophysical Properties of Mg(II), Zn(II), Cd(II), and Pd(II) Tetra-phenylporphyrin Complexes Proposed as Possible Drugs in Photodynamic Therapy. Molecules 2017, 22, 1093. [Google Scholar] [CrossRef] [Green Version]
- Vijay, D.; Varathan, E.; Subramanian, V. Theoretical design of core modified (oxa and thia) porphyrin based organic dyes with bridging thiophene linkers. J. Mater. Chem. A 2013, 1, 4358–4369. [Google Scholar] [CrossRef]
- Mazzone, G.; Alberto, M.E.; De Simone, B.C.; Marino, T.; Russo, N. Can Expanded Bacteriochlorins Act as Photosensitizers in Photodynamic Therapy? Good News from Density Functional Theory Computations. Mol. 2016, 21, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.D.; Xiao, H.M.; Tian, H. Comparative studies on the structures, infrared spectrum, and thermodynamic properties of phthalocyanine using ab initio Hartree–Fock and density functional theory methods. Int. J. Quantum Chem. 2002, 86, 531–540. [Google Scholar] [CrossRef]
- Bloch, F. Bemerkung zur Elektronentheorie des Ferromagnetismus und der elektrischen Leitfähigkeit. Z. Phys. 1929, 57, 545. [Google Scholar] [CrossRef]
- Huh, D.S.; Choe, S.J. Comparative DFT study for molecular geometries and spectra of methyl pheophorbides-a: Test of M06-2X and two other functionals. J. Porphyr. Phthalocyanines 2010, 14, 592–604. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen- bonded complexes. J. Chem. Phys. 2003, 119, 12129. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Tao, J.; Staroverov, V.N.; Scuseria, G.E. Meta-generalized gradient approximation: Explanation of a realistic nonempirical density functional. J. Chem. Phys. 2004, 120, 6898–6911. [Google Scholar] [CrossRef]
- Rydberg, P.; Olsen, L. The Accuracy of Geometries for Iron Porphyrin Complexes from Density Functional Theory. J. Phys. Chem. A 2009, 113, 11949–11953. [Google Scholar] [CrossRef]
- Wang, J.; Durbeej, B. How accurate are TD-DFT excited-state geometries compared to DFT ground-state geometries? J. Comput. Chem. 2020, 41, 1718–1729. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Grotjahn, R.; Kaupp, M. Validation of Local Hybrid Functionals for Excited States: Structures, Fluorescence, Phosphorescence, and Vibronic Spectra. J. Chem. Theory Comput. 2020, 16, 5821–5834. [Google Scholar] [CrossRef] [PubMed]
- Winter, N.O.C.; Graf, N.K.; Leutwyler, S.; Hättig, C. Benchmarks for 0–0 transitions of aromatic organic molecules: DFT/B3LYP, ADC(2), CC2, SOS-CC2 and SCS-CC2 compared to high-resolution gas-phase data. Phys. Chem. Chem. Phys. 2013, 15, 6623–6630. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of Porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Dev, P.; Agrawal, S.; English, N.J. Determining the appropriate exchange-correlation functional for time-dependent density functional theory studies of charge-transfer excitations in organic dyes. J. Chem. Phys. 2012, 136, 224301. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Balanay, M.P.; Kim, D.H. Molecular design of distorted push–pull porphyrins for dye-sensitized solar cells. Theor. Chem. Acc. 2012, 131, 1–12. [Google Scholar] [CrossRef]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334–14356. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Rafiq, M.I.; Zhang, Z.; Cao, J.; Geng, R.; Zhou, B.; Tang, W. TD-DFT benchmark for UV-visible spectra of fused-ring electron acceptors using global and range-separated hybrids. Phys. Chem. Chem. Phys. 2020, 22, 7864–7874. [Google Scholar] [CrossRef]
- Bhuyan, J. Metalloisoporphyrins: From synthesis to applications. Dalton Trans. 2015, 44, 15742–15756. [Google Scholar] [CrossRef]
- Körzdörfer, T.; Brédas, J.-L. Organic Electronic Materials: Recent Advances in the DFT Description of the Ground and Excited States Using Tuned Range-Separated Hybrid Functionals. Acc. Chem. Res. 2014, 47, 3284–3291. [Google Scholar] [CrossRef]
