
DeepNGlyPred: A Deep Neural Network-Based Approach for Human N-Linked Glycosylation Site Prediction

Abstract
:1. Introduction
2. Results and Discussion
2.1. Performance of DeepNGlyPred on N-GlyDE Dataset
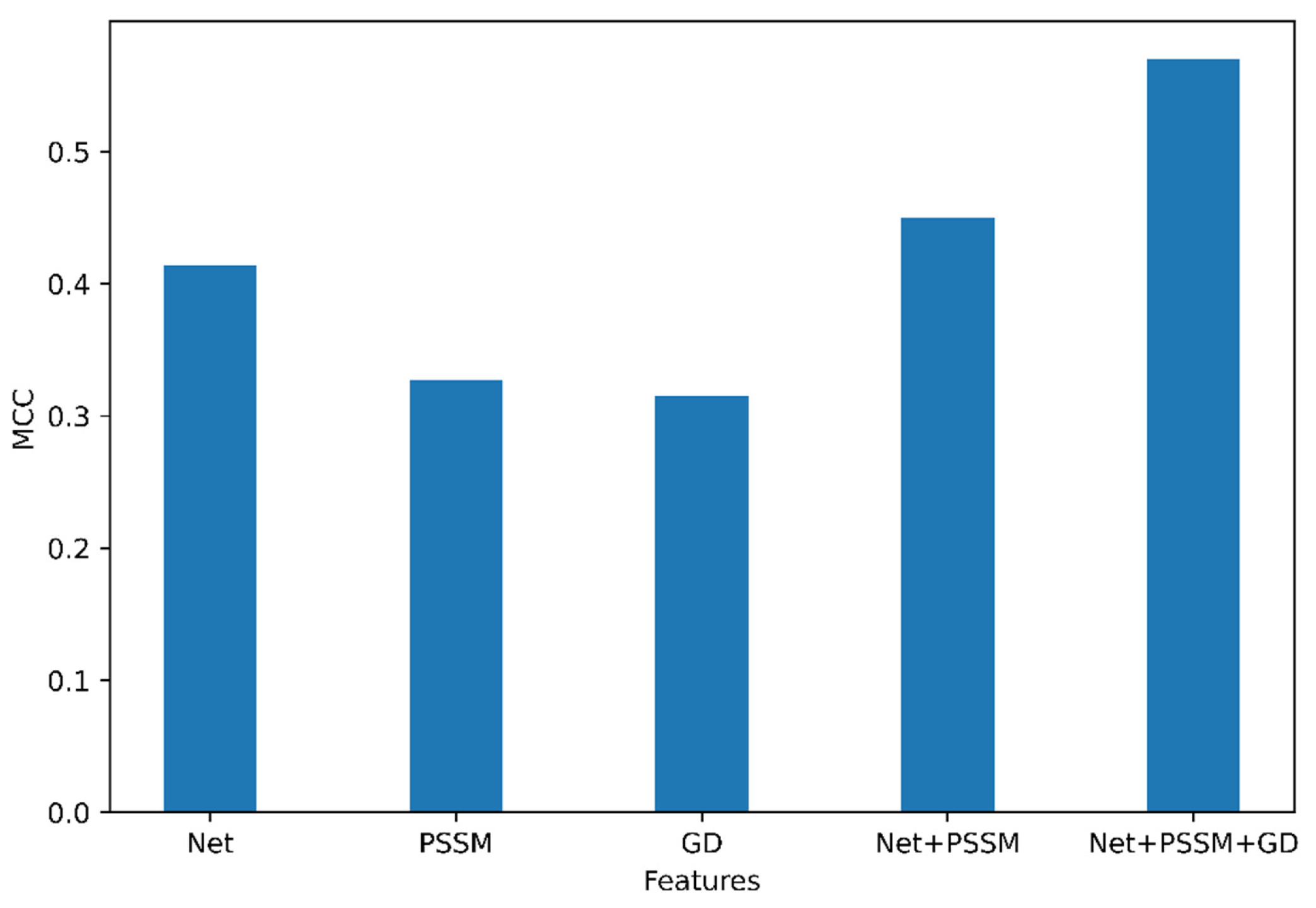
2.1.1. Optimal Feature Set
2.1.2. Selection of Window Size
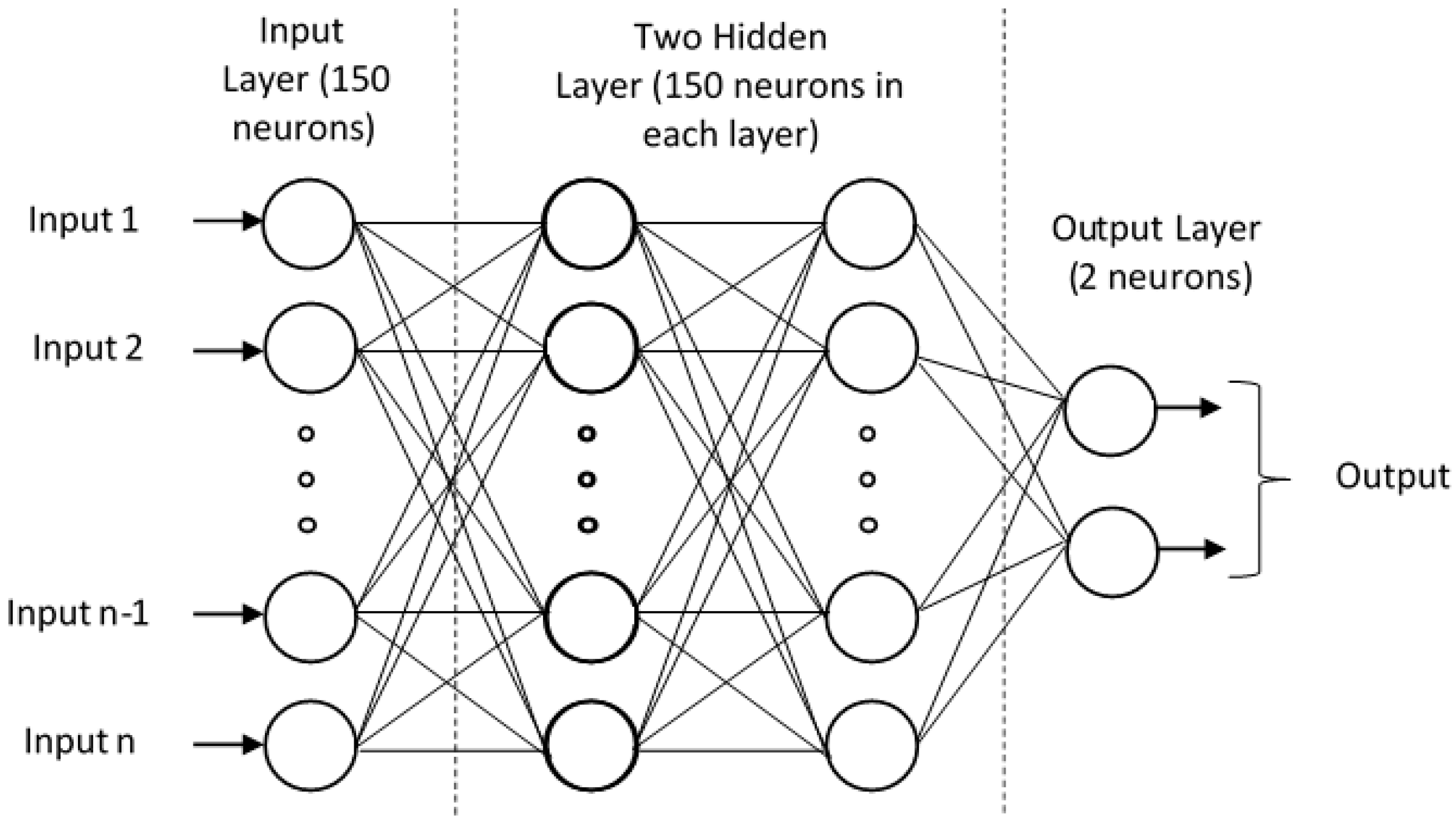
2.1.3. DNN Architecture
2.1.4. Cross-Validation Results
2.1.5. Independent Test Results
2.2. Performance of DeepNGlyPred on N-GlycositeAtlas Dataset
2.2.1. Optimal Feature Set
2.2.2. Selection of Window Size
2.2.3. DNN Architecture
2.2.4. Cross-Validation Results
2.2.5. Independent Test Results
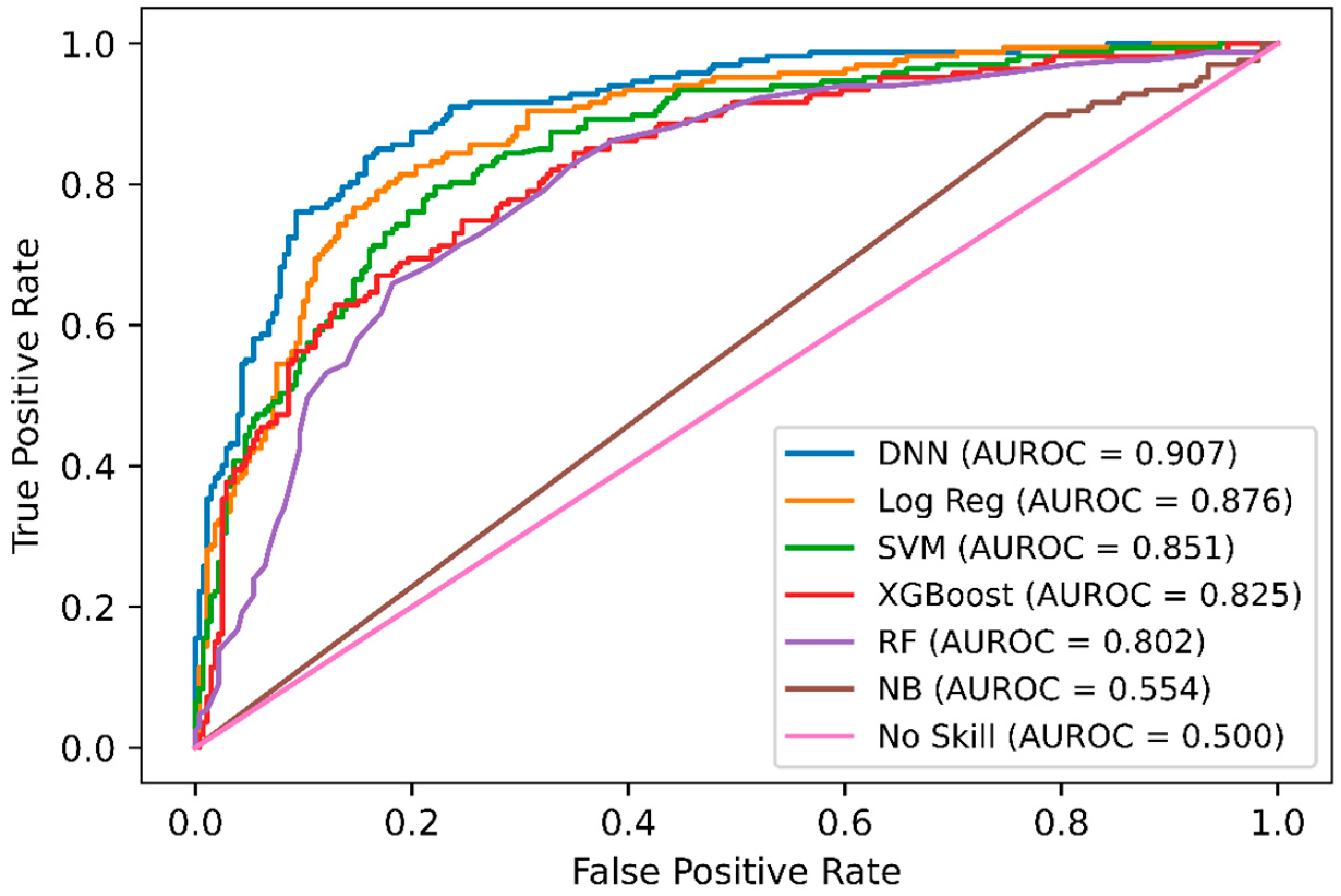
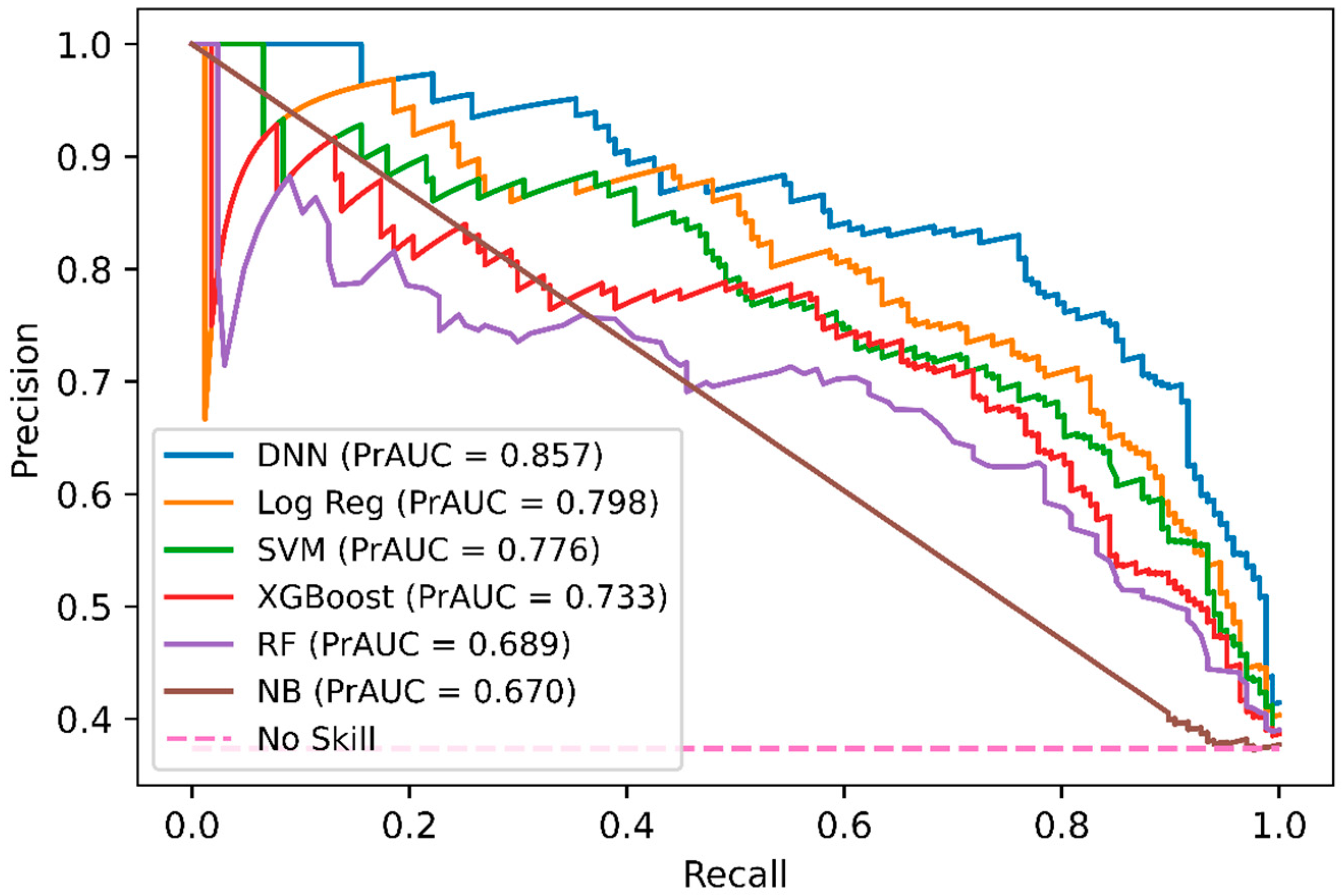
2.3. Comparison of DeepNGlyPred with Other Machine Learning Models
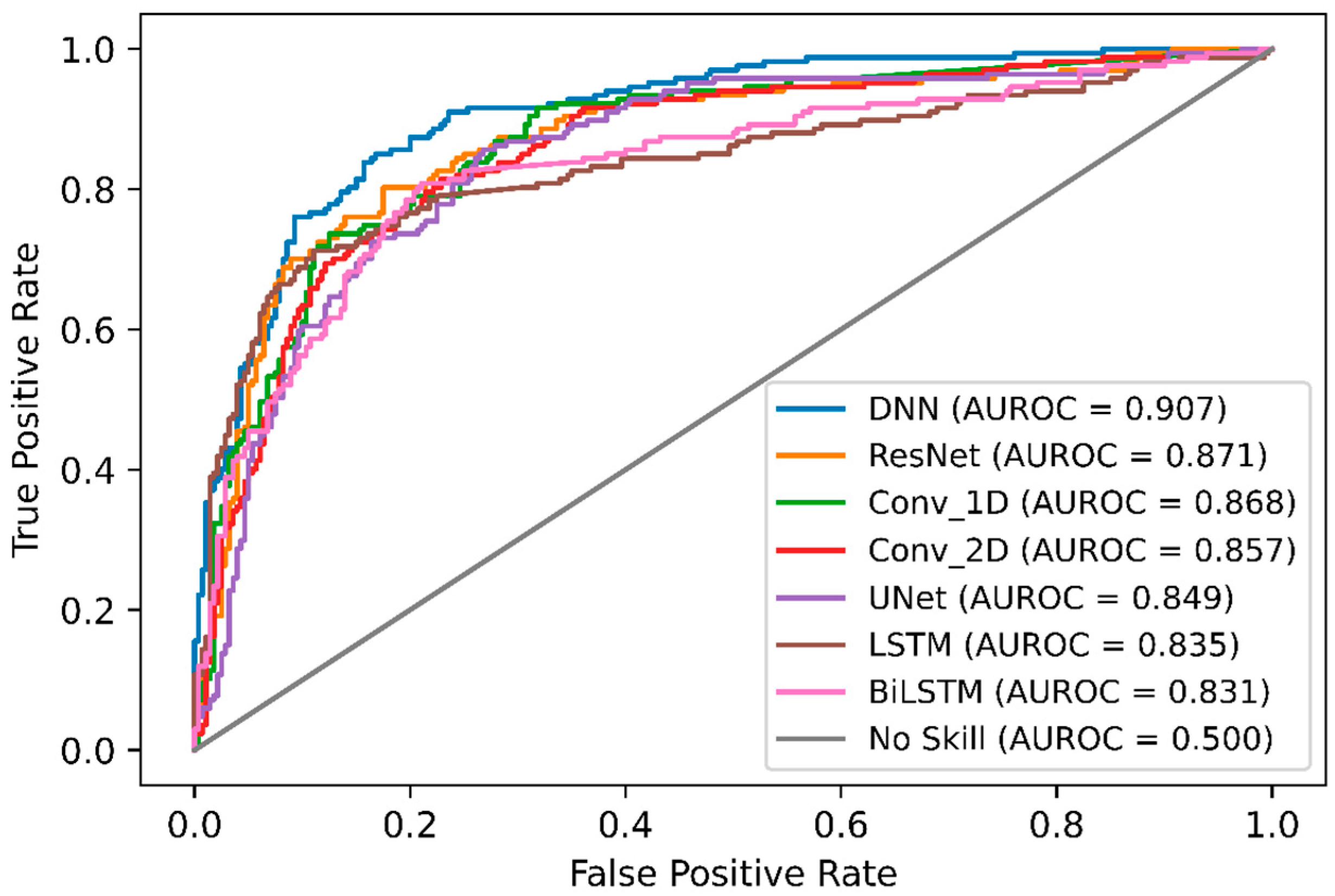
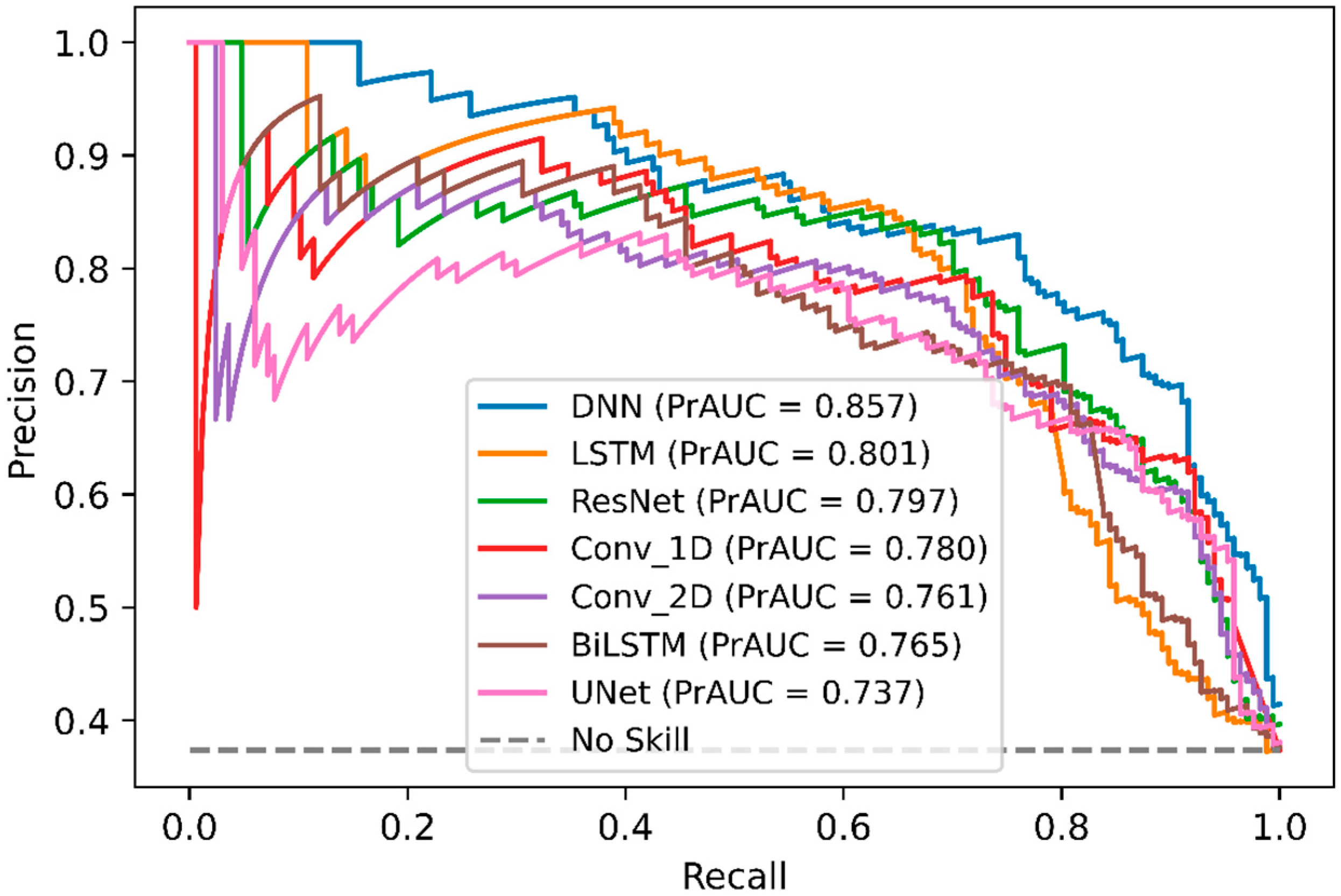
2.4. Comparison of DeepNGlyPred with Other Deep Learning Models
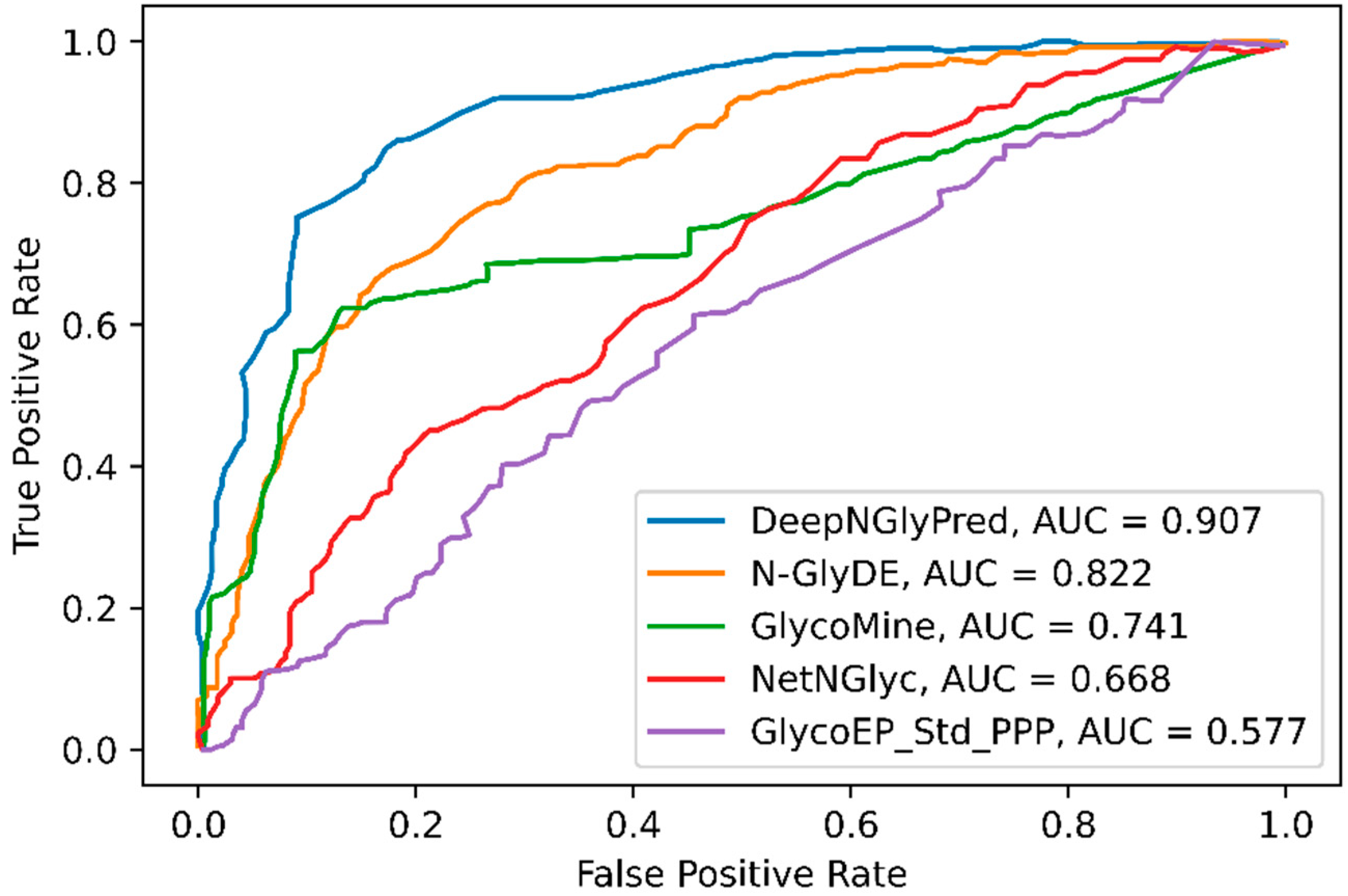
2.5. Comparison with Other Widely Available N-Linked Glycosylation Predictors
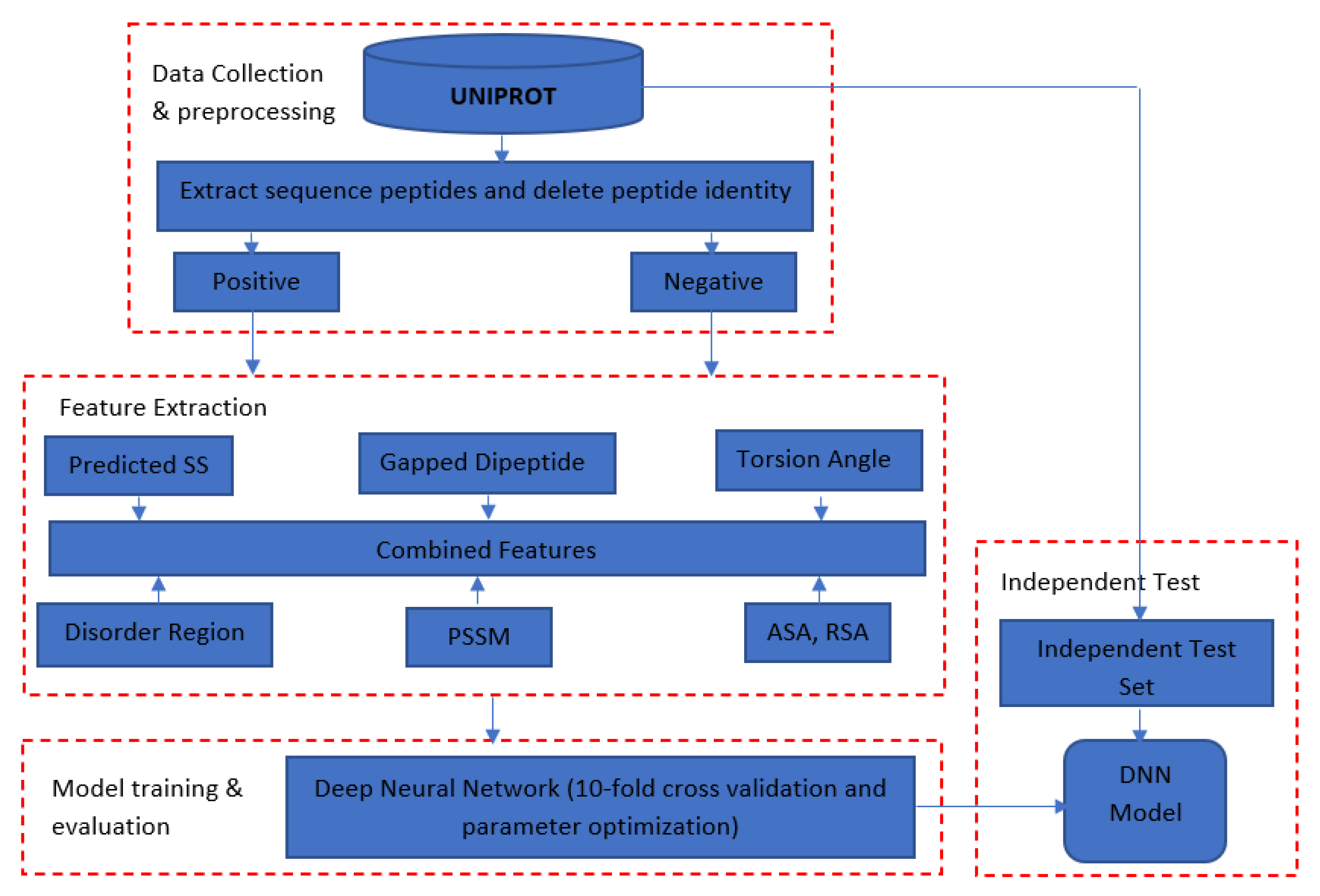
3. Materials and Methods
3.1. Datasets
3.1.1. N-GlyDE Dataset
3.1.2. N-GlycositeAtlas Dataset
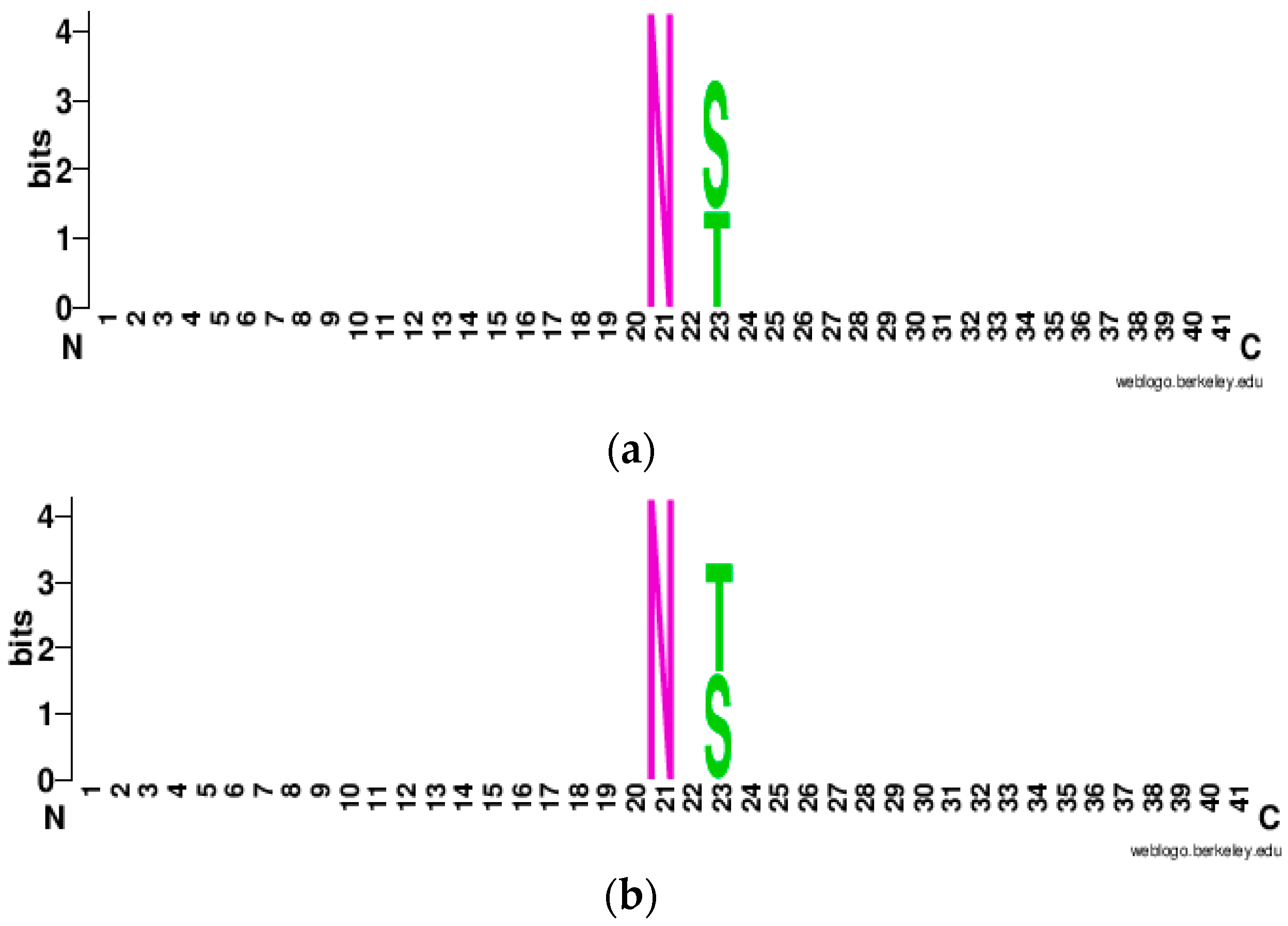
3.1.3. WebLogo Plot
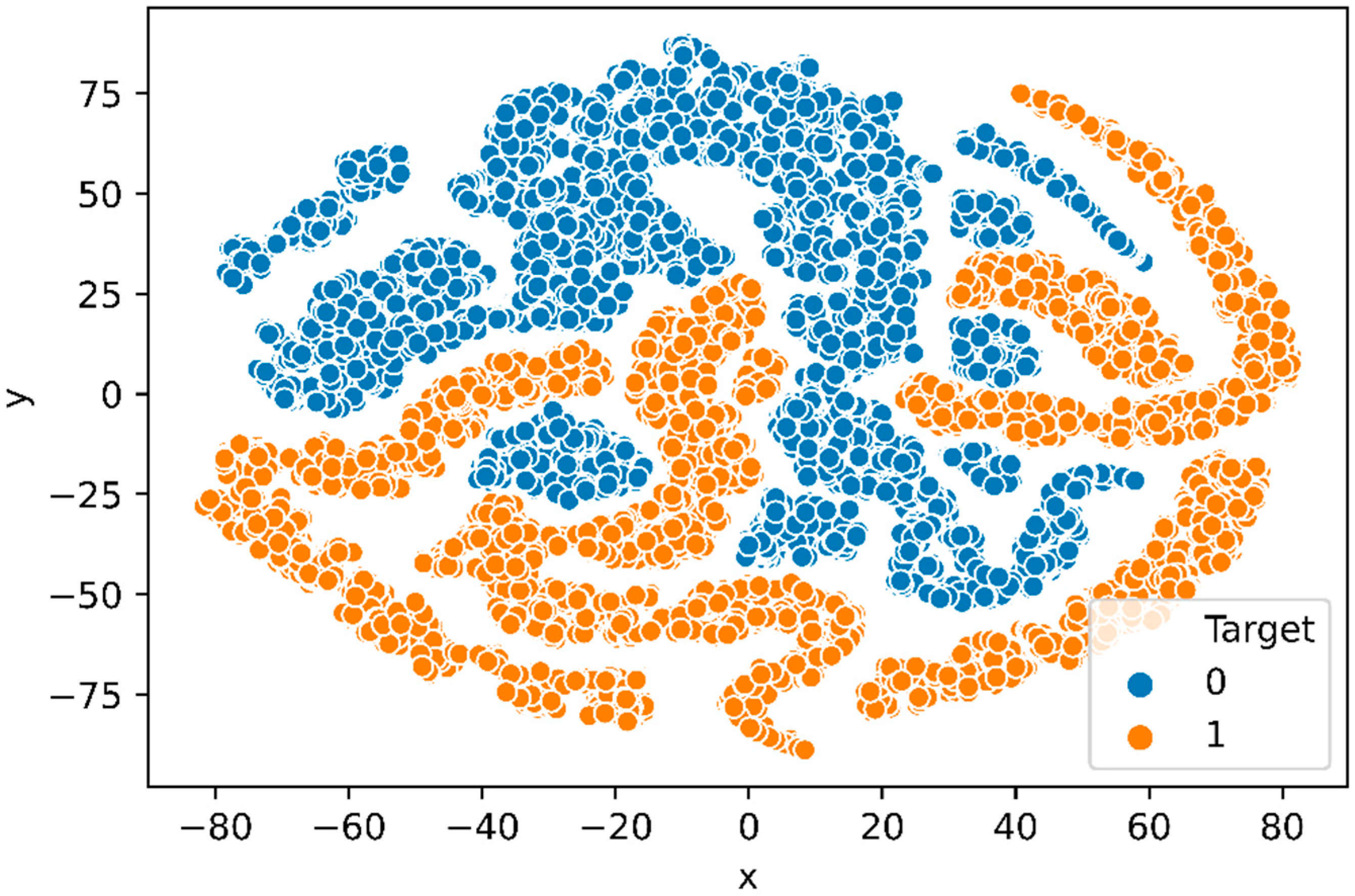
3.1.4. t-SNE Plot
3.2. Features Used in DeepNGlyPred
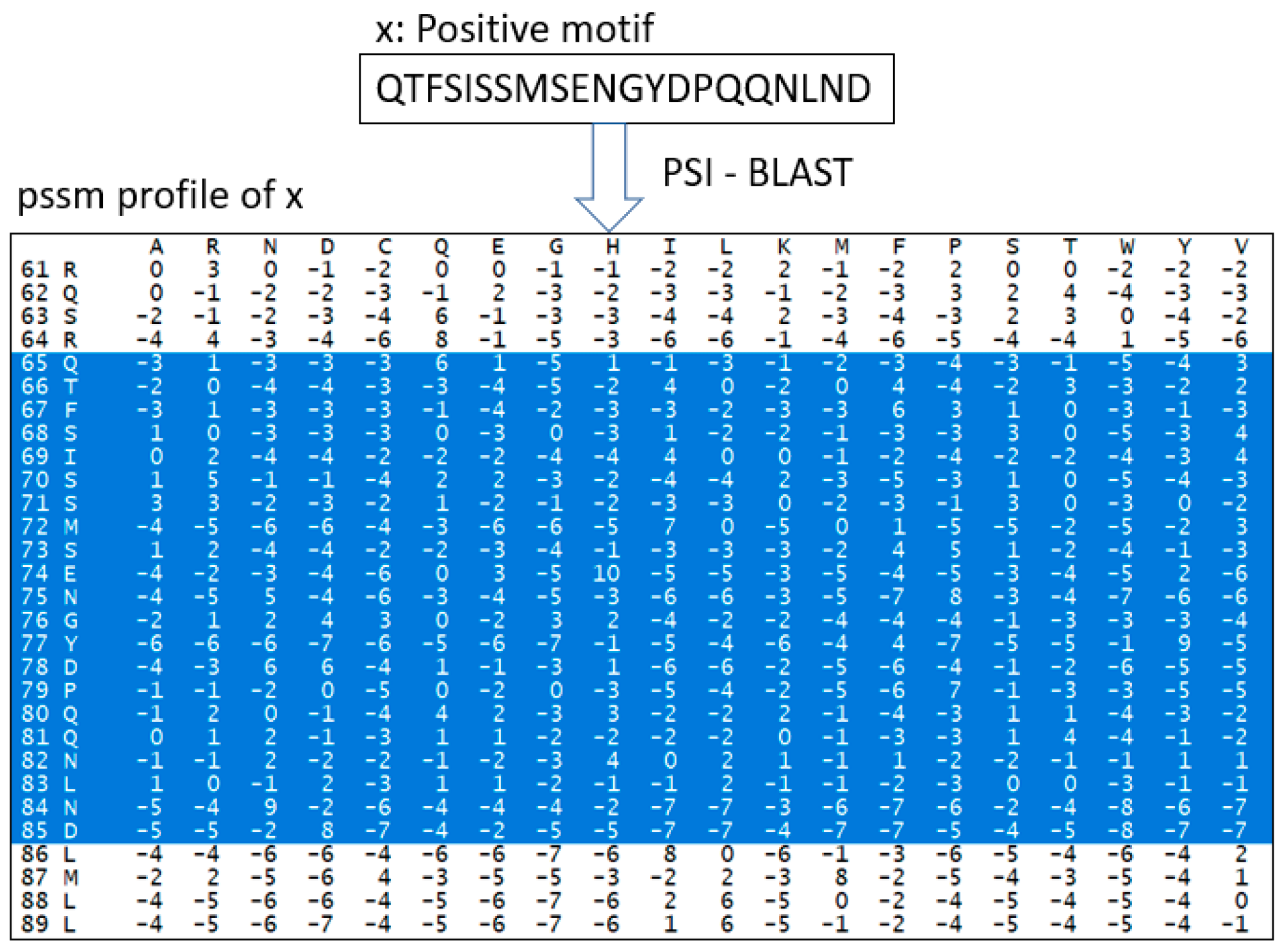
3.2.1. Position-Specific Scoring Matrix (PSSM)
3.2.2. Predicted Structural-Features
Predicted Secondary Structure (SS)
Predicted Accessible Surface Area (ASA), Relative Solvent Accessibility (RSA)
