3. Materials and Methods
3.1. Compound Characterization
All reagents were purchased from commercial sources and used without further purification. The
1H and
13C-NMR data were obtained Varian 500 MHz (Agilent, Santa Clara, CA, USA) and Bruker Ultrashield Avance 400 (Bruker, Billerica, MA, USA) spectrometers. Thin layer chromatography (TLC) and NMR were used to monitor reactions and determine compound purity. Compounds were purified by silica gel (Sortech, Norcross, GA, USA, 60 Å, 200–500 µm (35 × 70 mesh)) column chromatography or on pre-coated preparative TLC plates (SiliCycle Inc, Quebec City, Canada, 1000 μm, 20 × 20 cm). High-resolution mass spectrometric (HRMS) data was obtained on a Synapt G2 HDMS instrument operated in positive or negative ESI mode. Spectra are provided as supplementary information (
Supplementary Figure S1.1a–S1.16b).
3.2. Synthesis of (R)-(−)-Nopoic Acid Using Jones Reagent
(R)-(−)-Nopol (2.2 g, Sigma-Aldrich, St. Louis, MO, USA, 98% purity) was placed in a round bottom flask containing a magnetic stirring bar, and 20 mL of acetone was added. The solution was stirred in an ice-water bath for 10 min. To the solution, 10 mL of 3.88 M Jones reagent (Made from Fisher Scientific’s Chromium(VI) oxide, 99+% purity)in acetone was slowly added for 1 h. TLC was used to monitor the reaction progress. Once no more nopol was detected on analytical TLC, 1–2 mL of isopropanol was added to consume excess Jones reagent. The reaction turned brownish with the subsequent formation of dark green precipitate at the bottom of the flask. The reaction was washed with 20 mL of saturated NaHCO3 solution, and the crude product was extracted with 45 mL of ethyl acetate (EtOAc). The EtOAc extract was washed with brine and dried over MgSO4 and concentrated under vacuum. Pure nopoic acid was obtained by column chromatography using 85:1 CHCl3/ethyl acetate (Rf = 0.32). Pure nopoic acid was obtained in 30% yield. 1H-NMR (500 MHz, CDCl3) δ 11.48 (s, 1H, COOH), 5.42 (s, 1H, H-3′), 3.01 (dd, J = 15.7, 15.2 Hz, 2H, H-10′), 2.38 (m, 1H, H-6′), 2.33–2.19 (m, 2H, H-4′), 2.14 (td, J = 5.6, 1.6 Hz, 1H, H-5′), 2.09 (m, 1H, H-1′), 1.28 (s, 3H, H-9′), 1.22 (d, J = 8.7 Hz, 1H, H-6′), 0.83 (s, 3H, H-8′).13C-NMR (126 MHz, CDCl3) δ 178.0 (C=O), 140.5 (C-2′), 121.3 (C-3′), 45.9 (C-10′), 42.4 (C-5′), 40.5 (C-1′), 38.1 (C-7′), 31.7 (C-6′), 31.5 (C-4′), 26.2 (C-9′), 20.9 (C-8′).
3.3. Synthesis of (2-(6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)-N-(5-methoxyquinolin-8-yl Acetamide (1 and 2)
To a round bottom flask containing 3 mL of
N,
N-dimethylformamide (DMF, Fisher Scientific, >99.8%), nopoic acid (200 mg, 1.1 mmol, 1.0 eq.) was added, followed by 0.1 mL of
N,
N-diisopropylethylamine (DIEA, Sigma-Aldrich, 99.5% purity),
N,
N,
N′,
N′-Tetramethyl-
O-(1
H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, Sigma-Aldrich, 98% purity, 400.3 mg, 0.1 mmol), and HOBT (1-Hydroxybenzotriazole hydrate, 98% purity, 144.74 mg, 1.1 mmol, 1.0 eq.) [
24]. The reaction mixture was stirred at room temperature for 30 min, and 5-methoxy-8-aminoquinoline (186.3 mg, 1.1 mmol, and 1.0 eq, Sigma-Aldrich, 95% purity) was added to it. The reaction was allowed to run for 16 h. TLC results show some unreacted amine and activated nopoic acid with two prominent product spots. The compounds (
1 and
2) were purified using preparative TLC (10:1 hexane/ethyl acetate, R
f = 0.55 (
1) and 0.68 (
2)), and the overall product yield was 35%.
3.3.1. Compound 1
1H-NMR (500 MHz, CDCl3) δ 9.81 (s, 1H, NH), 8.79 (dd, J = 4.2, 1.7 Hz, 1H, H-2), 8.68 (d, J = 8.6 Hz, 1H, H-6), 8.56 (dd, J = 8.4 Hz, J = 1.7 Hz, 1H, H-4), 7.42 (dd, J = 8.4 Hz, J = 4.2 Hz, 1H, H-3), 6.83 (d, J = 8.6 Hz, 1H, H-7), 5.66 (m, 1H, H-3′), 3.98 (s, 3H, OMe), 3.32 (d, J = 14.2 Hz, 1H, H-10′), 3.13 (m, 1H, H-10′), 2.43–2.39 (m, 2H, H-6′, H-4′), 2.35–2.29 (m, 1H, H-1′), 2.22 (m, 1H, H-5′), 2.13 (m, 1H, H-4′), 1.48 (d, J = 8.7 Hz, 1H, H-6′), 1.26 (s, 3H, H-9′), 0.85 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 169.1 (C=O), 150.1 (C-5), 148.6 (C-2), 142.2 (C-2′), 139.2 (C-8), 131.1 (C-6), 128.1 (C-8a), 122.3 (C7), 120.6 (C-3), 120.4 (C-4a), 116.4 (C-3′), 104.3 (C-4), 55.8 (OMe), 46.9 (1′), 45.9 (C-10′), 40.4 (C-5′), 38.1 (C-6′), 31.9 (C-7′), 31.6 (C-4′), 26.1 (C-9′), 20.9 (C-8′). HRMS: [M + H]+: 337.1916 m/z calculated for C21H25N2O2, found 337.1910 m/z.