- Castro, A.; Marques, M.A.; Varsano, D.; Sottile, F.; Rubio, A. The challenge of predicting optical properties of biomolecules: What can we learn from time-dependent density-functional theory? Comptes Rendus Phys. 2009, 10, 469–490. [Google Scholar] [CrossRef] [Green Version]
- Jacquemin, D.; Planchat, A.; Adamo, C.; Mennucci, B. TD-DFT Assessment of Functionals for Optical 0–0 Transitions in Solvated Dyes. J. Chem. Theory Comput. 2012, 8, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Eriksson, E.S.E.; Eriksson, L.A. Can Range-Separated and Hybrid DFT Functionals Predict Low-Lying Excitations? A Tookad Case Study. J. Chem. Theory Comput. 2010, 6, 2086–2094. [Google Scholar] [CrossRef]
- Batra, K.; Zahn, S.; Heine, T. Benchmark of Simplified Time-Dependent Density Functional Theory for UV–Vis Spectral Properties of Porphyrinoids. Adv. Theory Simul. 2019, 3. [Google Scholar] [CrossRef]
- Mack, J.; Bunya, M.; Shimizu, Y.; Uoyama, H.; Komobuchi, N.; Okujima, T.; Uno, H.; Ito, S.; Stillman, M.J.; Ono, N.; et al. Application of MCD Spectroscopy and TD-DFT to Nonplanar Core-Modified Tetrabenzoporphyrins: Effect of Reduced Symmetry on Nonplanar Porphyrinoids. Chem. Eur. J. 2008, 14, 5001–5020. [Google Scholar] [CrossRef]
- Mack, J. Expanded, Contracted, and Isomeric Porphyrins: Theoretical Aspects. Chem. Rev. 2017, 117, 3444–3478. [Google Scholar] [CrossRef]
- Mack, J.; Stone, J.; Nyokong, T. Trends in the TD-DFT calculations of porphyrin and phthalocyanine analogs. J. Porphyr. Phthalocyanines 2014, 18, 630–641. [Google Scholar] [CrossRef]
- Petit, L.; Quartarolo, A.; Adamo, C.; Russo, N. Spectroscopic Properties of Porphyrin-Like Photosensitizers: Äâ Insights from Theory. J. Phys. Chem. B 2006, 110, 2398–2404. [Google Scholar] [CrossRef] [PubMed]
- Tessaro, A.L.; Batistela, V.R.; Pellosi, D.S.; Caetano, W.; Ueno, L.T.; Machado, A.E.D.H.; Van Der Linden, M.G.; Hioka, N. Electronic structures and spectroscopic properties of benzoporphyrin protolytic species: A TD-DFT study. Comput. Theor. Chem. 2013, 1020, 173–179. [Google Scholar] [CrossRef]
- Dulski, M.; Kempa, M.; Kozub, P.; Wójcik, J.; Rojkiewicz, M.; Kuś, P.; Szurko, A.; Ratuszna, A.; Wrzalik, R. DFT/TD-DFT study of solvent effect as well the substituents influence on the different features of TPP derivatives for PDT application. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 104, 315–327. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Pan, Y.; Li, L.; Qiu, F.; Wei, Y.; Hua, W.; Tian, G. On the spectral profile change in the Q band absorption spectra of metalloporphyrins (Mg, Zn, and Pd): A first-principles study. J. Chem. Phys. 2019, 150, 164308. [Google Scholar] [CrossRef]
- Improta, R.; Ferrante, C.; Bozio, R.; Barone, V. The polarizability in solution of tetra-phenyl-porphyrin derivatives in their excited electronic states: A PCM/TD-DFT study. Phys. Chem. Chem. Phys. 2009, 11, 4664–4673. [Google Scholar] [CrossRef]
- Wójcik, J.; Peszke, J.; Ratuszna, A.; Kuś, P.; Wrzalik, R. Theoretical investigation of porphyrin-based photosensitizers with enhanced NIR absorption. Phys. Chem. Chem. Phys. 2013, 15, 19651–19658. [Google Scholar] [CrossRef]
- Bettanin, F.; Antonio, F.C.T.; Honorio, K.M.; Homem-De-Mello, P. Quantum-chemistry descriptors for photosensitizers based on macrocycles. Chem. Biol. Drug Des. 2017, 89, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Alberto, M.E.; Marino, T.; Quartarolo, A.D.; Russo, N. Photophysical origin of the reduced photodynamic therapy activity of temocene compared to Foscan(R): Insights from theory. Phys. Chem. Chem. Phys. 2013, 15, 16167–16171. [Google Scholar] [CrossRef]
