
Fast Identification of Adverse Drug Reactions (ADRs) of Digestive and Nervous Systems of Organic Drugs by In Silico Models

Abstract
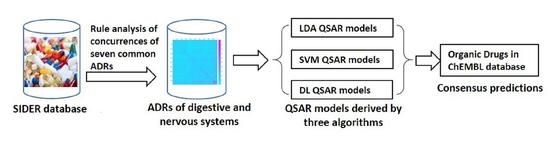
:
1. Introduction
2. Results and Discussions
2.1. Common ADRs of Marketed Organic Drugs
2.2. Associations between ADRs of Organic Drugs
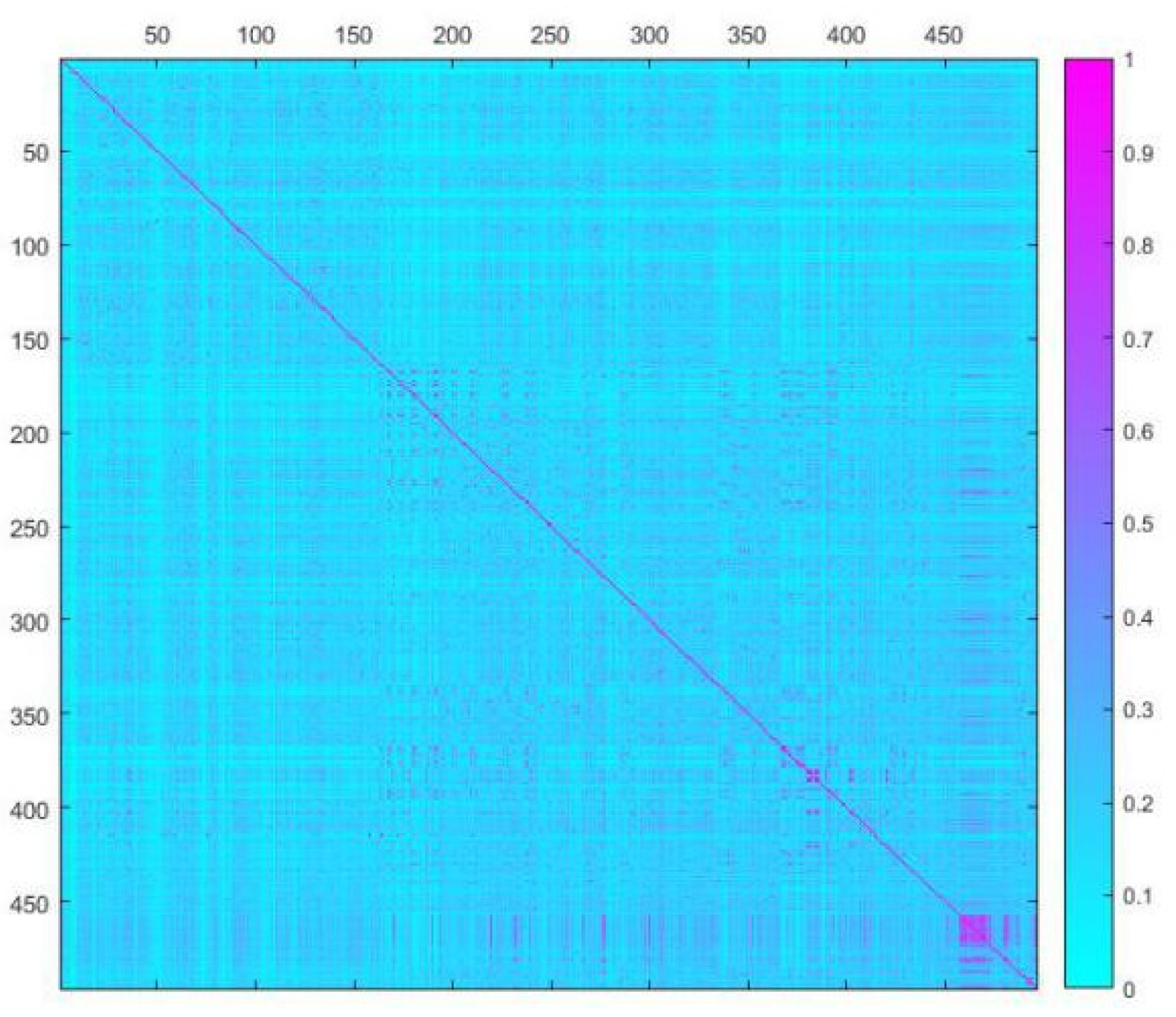
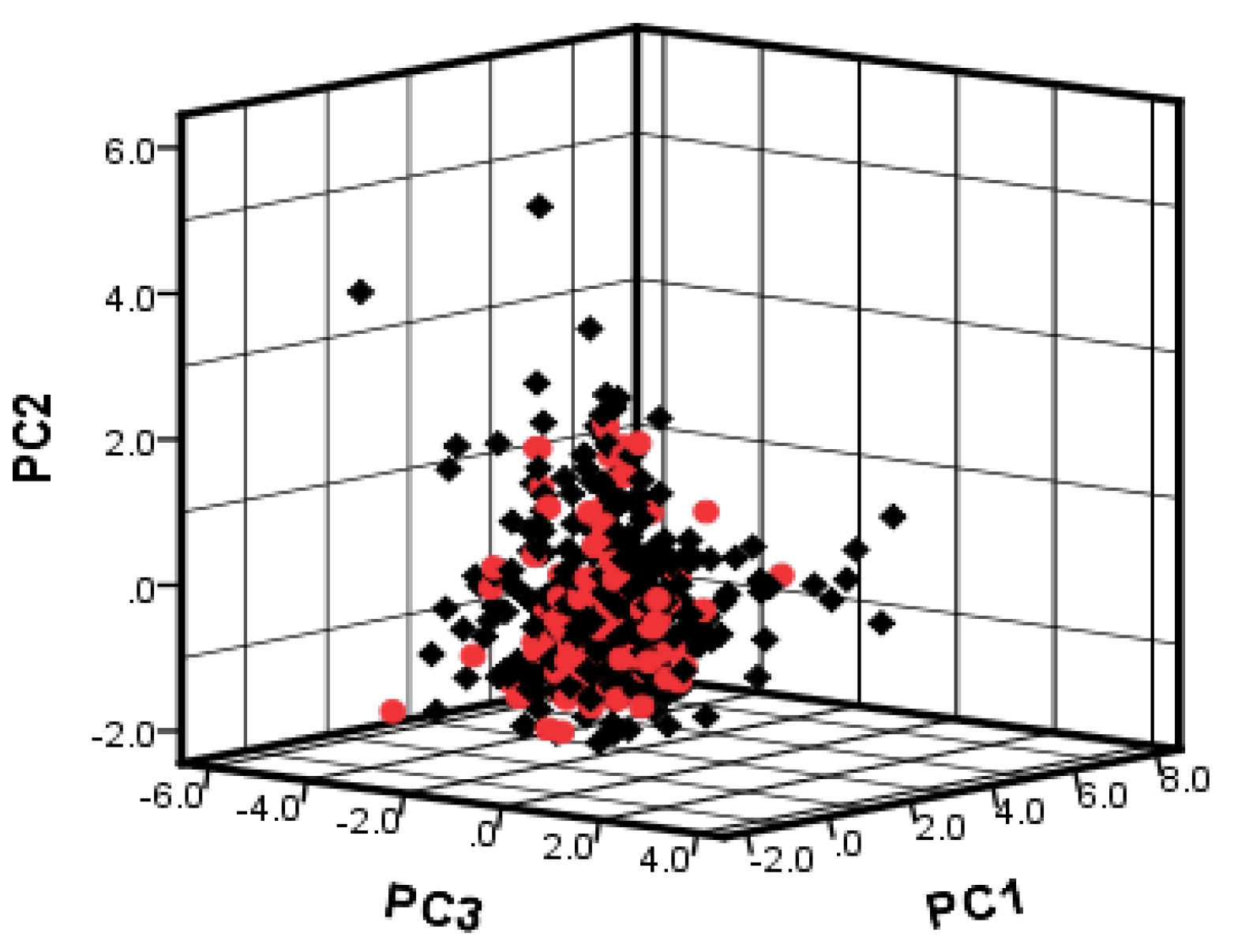
2.3. Dataset Splitting
2.4. SW-LDA Model Results of ADRs of Digestive System
0.07 × vsurf_DD13 − 1.472 × a_nP + 0.358 × MNDO_LUMO + 0.786 × reactive + 0.768 × a_nI −
0.296 × opr_violation − 2.593 × vsurf_CW1 − 0.007 × SlogP_VSA5 − 0.105 × vsurf_IW7 + 6.105
2.5. SW-LDA Model Results of ADRs of Nervous System
3.498 × Q_VSA_FPPOS + 0.153 × MNDO_dipole + 1.927
2.6. Interpretation of the Descriptors
2.7. Results of SVM Models
2.8. Results of Deep Learning Models
2.9. Comparison of Different Approaches and Consensus Prediction
3. Methods and Materials
3.1. Association Analysis of Common Side Effects of Drugs
3.2. Molecular Descriptor Calculation
3.3. Data Splitting
3.4. QSAR Model Approach
3.4.1. Stepwise Linear Discriminant Analysis
3.4.2. Support Vector Machine (SVM)
3.4.3. Deep Learning (DL)
3.5. Performance Evaluation
3.6. Model Application
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Michael, K.; Ivica, L.; Juhl, J.L.; Peer, B. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar]
- DrugBank. Available online: https://www.drugbank.ca/ (accessed on 10 May 2019).