3.3.2. Compound 2
1H-NMR (500 MHz, CDCl3) δ 9.49 (s, 1H, NH), 8.79 (dd, J = 4.3, 1.9 Hz, 1H, H-2), 8.76 (d, J = 8.6 Hz, 1H, H-6), 8.56 (dd, J = 8.5, 1.8 Hz, 1H, H-4), 7.43 (dd, J = 8.3, 4.2 Hz, 1H, H-3), 6.84 (d, J = 8.6 Hz, 1H, H-7), 5.76 (d, J = 2.2 Hz, 1H, H-3′), 3.98 (s, 3H, OMe), 3.52 (m, 1H, H-10′), 2.88 (m, 1H, H-10′), 2.54 (m, 1H, H-6′), 2.41 (m, 1H, H-4′), 2.09 (m, 1H, H-1′), 2.01-1.88 (m, 2H, H-5′, H4′), 1.46 (d, J = 10.0 Hz, 1H, H-6′), 1.29 (s, 3H, H-9′), 0.80 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 165.2 (C=O), 149.8 (C-5), 148.5 (C-2), 139.2 (C-2′), 131.3 (C-6), 128.7 (C-8), 120.7 (C-7), 120.6 (C-8a), 117.2 (C-4a), 116.2 (C-3′), 116.0 (C-3), 104.6 (C-4), 55.9 (OMe), 53.9 (C-1′), 41.1 (C-10′), 40.7 (C-5′), 27.4 (C-6′), 26.3 (C-9′), 23.9 (C-7′), 22.4 (C-4′), 22.30 (C-8′). HRMS: [M + H]+: 337.1916 m/z calculated for C21H25N2O2, found 337.1916 m/z.
3.4. Synthesis of 2-(6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)-N-(quinolin-8-yl) Acetamide (3 and 4)
Compounds 3 and 4 were synthesized as described for 1 and 2 above and separated by preparative TLC (85:1 CHCl3/ethyl acetate, Rf = 0.81 (3) and 0.85 (4)) with 25% yield. 8-Aminoquinoline was obtained from Oakwood Chemicals, Estill, SC, USA.
3.4.1. Compound 3
1H-NMR (500 MHz, CDCl3) δ 10.06 (s, 1H, NH), 8.78 (m, 1H, H-2), 8.76 (d, J = 7.8 Hz, 1H, H-4), 8.14 (dd, J = 8.2, 1.7 Hz, 1H, H-5), 7.53 (t, J = 7.8 Hz, 1H, H-6), 7.48 (dd, J = 8.3, 1.6 Hz, 1H, H-7), 7.43 (dd, J = 8.3, 4.2 Hz, 1H, H-3), 5.67 (s, 1H, H-3′), 3.34 (m, 1H, H-10′), 3.16 (dd, J = 15.3, 1.8 Hz, 1H, H-10′), 2.45 (m, 1H, H-4′), 2.40 (m, 1H, H-6′), 2.32 (m, 1H, H-4′), 2.22 (d, J = 5.6 Hz, 1H, H-1′), 2.12 (s, 1H, H-5′), 1.48 (d, J = 8.8 Hz, 1H, H-6′), 1.26 (s, 3H, H-9′), 0.85 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 169.6 (C=O), 148.3 (C-2), 142.2 (C-2′), 138.6 (C-8), 136.4 (C-5), 134.7 (C-8a), 128.0 (C-4a), 127.7 (C-6), 122.6 (C-7), 121.7 (C-3), 121.5 (C-4), 116.5 (C-3′), 47.2 (C-1′), 46.0 (C-10′), 40.5 (C-5′), 38.3 (C-6′), 32.1 (C-4′), 31.8 (C-7′), 26.2 (C-9′), 21.1 (C-8′). HRMS: [M + H]+: 307.1810 m/z calculated for C20H23N2O, found 307.1804 m/z.