- Cai, Z.-L.; Crossley, M.J.; Reimers, J.R.; Kobayashi, A.R.; Amos, R.D. Density Functional Theory for Charge Transfer: The Nature of the N-Bands of Porphyrins and Chlorophylls Revealed through CAM-B3LYP, CASPT2, and SAC-CI Calculations. J. Phys. Chem. B 2006, 110, 15624–15632. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 1993, 799–805. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Osadchuk, I.; Konrad, N.; Truong, K.-N.; Rissanen, K.; Clot, E.; Aav, R.; Kananovich, D.; Borovkov, V. Supramolecular Chirogenesis in Bis-Porphyrin: Crystallographic Structure and CD Spectra for a Complex with a Chiral Guanidine Derivative. Symmetry 2021, 13, 275. [Google Scholar] [CrossRef]
- Hasegawa, J.-Y.; Takata, K.; Miyahara, T.; Neya, S.; Frisch, M.J.; Nakatsuji, H. Excited States of Porphyrin Isomers and Porphycene Derivatives: A SAC-CI Study. J. Phys. Chem. A 2005, 109, 3187–3200. [Google Scholar] [CrossRef]
- Balanay, M.P.; Kim, D.H. DFT/TD-DFT molecular design of porphyrin analogues for use in dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2008, 10, 5121–5127. [Google Scholar] [CrossRef] [Green Version]
- Cammi, R.; Corni, S.; Mennucci, B.; Tomasi, J. Electronic excitation energies of molecules in solution: State specific and linear response methods for nonequilibrium continuum solvation models. J. Chem. Phys. 2005, 122, 104513. [Google Scholar] [CrossRef]
- Corni, S.; Cammi, R.; Mennucci, B.; Tomasi, J. Electronic excitation energies of molecules in solution within continuum solvation models: Investigating the discrepancy between state-specific and linear-response methods. J. Chem. Phys. 2005, 123, 134512. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Barone, V.; Scalmani, G.; Frisch, M.J. A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution. J. Chem. Phys. 2006, 125, 054103. [Google Scholar] [CrossRef]
- Fukuda, R.; Ehara, M. Mechanisms for Solvatochromic Shifts of Free-Base Porphine Studied with Polarizable Continuum Models and Explicit Solute–Solvent Interactions. J. Chem. Theory Comput. 2013, 9, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.-D.; Valiev, M.; Kowalski, K. Large-scale parallel calculations with combined coupled cluster and molecular mechanics formalism: Excitation energies of zinc–porphyrin in aqueous solution. Chem. Phys. Lett. 2008, 458, 205–209. [Google Scholar] [CrossRef]
- Shen, L. Theoretical investigation on the triplet excited state properties of the porphyrin-related photosensitizers and the implications in illustrating their photosensitization mechanisms. J. Mol. Struct. THEOCHEM 2008, 862, 130–132. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Adduct under Field—A Qualitative Approach to Account for Solvent Effect on Hydrogen Bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef] [Green Version]
- Shenderovich, I.G. Electric field effect on 31P NMR magnetic shielding. J. Chem. Phys. 2020, 153, 184501. [Google Scholar] [CrossRef] [PubMed]
- Chojecki, M.; Rutkowska-Zbik, D.; Korona, T. Dimerization Behavior of Methyl Chlorophyllide a as the Model of Chlorophyll a in the Presence of Water Molecules—Theoretical Study. J. Chem. Inf. Model. 2019, 59, 2123–2140. [Google Scholar] [CrossRef]
- Korona, T.; Rutkowska-Zbik, D. A theoretical study on elementary building blocks for organic solar cells—Influence of a donor molecule on electronic spectrum of PCBM. Comput. Theor. Chem. 2014, 1040, 243–258. [Google Scholar] [CrossRef]
- Park, Y.I.; Kuo, C.Y.; Martinez, J.S.; Park, Y.S.; Postupna, O.; Zhugayevych, A. Tailored Electronic Structure and Optical Properties of Conjugated Systems through Aggregates and Dipole–Dipole Interactions. ACS Appl. Mater. Interfaces 2013, 5, 4685–4695. [Google Scholar] [CrossRef]
- Mennucci, B.; Cappelli, C.; Guido, C.A.; Cammi, R.; Tomasi, J. Structures and Properties of Electronically Excited Chromophores in Solution from the Polarizable Continuum Model Coupled to the Time-Dependent Density Functional Theory. J. Phys. Chem. A 2009, 113, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Korona, K.; Korona, T.; Rutkowska-Zbik, D.; Grankowska-Ciechanowicz, S.; Iwan, A.; Kamińska, M. Polyazomethine as a component of solar cells-theoretical and optical study. J. Phys. Chem. Solids 2015, 86, 186–193. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Solvent effects on acid-base complexes. What is more important: A macroscopic reac-tion field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef]