Predicted Disordered Region
Torsion Angles (Φ, Ψ)
3.2.3. Gapped Dipeptide (GD)
3.3. Overall Approach
3.4. Model Training Using Deep Neural Network (DNN)
3.5. Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aebi, M.; Bernasconi, R.; Clerc, S.; Molinari, M. N-glycan structures: Recognition and processing in the ER. Trends Biochem. Sci. 2010, 35, 74–82. [Google Scholar] [CrossRef]
- Lederkremer, G.Z. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 2009, 19, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Lowe, J.B. Biological Roles of Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Gavel, Y.; von Heijne, G.; Creaser, E.; Murali, C.; Britt, K. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: Implications for protein engineering. Protein Eng. 1990, 3, 433–442. [Google Scholar] [CrossRef]
- Boscher, C.; Dennis, J.W.; Nabi, I.R. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [Google Scholar] [CrossRef]
- van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef]
- Pérez-Sala, D.; Mollinedo, F. Inhibition of N-linked glycosylation induces early apoptosis in human promyelocytic HL-60 cells. J. Cell. Physiol. 1995, 163, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.J.; Edge, C.J.; Dwek, R.A. Protein surface oligosaccharides and protein function. Nat. Genet. Mol. Biol. 1994, 1, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Wormald, M.R.; Dwek, R.A. Glycoproteins: Glycan presentation and protein-fold stability. Structure 1999, 7, R155–R160. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Hennet, T. Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim. Biophys. Acta 2012, 1820, 1306–1317. [Google Scholar] [CrossRef] [Green Version]