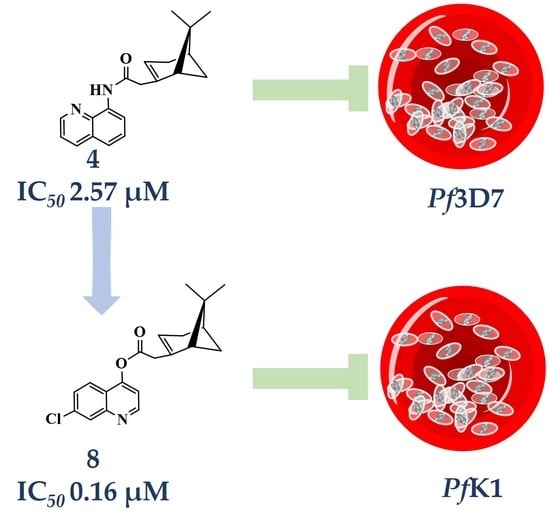
3.4.2. Compound 4
1H-NMR (500 MHz, CDCl3) δ 9.74 (s, 1H, NH), 8.85 (d, J = 7.8 Hz, 1H, H-2), 8.80 (dd, J = 4.3, 1.7 Hz, 1H, H-4), 8.16 (dd, J = 8.3, 1.7 Hz, 1H, H-5), 7.54 (t, J = 7.9 Hz, 1H, H-6), 7.49-7.47 (m, 1H, H-7), 7.45 (dd, J = 8.2, 4.2 Hz, 1H, H-3), 5.80 (s, 1H, H-3′), 3.55–3.48 (m, 1H, H-10′), 2.92–2.82 (m, 1H, H-10′), 2.57 (m, 1H, H-6′), 2.42 (m, 1H, H-4′), 2.09 (m, 1H, H-1′), 1.99 (m, 1H, H-5′), 1.93 (m, 1H, H-4′), 1.46 (d, J = 9.9 Hz, 1H, H-6′), 1.29 (s, 3H, H-9′), 0.81 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 166.4 (C=O), 165.5 (C-2), 148.1 (C-2′), 138.6 (C-8), 136.5 (C-5), 135.3 (C-8a), 128.1 (C-4a), 127.7 (C-6), 121.6 (C-7), 121.1 (C-3), 116.1 (C-3′), 115.9 (C-4), 54.0 (C-1′), 41.2 (C-5′), 40.7 (C-10′), 27.4 (C-6′), 26.3 (C-9′), 23.9 (C-7′), 22.5 (C-4′), 22.3 (C-8′). HRMS: [M + H]+: 307.1810 m/z calculated for C20H23N2O, found 307.1803 m/z.
3.5. Synthesis of 2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)-N-(quinolin-4-yl) Acetamide (5)
Compound 5 was synthesized as described for 1 and 2 above and separated by preparative TLC (20:1 CHCl3/ethyl acetate, Rf = 0.55) and 25% yield. 4-Aminoquinoline (95% purity) was obtained from Ark Pharm, Inc., Arlington Heights, IL, USA.

1H-NMR (500 MHz, CDCl3) δ 8.85 (d, J = 5.2 Hz, 1H, H-2), 8.34 (d, J = 5.2 Hz, 1H, H-3), 8.14 (d, J = 8.4 Hz, 1H, H-5), 7.74 (m, 1H, H-6), 7.73-7.69 (m, 1H, H-8), 7.58 (t, J = 7.58 Hz, 1H, H-7), 5.77 (s, 1H, H-3′), 3.37 (d, J = 15.5 Hz, 1H, H-10′), 3.18 (d, J = 15.4 Hz, 1H, H-10′), 2.53–2.48 (m, 1H, H-6′), 2.47 (m, 1H, H-5′), 2.42–2.36 (m, 1H, H-4′), 2.25–2.16 (m, 2H, H-4′), 2.19 (s, 1H, H-6′), 1.29 (s, 3H, H-9′), 1.26 (m, 1H, H-6′), 0.86 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 169.4 (C=O), 151.2 (C-2), 142.7 (C-2′), 140.3 (C-4a), 130.5 (C-8a), 129.4 (C-5), 126.6 (C-6), 123.6 (C-7), 119.7 (C-3), 118.6 (C-3′), 113.3 (C-8), 110.3 (C-4), 47.0 (C-1′), 45.8 (C-10′), 40.3 (C-5′), 38.2 (C-6′), 32.5 (C-4′), 31.7 (C-7′), 26.1 (C-9′), 21.1 (C-8′). HRMS: [M − H]-: 305.1654 m/z calculated for C20H21N2O, found 305.1655 m/z.
3.6. Synthesis of N-(7-chloroquinolin-4-yl)-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl) Acetamide (6)
Compound 6 was synthesized as described for 1 and 2 above and separated by preparative TLC (20:1 hexane/ethyl acetate, Rf = 0.78) and 36% yield. 7-Chloro-4-aminoquinoline (95% purity) was obtained from Life Chemicals, Ontario, Canada.

1H-NMR (500 MHz, CDCl3) δ 8.83 (d, J = 5.1 Hz, 1H, H-2), 8.31 (d, J = 5.2 Hz, 1H, H-3), 8.11 (s, J = 2.1 Hz, 1H, H-8), 7.65 (d, J = 9.0 Hz, 1H, H-5), 7.52 (dd, J = 9.0, 2.1 Hz, 1H, H-6), 5.79–5.75 (m, 1H, H-3′), 3.37 (d, J = 16.6 Hz, 1H, H-10′), 3.18 (d, J = 17.5 Hz, 1H, H-10′), 2.54–2.49 (m, 1H, H-6′), 2.49–2.44 (m, 1H, H-5′), 2.38 (m, 1H, H-1′), 2.21 (m, 2H, H-4′), 1.29 (s, 3H, H-9′), 1.23 (d, J = 8.8 Hz, 1H, H-6′), 0.86 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 169.5 (C=O), 152.4 (C-2), 142.8 (C-2′), 138.7 (C-4a) 136.7 (C-7), 135.6 (C-8), 129.5 (C-8a), 127.6 (C-5), 123.9 (C-6), 120.5 (C-3), 118.3 (C-3′), 110.7 (C-4), 47.1 (C-1′), 45.9 (C-10′), 40.4 (C-5′), 38.3 (C-6′), 32.7 (C-4′), 31.9 (C-7′), 26.1 (C-9′), 21.2 (C-8′). HRMS: [M − H]−: 339.1264 m/z calculated for C20H20ClN2O, found 339.1263 m/z.