- Watkins, P.B. Insight into hepatotoxicity: The troglitazone experience. Hepatology 2005, 41, 229–230. [Google Scholar] [CrossRef]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of drug induced liver injury (DILI)—Current issues and future perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef]
- Hosey, C.M.; Benet, L.Z. Experimental ADME and Toxicology. In Comprehensive Medicinal Chemistry III, 3rd ed.; Chackalamannil, S., Rotella, D., Ward, S., Eds.; Elsevier: Amsterdam, The Netherland, 2017. [Google Scholar]
- FDA. Guidance for industry integrated summaries of effectiveness and safety: Location within the common technical document; FDA: Rockville, MD, USA, 2009; pp. 532–536. [Google Scholar]
- Walker, R.M.; McElligott, T.F. Furosemide induced hepatotoxicity. J. Pathol. 1981, 135, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Markovic, M.; Zur, M.; Fine-Shamir, N.; Haimov, E.; González-Álvarez, I.; Dahan, A. Segmental-dependent solubility and permeability as key factors guiding controlled release drug product development. Pharmaceutics 2020, 12, 295. [Google Scholar] [CrossRef] [Green Version]
- Lo Piparo, E.; Fratev, F.; Lemke, F.; Mazzatorta, P.; Smiesko, M.; Fritz, J.I.; Benfenati, E. QSAR models for Daphnia magna toxicity prediction of benzoxazinone allelochemicals and their transformation products. J. Agric. Food Chem. 2016, 54, 1111–1115. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, B.; Wang, C. Hepatotoxicity evaluation of traditional Chinese medicines using a computational molecular model. Clin. Toxicol. 2017, 55, 996–1000. [Google Scholar] [CrossRef]
- Huang, S.H.; Tung, C.W.; Fülöp, F.; Li, J.H. Developing a qsar model for hepatotoxicity screening of the active compounds in traditional Chinese medicines. Food Chem Toxicol. 2015, 78, 71–77. [Google Scholar] [CrossRef]
- Ancuceanu, R.; Hovanet, M.V.; Anghel, A.I.; Furtunescu, F.; Dinu, M. Computational models using multiple machine learning algorithms for predicting drug hepatotoxicity with the dilirank dataset. Int. J. Mol. Sci. 2020, 21, 2114. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Guo, P.; Zhou, Y.; Zhou, J.; Wang, Q.; Zhang, F.; Fang, J.; Cheng, F. Deep learning-based prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model 2019, 59, 1073–1084. [Google Scholar] [CrossRef]
- Satalkar, V.; Kulkarni, S.; Joshi, D. QSAR based analysis of fatal drug induced renal toxicity. J. Comput. Methods Mol. Des. 2015, 5, 24–32. [Google Scholar]
- Sun, Y.; Shi, S.; Li, Y.; Wang, Q. Development of quantitative structure-activity relationship models to predict potential nephrotoxic ingredients in traditional chinese medicines. Food Chem. Toxicol. 2019, 128, 163–170. [Google Scholar] [CrossRef]
- Ordonez, C.; Zhao, K. Evaluating association rules and decision trees to predict multiple target attributes. Intell. Data Anal. 2011, 15, 173–192. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Yang, X.; Lai, X.; Gao, Y. Structural Investigation for optimization of anthranilic acid derivatives as partial fxr agonists by in silico approaches. Int. J. Mol. Sci. 2016, 17, 536. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Yang, F.; Kang, J.; Gan, H.; Lai, X.; Gao, Y. Discovery of molecular mechanism of a clinical herbal formula upregulating serum HDL-c levels in treatment of metabolic syndrome by in vivo and computational studies. Bioorg. Med. Chem. Lett. 2018, 28, 174–180. [Google Scholar] [CrossRef]