- Jaeken, J.; Rymen, D.; Matthijs, G. Congenital disorders of glycosylation: Other causes of ichthyosis. Eur. J. Hum. Genet. 2013, 22, 444. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chan, D.W. Cancer Biomarker Discovery in Plasma Using a Tissue-targeted Proteomic Approach. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1915–1917. [Google Scholar] [CrossRef] [Green Version]
- Kowarik, M.; Young, N.M.; Numao, S.; Schulz, B.L.; Hug, I.; Callewaert, N.; Mills, D.C.; Watson, D.C.; Hernandez, M.; Kelly, J.F.; et al. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 2006, 25, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Petrescu, A.-J.; Milac, A.-L.; Petrescu, S.M.; Dwek, R.A.; Wormald, M.R. Statistical analysis of the protein environment of N-glycosylation sites: Implications for occupancy, structure, and folding. Glycobiology 2003, 14, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Zielinska, D.F.; Gnad, F.; Wiśniewski, J.R.; Mann, M. Precision Mapping of an In Vivo N-Glycoproteome Reveals Rigid Topological and Sequence Constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Schulz, B.L. Beyond the Sequon: Sites of N-Glycosylation. In Glycosylation; Petrescu, S., Ed.; InTech: Rijeka, Croatia, 2012; pp. 21–40. [Google Scholar] [CrossRef] [Green Version]