3.7. Synthesis of quinolin-4-yl-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl) Acetate (7)

N-(3-Dimethylaminopropyl)-
N′-ethylcarbodiimide hydrochloride (EDCI, Sigma-Aldrich, 99% purity, 171.7 mg, 0.90 mmol, 1.3 eq.), DMAP (4-Dimethylaminopyridine, 99%, ACROS Organics™, 98.5% purity, 25.7 mg, 0.21 mmol, 0.5 eq.), and DIEA (2.04 mmol, 0.36 mL, and 3eq.) were added to a solution of 4-hydroxyquinoline (100 mg, 0.68 mmol, 1.0 eq., Ark Pharm, Inc., 97% purity) and nopoic acid (0.42 mmol, 1.2 eq.) in tetrahydrofuran (THF, Fisher Scientific, 99.9%, 2.0 mL) at 0 °C and stirred for 15 min. [
25]. The temperature was raised to 23 °C, and the reaction mixture was stirred for 3 h while monitoring by analytical TLC. The reaction appeared cloudy at first but after stirring for 1 h at room temperature, it became clear, with the formation of colorless solid precipitate. The reaction mixture was quenched with brine, extracted with EtOAc, concentrated, and separated by preparative TLC (20:1 CHCl
3/ethyl acetate, R
f = 0.55) with 25% yield.
1H-NMR (500 MHz, CD3OD) δ 8.84 (d, J = 5.0 Hz, 1H, H-2), 8.05 (d, J = 8.5 Hz, 1H, H-8), 8.02 (d, J = 8.5, 1.4 Hz, 1H, H-5), 7.81 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H, H-6), 7.64 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H, H-7), 7.44 (d, J = 5.0 Hz, 1H, H-3), 5.97 (m, 1H, H-3′), 3.34–3.28 (m, 1H, H-10′), 2.82-2.70 (m, 2H, H-10′, H-1′), 2.52 (m, 1H, H-6′), 2.11 (m, 1H, H-5′), 2.02 (m, 1H, H-4′), 1.96 (m, 1H, H-4′), 1.48 (d, J = 10.2 Hz, 1H, H-6′), 1.34 (s, 3H, H-9′), 0.84 (s, 3H, H-8′). 13C-NMR (126 MHz, CD3OD) δ 177.2 (C=O), 164.3 (C-4a), 156.5 (C-2), 151.9 (C-7), 150.5 (C-2), 131.7 (C-8), 129.2 (C-8a), 124.2 (C-6), 122.8 (C-3), 114.6 (C-3′), 111.4 (C-4′), 42.1 (C-1′), 41.6 (C-10′), 34.7 (C-5′), 28.0 (C-6′), 26.7 (C-4′), 26.4 (C-7′), 26.1 (C-9′), 24.5 (C-8′). [M + H]+: 308.1650 m/z calculated for C20H22NO2, found 308.1651 m/z.
3.8. Synthesis of 7-chloroquinolin-4-yl-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl) Acetate (8)
Compound 8 was synthesized as described for 7 above and separated by preparative TLC (20:1 hexane/ethyl acetate and Rf = 0.85) with 40% yield. 7-Chloro-4-hydroxyquinoline (99% purity) was obtained from Sigma-Aldrich.

1H-NMR (500 MHz, (CD3)2CO) δ 8.95 (d, J = 4.8 Hz, 1H, H-2), 8.10 (d, J = 2.2 Hz, 1H, H-8), 8.06 (d, J = 9.0 Hz, 1H, H-5), 7.62 (dd, J = 8.9, 2.0 Hz, 1H, H-6), 7.49 (d, J = 4.9 Hz, 1H, H-3), 5.99 (m, 1H, H-3′), 3.31 (dd, J = 20.4, 9.2 Hz, 1H, H-10′), 2.84–2.78 (m, 1H, H-10′), 2.75 (m, 1H, H-1′), 2.51 (dt, J = 10.8, 5.8 Hz, 1H, H-6′), 2.14–2.10 (m, 1H, H-5′), 2.02 (ddt, J = 10.1, 3.9, 1.9 Hz, 1H, H-4′), 1.94 (m, 1H, H-4′), 1.49 (d, J = 10.1 Hz, 1H, H-6′), 1.33 (s, 3H, H-9′), 0.84 (s, 3H, H-8′). 13C-NMR (126 MHz, (CD3)2CO) δ 176.1 (C=O), 163.4 (C-4a), 155.3 (C-2), 153.4 (C-7), 151.2 (C-2′), 136.1 (C-8), 129.0 (C-8a), 128.3 (C-5), 124.5 (C-6), 122.2 (C-3), 114.6 (C-3′), 111.3 (C-4), 54.7 (C-1′), 41.7 (C-10′), 41.1(C-5′), 27.6 (C-6′), 26.2 (C-9′), 24.1 (C-4′), 23.6 (C-7′), 22.4 (C-8′). [M + H]+: 342.1261 m/z for calculated C20H21ClNO2, found 342.1259 m/z.