- Guo, J. Simultaneous variable selection and class fusion for high-dimensional linear discriminant analysis. Biostatistics 2010, 11, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vapnik, V. The Support Vector Method of Function Estimation. In Nonlinear Modeling; Springer: Boston, MA, USA, 1998; pp. 55–85. [Google Scholar]
- Cheng, F.; Guo, T.; Liu, C.; Wang, Y.; Huang, B. Identification of the thief zone using a support vector machine method. Processes 2019, 7, 373. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Khumprom, P.; Yodo, N. A data-driven predictive prognostic model for lithium-ion batteries based on a deep learning algorithm. Energies 2019, 12, 660. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, S.; Liu, Y.; Zhang, Y.; Zhou, F. Integration of 24 feature types to accurately detect and predict seizures using scalp EEG signals. Sensors 2018, 18, 1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, A. Measuring diagnostic and predictive accuracy in disease management: An introduction to receiver operating characteristic (ROC) analysis. J. Eval. Clin. Pract. 2006, 12, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | ADR Categories | Main Items of ADRs | No. of Drugs |
|---|---|---|---|
| 1 | Digestive system reactions | Abdominal pain; diarrhea; abdominal bloating; constipation; gastrointestinal disorder; nausea; anorexia; digestion impaired; gastrointestinal hemorrhage; flatulence; abnormal feces; abdominal pain upper; vomiting; gastroesophageal reflux disease, etc. | 418 |
| 2 | Nervous reactions | Abnormal involuntary movements; convulsion; headache; balance disorder; dizziness; arthralgia; depression; paresthesia; ataxia; amnesia; disorder sight; feeling abnormal; deafness; abnormal behavior; nervous symptoms; etc. | 442 |
| 3 | Allergy reactions | Pruritus; dermatitis; dyspnea; injection site pain; rash maculo-papular; erythema; acne; application site irritation; anaphylactic shock; injection site reaction; application site erythema; allergic contact dermatitis; seborrheic dermatitis; blisters; etc. | 259 |
| 4 | Hepatotoxic reactions | Alanine aminotransferase increased; liver function test abnormal; hepatic encephalopathy; hepatic enzyme abnormal; transaminases increased; hepatitis; hepatotoxicity; hepatic failure; etc. | 75 |
| 5 | Cardiovascular reactions | Atrial fibrillation; cardiac output decreased; bradycardia; angina pectoris; arrhythmia; acute coronary syndrome; arterial insufficiency; cardiac disorder; angiopathy; atrial fibrillation; cardiac murmur; cardiotoxicity; blood triglycerides increased; ventricular arrhythmia; etc. | 85 |
| 6 | Urinary reactions | Urinary tract infection; dysuria; bladder pain; micturition disorder; urinary hesitation; nephropathy toxic; renal failure; renal tubular acidosis; blood creatinine increased; urinary retention; albuminuria; hematuria; urethral disorder; chronic kidney disease; protein in urine; etc. | 99 |
| 7 | Hematologic reactions | Thrombocytopenia; anemia; coagulopathy; agranulocytosis; eosinophilia; hemoglobin decreased; platelet count decreased; activated partial thromboplastin time prolonged; white blood cell count decreased; etc. | 108 |
| Consequent | Antecedent | Instances | Support% | Confidence% | Rule Support% | Lift |
|---|---|---|---|---|---|---|
| ADRs of nervous system | ADRs of digestive system | 418 | 73.85 | 81.1 | 59.89 | 1.04 |