- Nita-Lazar, M.; Wacker, M.; Schegg, B.; Amber, S.; Aebi, M. The N-X-S/T consensus sequence is required but not sufficient for bacterial N-linked protein glycosylation. Glycobiology 2004, 15, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Wacker, M.; Feldman, M.; Callewaert, N.; Kowarik, M.; Clarke, B.R.; Pohl, N.L.; Hernandez, M.; Vines, E.D.; Valvano, M.; Whitfield, C.; et al. Substrate specificity of bacterial oligosaccharyltransferase suggests a common transfer mechanism for the bacterial and eukaryotic systems. Proc. Natl. Acad. Sci. USA 2006, 103, 7088–7093. [Google Scholar] [CrossRef] [Green Version]
- Medzihradszky, K.F. Peptide Sequence Analysis. Methods Enzymol. 2005, 402, 209–244. [Google Scholar] [CrossRef]
- Agarwal, K.L.; Kenner, G.W.; Sheppard, R.C. Feline gastrin. An example of peptide sequence analysis by mass spectrometry. J. Am. Chem. Soc. 1969, 91, 3096–3097. [Google Scholar] [CrossRef]
- Slade, D.; Subramanian, V.; Fuhrmann, J.; Thompson, P.R. Chemical and biological methods to detect post-translational modifications of arginine. Biopolymers 2013, 101, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2001, 7, 310–322. [Google Scholar]
- Caragea, C.; Sinapov, J.; Silvescu, A.; Dobbs, D.; Honavar, V. Glycosylation site prediction using ensembles of Support Vector Machine classifiers. BMC Bioinform. 2007, 8, 438. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, J.S.; Bhat, A.H.; Raghava, G.P.S.; Rao, A. GlycoPP: A Webserver for Prediction of N- and O-Glycosites in Prokaryotic Protein Sequences. PLoS ONE 2012, 7, e40155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.-H.; Chang, C.-C.; Lin, S.-H.; Chen, C.-W.; Chang, Z.-H.; Chu, Y.-W. N-GlycoGo: Predicting Protein N-Glycosylation Sites on Imbalanced Data Sets by Using Heterogeneous and Comprehensive Strategy. IEEE Access 2020, 8, 165944–165950. [Google Scholar] [CrossRef]