- Alam, M.; Daniel, C. One- and two-photon activity of diketopyrrolopyrrole-Zn-porphyrin conjugates: Linear and quadratic density functional response theory applied to model systems. Theor. Chem. Acc. 2016, 135, 1–13. [Google Scholar] [CrossRef]
- Olsen, J.; Jo/rgensen, P. Linear and nonlinear response functions for an exact state and for an MCSCF state. J. Chem. Phys. 1985, 82, 3235–3264. [Google Scholar] [CrossRef] [Green Version]
- Furche, F. On the density matrix based approach to time-dependent density functional response theory. J. Chem. Phys. 2001, 114, 5982–5992. [Google Scholar] [CrossRef]
- Sałek, P.; Vahtras, O.; Guo, J.; Luo, Y.; Helgaker, T.; Ågren, H. Calculations of two-photon absorption cross sections by means of density-functional theory. Chem. Phys. Lett. 2003, 374, 446–452. [Google Scholar] [CrossRef]
- Esipova, T.V.; Rivera-Jacquez, H.J.; Weber, B.; Masunov, A.E.; Vinogradov, S.A. Two-Photon Absorbing Phosphorescent Metalloporphyrins: Effects of pi-Extension and Peripheral Substitution. J. Am. Chem. Soc. 2016, 138, 15648–15662. [Google Scholar] [CrossRef]
- Esipova, T.V.; Rivera-Jacquez, H.J.; Weber, B.; Masunov, A.E.; Vinogradov, S.A. Stabilizing g-States in Centrosymmetric Tetrapyrroles: Two-Photon-Absorbing Porphyrins with Bright Phosphorescence. J. Phys. Chem. A 2017, 121, 6243–6255. [Google Scholar] [CrossRef] [PubMed]
- Day, P.N.; Nguyen, K.A.; Pachter, R. Calculation of One-Photon and Two-Photon Absorption Spectra of Porphyrins Using Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2008, 4, 1094–1106. [Google Scholar] [CrossRef]
- Greco, J.A.; Shima, S.; Wagner, N.L.; McCarthy, J.R.; Atticks, K.; Brückner, C.; Birge, R.R. Two-Photon Spectroscopy of the Q-Bands of meso-Tetraphenyl-Porphyrin and -Chlorin Framework Derivatives. J. Phys. Chem. C 2015, 119, 3711–3724. [Google Scholar] [CrossRef]
- Chandra Jha, P.; Minaev, B.; Agren, H. One- and two-photon absorptions in asymmetrically substituted free-base por-phyrins: A density functional theory study. J. Chem. Phys. 2008, 128, 74302. [Google Scholar] [CrossRef]
- Ravotto, L.; Meloni, S.L.; Esipova, T.V.; Masunov, A.E.; Anna, J.M.; Vinogradov, S.A. Three-Photon Spectroscopy of Porphyrins. J. Phys. Chem. A 2020, 124, 11038–11050. [Google Scholar] [CrossRef] [PubMed]
- Cohanoschi, I.; Echeverría, L.; Hernández, F.E. Three-photon absorption measurements in hematoporphyrin IX: “Ground-breaking opportunities in deep photodynamic therapy”. Chem. Phys. Lett. 2006, 419, 33–36. [Google Scholar] [CrossRef]
- Morisue, M.; Ogawa, K.; Kamada, K.; Ohta, K.; Kobuke, Y. Strong two-photon and three-photon absorptions in the antiparallel dimer of a porphyrin–phthalocyanine tandem. Chem. Commun. 2010, 46, 2121–2123. [Google Scholar] [CrossRef] [Green Version]
- Ågren, H.; Vahtras, O.; Minaev, B. Response Theory and Calculations of Spin-Orbit Coupling Phenomena in Molecules. Quantum Boundaries Life 1996, 27, 71–162. [Google Scholar] [CrossRef]
- Gao, X.; Bai, S.; Fazzi, D.; Niehaus, T.; Barbatti, M.; Thiel, W. Evaluation of Spin-Orbit Couplings with Linear-Response Time-Dependent Density Functional Methods. J. Chem. Theory Comput. 2017, 13, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, P.; Autschbach, J. Variational versus Perturbational Treatment of Spin–Orbit Coupling in Relativistic Density Functional Calculations of Electronic g Factors: Effects from Spin-Polarization and Exact Exchange. J. Chem. Theory Comput. 2013, 9, 1052–1067. [Google Scholar] [CrossRef]