3.9. Synthesis of 7-chloro-2,8-dimethylquinolin-4-yl-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl) Acetate (9)
Compound 9 was synthesized as described for 7 above and separated by preparative TLC (4:1 hexane/ethyl acetate, Rf = 0.89) with 39% yield. 7-Chloro-2,8-dimethyl-4-hydroxyquinoline (98% purity) was obtained from Sigma-Aldrich.

1H-NMR (500 MHz, (CD3)2CO) δ 7.78 (d, J = 9.0 Hz, 1H, H-6), 7.50 (d, J = 8.9 Hz, 1H, H-5), 7.31 (s, 1H, H-3), 5.96 (m, 1H, H-3′), 3.34–3.27 (m, 1H, H-10′), 2.82 (s, 3H, C-3Me), 2.77 (m, 1H, H-10′) 2.74 (m, 1H, H-1′), 2.71 (s, 3H, C-8Me), 2.51 (m, 1H, H-6′), 2.11 (m, 1H, H-5′), 2.03–1.99 (m, 1H, H-4′), 1.97–1.91 (m, 1H, H-4′), 1.48 (d, J = 10.1 Hz, 1H, H-6′), 1.33 (s, 3H, H-9′), 0.83 (s, 3H, H-8′). 13C-NMR (126 MHz (CD3)2CO) δ 175.6 (C=O), 163.5 (C-4a), 160.8 (C-2), 155.6 (C-7), 149.6 (C-2′), 135.6 (C-8), 134.9 (C-8a), 127.6 (C-5), 120.9 (C-6), 120.5 (C-3), 114.6 (C-3′), 111.4 (C-4), 54.7 (C-1′), 41.7 (C-10′), 41.1 (C-5′), 27.6 (C-6′), 26.2 (C-9′), 25.8 (C-4′), 24.1 (C-2Me), 23.5 (C-7′), 22.4 (C-8′), 14.7 (C-8Me). [M + H]+: 370.1574 m/z for calculated C22H25ClNO2, found 370.1574 m/z.
3.10. Synthesis of 6-fluoro-2-(trifluoromethyl)quinolin-4-yl-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2yl) Acetate (10)
Compound 10 was synthesized as described for 7 above and separated by preparative TLC (4.5:1 hexane/ethyl acetate, Rf = 0.78) with 33% yield. 6-Fluoro-4-hydroxy-2-(trifluoromethyl)quinoline (98% purity) was obtained from Sigma-Aldrich.
1H-NMR (500 MHz, CDCl3) δ 8.25 (m, 1H, H-7), 7.78 (s, 1H, H-3), 7.67 (m, 1H, H-8), 7.60 (s, 1H, H-5), 5.91 (s, 1H, H-3′), 3.33 (dd, J = 20.5, 9.3 Hz, 1H, H-10′), 2.88–2.73 (m, 1H, H-10′), 2.71 (t, J = 5.4 Hz, 1H, H-1′), 2.50 (m, 1H, H-6′), 2.16 (m, 1H, H-5′), 2.05–1.93 (m, 2H, H-4′), 1.47 (d, J = 10.2 Hz, 1H, H-6′), 1.34 (s, 3H, H-9′), 0.85 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 177.7 (C=O), 162.4 (C-6), 146.2 (C-2), 142.2 (C-2′), 133.1 (C-8), 133.0 (C-4a), 124.2 (CF3), 121.9 (C-7), 121.7 (C-8a), 110.1 (C-3′), 109.8 (C-5), 105.8 (C-3), 105.6 (C-4), 54.4 (C-1′), 41.4 (C-10′), 40.4 (C-5′), 27.4 (C-6′), 26.2 (C-9′), 23.7 (C-4′), 23.4 (C-7′), 22.4 (C-8′). [M + Na]+: 416.1250 m/z calculated for C21H19F4NO2Na, found: 416.1245 m/z.
3.11. Synthesis of 7-chloro-8-methylquinolin-4-yl-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl) Acetate (11)
Compound 11 was synthesized as described for 7 above and separated by preparative TLC (5:1 hexane/ethyl acetate, Rf = 0.82) with 38% yield. 7-Chloro-4-hydroxy-8-methylquinoline (98% purity) was obtained from Sigma-Aldrich.

1H-NMR (500 MHz, CDCl3) δ 8.92 (d, J = 4.9 Hz, 1H, H-2), 7.79 (d, J = 8.9 Hz, 1H, H-6), 7.51 (d, J = 9.0 Hz, 1H, H-5), 7.33 (d, J = 4.9 Hz, 1H, H-3), 5.90 (t, J = 2.3 Hz, 1H, H-3′), 3.36–3.27 (m, 1H, H-10′), 2.88 (s, 3H, C-8Me), 2.76 (m, 1H, H-10′), 2.68 (t, J = 5.3 Hz, 1H, H-1′), 2.48 (m, 1H, H-6′), 2.13 (s, 1H, H-5′), 2.03-1.98 (m, 1H, H-4′), 1.93 (m, 1H, H-4′), 1.46 (d, J = 10.1 Hz, 1H, H-6′), 1.25 (s, 3H, H-9′), 0.84 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 175.8 (C=O), 163.0 (C-4a), 154.6 (C-2), 150.5 (C-7), 149.8 (C-2′), 135.4 (C-8), 135.0 (C-8a), 128.0 (C-5), 121.0 (C-6), 119.9 (C-3), 112.8 (C-3′), 110.5 (C-4), 54.2 (C-1′), 41.1 (C-10′), 40.35 (C-5′), 31.92 (C-7′), 27.24 (C-6′), 26.02 (C-9′), 23.53 (C-4′), 22.23 (C-8′), 14.77 (C-8Me). [M + Na]+: 378.1237 m/z for calculated for C21H22ClNO2Na found 378.1234 m/z.