- Pugalenthi, G.; Nithya, V.; Chou, K.-C.; Archunan, G. Nglyc: A Random Forest Method for Prediction of N-Glycosylation Sites in Eukaryotic Protein Sequence. Protein Pept. Lett. 2020, 27, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Wang, M.; Webb, G.; Zhang, Y.; Whisstock, J.C.; Song, J. GlycoMine: A machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015, 31, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Taherzadeh, G.; Dehzangi, A.; Golchin, M.; Zhou, Y.; Campbell, M.P. SPRINT-Gly: Predicting N- and O-linked glycosylation sites of human and mouse proteins by using sequence and predicted structural properties. Bioinformatics 2019, 35, 4140–4146. [Google Scholar] [CrossRef] [PubMed]
- Adamczak, R.; Porollo, A.; Meller, J. Accurate prediction of solvent accessibility using neural networks-based regression. Proteins 2004, 56, 753–767. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, R.; Yang, Y.; Paliwal, K.K.; Zhou, Y. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics 2017, 33, 2842–2849. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; McGuffin, L.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Sodhi, J.; McGuffin, L.; Buxton, B.; Jones, D. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Hanson, J.; Yang, Y.; Paliwal, K.K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics 2016, 33, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, C.; Revote, J.; Zhang, Y.; Webb, G.I.; Li, J.; Song, J.; Lithgow, T. GlycoMinestruct: A new bioinformatics tool for highly accurate mapping of the human N-linked and O-linked glycoproteomes by incorporating structural features. Sci. Rep. 2016, 6, 34595. [Google Scholar] [CrossRef] [Green Version]
- Pitti, T.; Chen, C.-T.; Lin, H.-N.; Choong, W.-K.; Hsu, W.-L.; Sung, T.-Y. N-GlyDE: A two-stage N-linked glycosylation site prediction incorporating gapped dipeptides and pattern-based encoding. Sci. Rep. 2019, 9, 15975. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Hu, Y.; Ao, M.; Shah, P.; Chen, J.; Yang, W.; Jia, X.; Tian, Y.; Thomas, S.; Zhang, H. N-GlycositeAtlas: A database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin. Proteom. 2019, 16, 35. [Google Scholar] [CrossRef] [Green Version]
- Do, D.T.; Le, T.Q.; Le, N.Q. Using deep neural networks and biological subwords to detect protein S-sulfenylation sites. Brief. Bioinform. 2020, 22, bbaa128. [Google Scholar] [CrossRef]
- Thapa, N.; Chaudhari, M.; McManus, S.; Roy, K.; Newman, R.H.; Saigo, H.; Kc, D.B. DeepSuccinylSite: A deep learning based approach for protein succinylation site prediction. BMC Bioinform. 2020, 21 (Suppl. S3), 63. [Google Scholar] [CrossRef]
- Thapa, N.; Chaudhari, M.; Iannetta, A.A.; White, C.; Roy, K.; Newman, R.H.; Hicks, L.M.; Kc, D.B. A deep learning based approach for prediction of Chlamydomonas reinhardtii phosphorylation sites. Sci. Rep. 2021, 11, 12550. [Google Scholar] [CrossRef]