- Plotnikov, V.G. Regularities of the processes of radiationless conversion in polyatomic molecules. Int. J. Quantum Chem. 1979, 16, 527–541. [Google Scholar] [CrossRef]
- De Simone, B.C.; Mazzone, G.; Russo, N.; Sicilia, E.; Toscano, M. Computational Investigation of the Influence of Halogen Atoms on the Photophysical Properties of Tetraphenylporphyrin and Its Zinc(II) Complexes. J. Phys. Chem. A 2018, 122, 2809–2815. [Google Scholar] [CrossRef]
- Alberto, M.E.; De Simone, B.C.; Mazzone, G.; Marino, T.; Russo, N. Photophysical properties of free and metallated meso-substituted tetrabenzotriazaporphyrin from density functional theory investigation. Dye. Pigment. 2015, 120, 335–339. [Google Scholar] [CrossRef]
- Yamamoto, S.; Diercksen, G.H.; Karelson, M. An ab initio CI study of electronic spectra of substituted free-base porphyrins. Chem. Phys. Lett. 2000, 318, 590–596. [Google Scholar] [CrossRef]
- Dougherty, T.J. Photosensitizers: Therapy and detection of malignant tumors. Photochem. Photobiol. 1987, 45, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Scurlock, R.D.; Nonell, S.; Braslavsky, S.E.; Ogilby, P. Effect of Solvent on the Radiative Decay of Singlet Molecular Oxygen (a1.DELTA.g). J. Phys. Chem. 1995, 99, 3521–3526. [Google Scholar] [CrossRef]
- Alberto, M.E.; Marino, T.; Russo, N.; Sicilia, E.; Toscano, M. The performance of density functional based methods in the description of selected biological systems and processes. Phys. Chem. Chem. Phys. 2012, 14, 14943–14953. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, Z.H.; Sharma, D.; Mukherjee, A.; Basu, S.; Shukla, P.K.; Jha, T.; Sengupta, D. meso-Thiophenium Porphyrins and Their Zn(II) Complexes: A New Category of Cationic Photo-sensitizers. ACS Med. Chem. Lett. 2020, 11, 2041–2047. [Google Scholar] [CrossRef]
- Henderson, B.W.; A Bellnier, D.; Greco, W.R.; Sharma, A.; Pandey, R.K.; A Vaughan, L.; Weishaupt, K.R.; Dougherty, T.J. An in vivo quantitative structure-activity relationship for a congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy. Cancer Res. 1997, 57, 4000–4007. [Google Scholar]
- Potter, W.R.; Henderson, B.W.; Bellnier, D.A.; Pandey, R.K.; Vaughan, L.A.; Weishaupt, K.R.; Dougherty, T.J. Parabolic quantitative structure-activity relationships and photodynamic therapy: Application of a three-compartment model with clearance to the in vivo quantitative structure-activity relationships of a congeneric series of pyropheophorbide derivatives used as photosensitizers for photodynamic therapy. Photochem. Photobiol. 1999, 70, 781–788. [Google Scholar]
- Banfi, S.; Caruso, E.; Buccafurni, L.; Murano, R.; Monti, E.; Gariboldi, M. Comparison between 5,10,15,20-Tetraaryl- and 5,15-Diarylporphyrins as Photosensitizers: Synthesis, Pho-todynamic Activity, and Quantitative Structure-Activity Relationship Modeling. J. Med. Chem. 2006, 49, 3293–3304. [Google Scholar] [CrossRef]
- Geary, R.C. The Contiguity Ratio and Statistical Mapping. Inc. Stat. 1954, 5, 115. [Google Scholar] [CrossRef]
- Randić, M. Novel Shape Descriptors for Molecular Graphs. J. Chem. Inf. Comput. Sci. 2001, 41, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Todeschini, R.; Pavan, M. Structure/Response Correlation and Similarity/Diversity Analysis by GETA-WAY descriptors. Part 1. Theory of the Novel 3D Molecular Descriptors. J. Chem. Inf. Comput. Sci. 2002, 42, 682–692. [Google Scholar] [CrossRef]
- Rutkowska-Zbik, D.; Witko, M. Metallobacteriochlorophylls as potential dual agents for photodynamic therapy and chemotherapy. J. Mol. Model. 2013, 19, 4155–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falahati, K.; Hamerla, C.; Huix-Rotllant, M.; Burghardt, I. Ultrafast photochemistry of free-base porphyrin: A theoretical investigation of B → Q internal conversion mediated by dark states. Phys. Chem. Chem. Phys. 2018, 20, 12483–12492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).