3.12. Synthesis of 2,8-bis(trifluoromethyl)quinolin-4-yl-2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl) Acetate (12)
Compound 12 was synthesized as described for 7 above and separated by preparative TLC (4.5:1 hexane/ethyl acetate, Rf = 0.76) with 31% yield. 2,8-Bis(trifluoromethyl)-4-quinolinol (99% purity, Acros Organics) was obtained from Thermo Fisher Scientific, Waltham, MA.

1H-NMR (500 MHz, CDCl3) δ 8.28 (d, J = 8.6 Hz, 1H, H-7), 8.18 (d, J = 7.4 Hz, H-5), 7.84 (s, 1H, H-3), 7.74–7.67 (m, 1H, H-6), 5.92 (s, 1H, H-3′), 3.38–3.28 (m, 1H, H-10′), 2.80 (m, 1H, H-10′), 2.71 (m, 1H, H-1′), 2.50 (m, 1H, H-6′), 2.16 (m, 1H, H-5′), 2.08–2.00 (m, 1H, H-4′), 1.96 (m, 1H, H-4′), 1.47 (d, J = 10.3 Hz, 1H, H-6′), 1.34 (s, 3H, H-9′), 0.85 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 178.0 (C=O), 162.4 (C-6), 156.2 (C-2), 149.3 (C-8a), 145.4 (C-2′), 129.6 (C-8), 129.6 (C-4a), 127.21 (C-4), 126.3 (C-5), 124.7 (CF3), 124.0 (CF3), 122.6 (C-7), 122.5 (C-3), 110.1 (C-3′), 54.5 (C-1′), 41.4 (C-10′), 40.4 (C-5′), 27.4 (C-6′), 26.2 (C-9′), 23.7 (C-4′), 23.5 (C-8′), 22.4 (C-8′). [M + Na]+: 466.1218 m/z calculated for C22H19F6NO2Na, found 466.1220 m/z.
3.13. Synthesis of 2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl-7-chloro-2-methylquinoline-4-carboxylate (13)
EDCI (112.1 mg, 0.58 mmol, and 1.3 eq.) was added to a solution of 7-chloro-2 methyl-4-quinolinecarboxylic acid (99.7 mg, 0.45 mmol, 1.0 eq., Sigma-Aldrich, 98% purity) in THF (1.0 mL) at 0 °C and stirred for 15 min. Nopol (90.0 mg, 0.54 mmol, 1.2 eq.) in THF (1.0 mL) was added, followed by DIEA (2.04 mmol, 0.36 mL, 3 eq.). The reaction mixture was stirred at room temperature for 3 h while monitoring by analytical TLC. The reaction mixture was quenched with brine, extracted with EtOAc, concentrated, and separated by preparative TLC (3:1 hexane/ethyl acetate, Rf = 0.92) with 35% yield.

1H-NMR (500 MHz, CDCl3) δ 8.69 (dd, J = 9.1, 1.5 Hz, 1H, H-6), 8.06 (d, J = 1.9 Hz 1H, H-8), 7.78 (d, J = 1.6 Hz 1H, H-3), 7.51 (d, J = 9.1 Hz, 1H, H-5), 5.40 (m, 1H, H-3′), 4.45 (m, 2H, H-11′), 2.77 (d, J = 1.7 Hz, 3H, C-2Me), 2.49 (s, 2H, H-10′), 2.43–2.38 (m, 1H, H-6′), 2.33-2.20 (m, 2H, H-4′), 2.15–2.09 (m, 2H, H-5′, H-1′), 1.28 (s, 3H, H-9′), 1.18 (d, J = 8.6 Hz, 1H, H-6′), 0.85 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 166.1 (C=O), 160.0 (C-3), 149.5 (C-2), 144.0 (C-2′), 135.8 (C-8), 135.3 (C-8a), 128.3 (C-5), 128.2 (C-6), 127.1 (C-4), 123.6 (C-4a), 122.0 (C-3), 119.5 (C-3′), 64.3 (C-11′), 45.9 (C-10′), 40.8 (C-1′), 38.2 (C-5′), 36.1 (C-7′), 31.9 (C-6′), 31.6 (C-4′), 26.4 (C-9′), 25.3 (C-2Me), 21.3 (C-8′). [M + H]+: 370.1574 m/z calculated for C22H25ClNO2, found 370.1575 m/z.
3.14. Synthesis of 2-((1S,5R)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl-2-chloroquinoline-3-carboxylate (14)
Compound 14 was synthesized as described for 13 above and separated by preparative TLC preparative TLC by preparative TLC (40:1 CHCl3/Et2O, Rf = 0.72) with 50% yield. The acid, 2-chloroquinoline-3-carboxylic acid (97% purity) was obtained from Sigma-Aldrich.