- Pakhrin, S.; Shrestha, B.; Adhikari, B.; Kc, D. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci. 2021, 22, 5553. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins 2019, 87, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Lemaitre, G.; Nogueira, F.; Aridas, C.K. Imbalanced-learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. 2017, 18, 559–563. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Parameters | Parameters Used |
|---|---|
| No. of layers | 4 |
| No. of neuron in three layers | 150 |
| No. neuron in the output layer | 2 |
| Activation Function | sigmoid |
| Activation Function at output layer | softmax |
| Optimizer | Adam |
| Learning rate | 0.001 |
| Objective/loss function | Binary_crossentropy |
| Model Checkpoint | Monitor = ‘val_accuracy’ |
| Reduce learning rate on plateau | Factor = 0.001 |
| Early stopping | patience = 5 |
| Dropout | 0.3 |
| Batch_size | 256 |
| Epochs | 400 |
| Name of the Parameters | Parameters Used |
|---|---|
| No. of layers | 5 |
| No. neuron in four layers | 1024 |
| No. of neuron in the output layer | 2 |
| Activation Function | sigmoid |
| Activation Function at output layer | softmax |
| Optimizer | Adam |
| Learning rate | 0.001 |
| Objective/loss function | Binary_crossentropy |
| Model Checkpoint | Monitor = ‘val_accuracy’ |
| Reduce learning rate on plateau | Factor = 0.001 |
| Early stopping | patience = 5 |
| Dropout | 0.3 |
| Batch_size | 256 |
| Epochs | 400 |
| Predictors | Accuracy (%) | Precision (%) | Sensitivity (%) | Specificity (%) | MCC |
|---|---|---|---|---|---|