1H-NMR (500 MHz, CD3OD) δ 8.76 (s, 1H, H-4), 8.01 (d, J = 8.1 Hz, 1H, H-5), 7.94 (d, J = 8.5 Hz, 1H, H-8), 7.88 (dd, J = 8.5, 6.9 Hz, 1H, H-7), 7.68 (t, J = 7.6 Hz, 1H, H-6), 5.39 (s, 1H, H-3′), 4.38 (qt, J = 10.9, 6.8 Hz, 2H, H-11′), 2.46 (m, 2H, H-10′), 2.40 (dt, J = 8.7, 5.7 Hz, 1H, H-6′), 2.30-2.17 (m, 2H, H-4′), 2.17-2.12 (m, 1H, H-5′), 2.06 (m, 1H, H-1′), 1.27 (s, 3H, H-9′), 1.17 (d, J = 8.6 Hz, 1H, H-6′), 0.84 (s, 3H, H-8′). 13C-NMR (126 MHz, CD3OD) 165.70 (C=O), 149.2 (C-2), 148.4 (C-4a), 145.5 (C-3), 143.0 (C-3′), 134.1 (C-6), 129.9 (C-8), 129.3 (C-8a), 128.7 (C-4), 127.3 (C-5), 125.9 (C-7), 120.2 (C-3′), 65.3 (C-11′), 46.9 (C-10′), 42.0 (C-5′), 38.9 (C-1′), 36.9 (C-6′), 32.6 (C-7′), 32.3 (C-4′), 26.7 (C-9′), 21.6 (C-8′). [M + H]+: 356.1417 m/z calculated for C21H23ClNO2, found 356.1409 m/z.
3.15. Synthesis of 2-((1R,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl-1H-indole-2-carboxylate (15)
Compound 15 was synthesized as described for 13 above and separated by preparative TLC (40:1 CHCl3/Et2O, Rf = 0.72) with 50% yield. Indole 2-carboxylic acid (98% purity) was obtained from Sigma-Aldrich.
1H-NMR (500 MHz, CD3OD) δ 7.61 (d, J = 8.1 Hz, 1H, H-7), 7.43 (d, J = 8.4 Hz, 1H, H-4), 7.24 (t, J = 7.7 Hz, 1H, H-5), 7.11 (s, 1H, H-3), 7.06 (t, J = 7.6 Hz, 1H, H-6), 5.39 (s, 1H, H-3′), 4.38–4.27 (m, 2H, H-11′), 2.47–2.39 (m, 3H, H-10′, H-6′), 2.31–2.19 (m, 2H, H-4′), 2.17 (t, J = 5.6 Hz, 1H, H-5′), 2.07 (s, 1H, H-1′), 1.28 (s, 3H, H-9′), 1.19 (d, J = 8.6 Hz, 1H, H-6′), 0.86 (s, 3H, H-8′). 13C-NMR (126 MHz, CD3OD) δ 163.5 (C=O), 145.8 (C-3′), 139.0 (C-7a), 128.6 (C-3a), 126.0 (C-2), 123.1 (C-4), 121.3 (C-6), 120.2 (C-5), 113.3 (C-2′), 109.2 (C-7), 64.2 (C-11′), 47.0 (C-10′), 42.0 (C-1′), 40.0 (C-5′), 37.1 (C-7′), 32.6 (C-6′), 32.4 (C-4′), 26.7 (C-9′), 24.2 (C-9), 21.6 (C-8) HRMS: [M + H]+: 310.1807 m/z calculated for C20H24NO2, found 310.1806 m/z.
3.16. Synthesis of 2-((1S,2S,4R,6S)-7,7-dimethyl-3-oxatricyclo[4.1.1.0 2,4]octan-2-yl)ethyl-7-chloro-2-methylquinoline-4-carboxylate (16)
Nopol (500 mg, 3.0 mmol, 1.eq.) was added to a round 100 mL bottom flask, followed by 4.0 mL of ethyl acetate, 4.0 mL of deionized H
2O, and 8.0 mL of acetone, and stirred at room temperature. NaHCO
3(s) (9.0 mmol, 756 mg, 3.0 eq.) was added to the mixture and stirred for 15 min. Oxone salt (3.6 mmol, 2.4 g, 1.2 eq.), dissolved in 1.0 mL of water, was added dropwise for 1 h, and analytical TLC was used to monitor the reaction for 2.5 h [
26]. The reaction was quenched with 3.0 mL of H
2O. The organic layer was extracted with 15.0 mL CHCl
3 (3 times), dried over MgSO
4(s), and concentrated. The crude colorless solid obtained was separated by preparative TLC (10:1 CHCl
3/ethyl acetate, R
f = 0.37) with 40% yield of nopol oxide. EDCI (112.1 mg, 0.58 mmol, 1.3 eq.), DMAP (27.5 mg, 0.23 mmol, 0.5 eq.), and DIEA (2.04 mmol, 0.36 mL, 3 eq.) were added to a solution of 7-chloro-2-methyl-4-quinolinecarboxylic acid (99.7 mg, 0.45 mmol, 1.0 eq., Sigma-Aldrich, 98%) and nopol oxide (151.9 mg, 0.54 mmol, and 1.2 eq.) in THF (2.0 mL) at 0 °C and stirred for 15 min. The temperature was raised to 23 °C, and the mixture was stirred for 3 h while monitoring by analytical TLC, quenched with brine, extracted with EtOAc, and separated by preparative TLC (4.5:1 hexane/ethyl acetate, R
f = 0.59) with 49% yield.

1H-NMR (500 MHz, CDCl3) δ 8.68 (d, J = 9.1 Hz, 1H, H-5), 8.06 (d, J = 2.2 Hz, 1H, H-8), 7.79 (s, 1H, H-3), 7.53 (d, J = 9.1, 2.2 Hz, 1H, H-6), 4.57–4.44 (m, 2H, H-11′), 3.21 (d, J = 4.2, 1.5 Hz, 1H, H-3′), 2.78 (s, 3H, C-2Me), 2.29 (dt, J = 14.8, 6.0 Hz, 1H, H-6′), 2.16 (s, 1H, H-5′), 2.08–2.02 (m, 2H, H-4′, H-1′), 1.94 (m, 1H, H-10′), 1.77 (m, 1H, H-10′), 1.67 (d, J = 9.8 Hz, 1H, H-6′), 1.32 (s, 3H, H-9′), 0.98 (s, 3H, H-8′). 13C-NMR (126 MHz, CDCl3) δ 166.0 (C=O), 160.0 (C-3), 149.5 (C-2), 135.9 (C-8), 135.1 (C-8a), 128.3 (C-5), 128.2 (C-6), 127.1 (C-4), 123.6 (C-4a), 121.9 (C-3), 61.9 (C-3′), 62.0 (C-11′), 55.5 (C-2′), 43.7 (C-10′), 40.8 (C-1′), 40.0 (C-5′), 34.0 (C-7′), 27.7 (C-6′), 26.9 (C-4′), 25.9 (C-9′), 25.5 (C-2Me), 20.3 (C-8′). [M + H]+: 386.1523 m/z for calculated C22H25ClNO3, found 386.1525 m/z.
3.17. Plasmodium falciparum Assay
a.
Pf3D7: 100 μL of complete media (Sigma-Aldrich’s RPMI 1640, 25 mM HEPES, 10 μg/mL gentamicin, 0.5 mM hypoxanthine supplemented with NaHCO
3 and 0.5% Albumax II, 37 °C) containing compounds (50–0.3 µM) or 100 µM chloroquine were added to 96 well plates in triplicates. This was followed by the addition of 100 µL erythrocytic asexual culture of
Pf3D7 (10% hematocrit and 0.25% ring stage parasitemia), maintained at atmospheric conditions of 1% oxygen, 5% carbon dioxide, and 94% nitrogen, and allowed to grow for 96 h at 37 °C. The cultures were then frozen at −80 °C overnight and thawed at 37 °C for 4 h. Aliquots (100 µL) from each well were transferred to black 96-well plates, 100 μL of 2× SYBR Green (Molecular Probes) in lysis buffer (20 mM Tris at pH 7.5, 5 mM EDTA, 0.008% saponin, 0.08% Triton X-100) was added to each well and mixed thoroughly with a pipette. The plates were incubated in the dark at room temperature for 1 h and read at λ
ex 485 nm and λ
em 530 nm [
27]. Infected and untreated red blood cells (iRBC) cultures served as negative control. Percent inhibitory activities were calculated using (=100 − 100*((Sample − 100 µM CQ)/(iRBC − 100 µM CQ)) and used to plot sigmoidal inhibition curves to obtain the EC
50 values.
b.
PfNH54 and PfK1: The growth inhibitory activities of the compounds against erythrocytic stages of
PfNH54 and
PfK1 were determined using the [
3H]-hypoxanthine incorporation assay as previously reported [
28]. Parasites in RPMI 1640 medium containing 5% Albumax (without hypoxanthine) were treated with compounds (50–0.3 µM) in 96 well plates for 48 h at 37 °C in 92% N
2, 5% CO
2, and 3% O
2.
3H-hypoxanthine (0.5 μCi) was added to each well, incubated for an additional 24 h, harvested onto glass-fiber filters, and washed with distilled water. Radioactivity at each compound concentration was counted using a Betaplate™ liquid scintillation counter (Wallac, Zurich), and the results were recorded as counts per minute (CPM) per well. The CPMs were expressed as a percentage of the CPMs from the untreated controls and used to estimate the EC
50 from sigmoidal inhibition curves. Artemisinin and CQ were used as positive controls.
3.18. Cytotoxicity Assay
Human hepatocarcinoma cell line (Hep G2, ATCC CRL-11997TM) was used for cytotoxicity studies as previously described [
29]. The cells were grown in complete medium (DMEM:F12 containing
l-glutamine and sodium bicarbonate, 10% FBS, 1% penicillin/streptomycin, Thermo Fisher Scientific) at 37 °C in 5% CO
2 environment. Cells (198 µL, 5 × 105/mL) were seeded into 96-well plates and incubated overnight. The cells were treated with the compounds (160–1.25 µM, in triplicates) or DMSO (1%) for 72 h. The cell medium was removed and replaced with DMEM:F12 medium containing MTT (0.5 mg/mL) was added to the cells and incubated for 1.5 h. The MTT-containing medium was gently removed and replaced with DMSO (200 µL/well). The contents of each well were then repeatedly mixed with a multichannel pipette and incubated for 10 min. The plates were read at 570 nm. DMSO-treated (1%) cells were used as negative assay control and SDS (10%) was used as assay positive control. Percent cell viabilities were expressed as a percentage of the mean viability of DMSO-treated (1%) cells.






















{kind=link}
{kind=link}