| DeepNGlyPred (N-GlycositeAtlas) | 79.4 | 66.9 | 88.6 | 73.9 | 0.605 |
| DeepNGlyPred (N-GlyDE) | 77.8 | 69.5 | 72.4 | 81.0 | 0.531 |
| N-GlyDE | 74.0 | 61.3 | 82.6 | 68.9 | 0.499 |
| GlycoMine | 72.5 | 61.6 | 70.0 | 73.9 | 0.43 |
| NetNGlyc | 57.2 | 46.0 | 84.4 | 41.1 | 0.265 |
| GlycoEP_Std_PPP | 57.4 | 43.7 | 51.2 | 61.0 | 0.119 |
| Name of Dataset | Positive Site | Negative Site | Total |
|---|---|---|---|
| N-GlyDE (training) | 1030 | 2050 | 3080 |
| N-GlyDE (independent test) | 167 | 280 | 447 |
| N-GlycositeAtlas (training, CD-HIT) | 9450 | 9450 | 18,900 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakhrin, S.C.; Aoki-Kinoshita, K.F.; Caragea, D.; KC, D.B. DeepNGlyPred: A Deep Neural Network-Based Approach for Human N-Linked Glycosylation Site Prediction. Molecules 2021, 26, 7314. https://doi.org/10.3390/molecules26237314
Pakhrin SC, Aoki-Kinoshita KF, Caragea D, KC DB. DeepNGlyPred: A Deep Neural Network-Based Approach for Human N-Linked Glycosylation Site Prediction. Molecules. 2021; 26(23):7314. https://doi.org/10.3390/molecules26237314
Chicago/Turabian StylePakhrin, Subash C., Kiyoko F. Aoki-Kinoshita, Doina Caragea, and Dukka B. KC. 2021. "DeepNGlyPred: A Deep Neural Network-Based Approach for Human N-Linked Glycosylation Site Prediction" Molecules 26, no. 23: 7314. https://doi.org/10.3390/molecules26237314
APA StylePakhrin, S. C., Aoki-Kinoshita, K. F., Caragea, D., & KC, D. B. (2021). DeepNGlyPred: A Deep Neural Network-Based Approach for Human N-Linked Glycosylation Site Prediction. Molecules, 26(23), 7314. https://doi.org/10.3390/molecules26237314