| Descriptors | Chemical Meaning | Tolerance | Wilks’ Lambda | VIF | F to Remove | Non-Standardized Coefficient | p-Value |
|---|---|---|---|---|---|---|---|
| PEOE_VSA-4 | Total negative van der Waals surface area | 0.771 | 0.853 | 1.297 | 71.489 | 0.078 | 0.00 |
| PEOE_VSA_FNEG | Fractional negative van der Waals surface area | 0.773 | 0.736 | 1.294 | 11.259 | 3.495 | 0.00 |
| vsurf_CP | Critical packing parameter | 0.697 | 0.745 | 1.435 | 15.798 | −3.235 | 0.00 |
| vsurf_DD13 | Contact distances of lowest hydrophobic energy | 0.908 | 0.737 | 1.101 | 11.578 | 0.07 | 0.00 |
| a_nP | Number of phosphorus atoms | 0.898 | 0.74 | 1.114 | 13.367 | −1.472 | 0.00 |
| MNDO_LUMO | The energy (eV) of the lowest unoccupied molecular orbital calculated using the MNDO Hamiltonian | 0.757 | 0.75 | 1.321 | 18.153 | 0.358 | 0.00 |
| reactive | Indicator of the presence of reactive groups | 0.95 | 0.733 | 1.052 | 9.782 | 0.786 | 0.00 |
| a_nI | Number of iodine atoms | 0.972 | 0.728 | 1.029 | 7.317 | 0.768 | 0.00 |
| opr_violation | The number of violations of Oprea’s lead-like test | 0.418 | 0.739 | 2.392 | 12.911 | −0.296 | 0.00 |
| vsurf_CW1 | Capacity factor | 0.417 | 0.74 | 2.392 | 13.506 | −2.593 | 0.00 |
| SlogP_VSA5 | The subdivided surface areas | 0.715 | 0.725 | 1.399 | 5.435 | −0.007 | 0.00 |
| vsurf_IW7 | Hydrophilic integy moment | 0.946 | 0.723 | 1.057 | 4.663 | −0.105 | 0.00 |
| Constant | 6.105 |
| QSAR Models | Accuracy Training Set | Accuracy Test Set | Accuracy 10-Fold CV | Total Accuracy | BACC | Sensitivity | Specificity | ROC AUC | |
|---|---|---|---|---|---|---|---|---|---|
| Digestive system | LDA | 76.58% | 72.65% | 72.89% | 75.65% | 73.9% | 80.5% | 67.3% | 0.815 |
| SVM | 98.42% | 76.92% | 75.79% | 93.36% | 97.61% | 99.63% | 95.58% | 0.989 | |
| DL | 87.89% | 78.63% | 78.42% | 85.71% | 83.73% | 94% | 73.45% | 0.915 | |
| Nervous system | LDA | 69.21% | 64.1% | 68.68% | 68% | 67.4% | 70.8% | 64% | 0.695 |
| SVM | 80.26% | 83.76% | 76.05% | 81.09% | 62.94% | 95.53% | 30.34% | 0.784 | |
| DL | 82.89% | 81.2% | 73.68% | 82.49% | 72.84% | 91.75% | 53.93% | 0.788 | |
| Descriptors | Chemical Meaning | Tolerance | Wilks’ Lambda | VIF | F to Remove | Non-standardized Coefficient | p-Value |
|---|---|---|---|---|---|---|---|
| PEOE_VSA+5 | Total positive van der Waals surface area | 0.53 | 0.932 | 1.887 | 7.712 | 0.03 | 0.004 |
| vsurf_IW7 | Hydrophilic integy moment | 0.949 | 0.932 | 1.054 | 7.875 | −0.244 | 0.001 |
| std_dim2 | Standard dimension 2 | 0.311 | 0.939 | 3.215 | 10.599 | −0.697 | 0.00 |
| SMR_VSA3 | Subdivided surface areas | 0.304 | 0.933 | 3.289 | 8.231 | 0.024 | 0.00 |
| Q_VSA_FPPOS | Fractional positive polar van der Waals surface area | 0.917 | 0.925 | 1.091 | 4.821 | −3.498 | 0.00 |
| MNDO_dipole | The dipole moment calculated using the MNDO Hamiltonian | 0.888 | 0.923 | 1.126 | 4.261 | 0.153 | 0.00 |
| Constant | 1.927 |
| Descriptors | PEOE_ VSA+5 | Q_VSA_ FPPOS | MNDO_ dipole | SlogP_ VSA3 | vsurf_ IW7 | std_ dim2 |
|---|---|---|---|---|---|---|
| reactive | 0.127 | −0.027 | 0.058 | 0.169 | −0.078 | 0.129 |
| a_nI | 0.108 | 0.056 | −0.073 | 0.075 | −0.054 | 0.049 |
| a_nP | −0.044 | 0.166 | -0.022 | 0.036 | −0.021 | 0.038 |
| PEOE_VSA-4 | −0.047 | 0.173 | 0.143 | 0.014 | 0.002 | −0.018 |
| PEOE_VSA_FNEG | −0.071 | −0.714 | −0.076 | −0.244 | 0.131 | −0.131 |
| opr_violation | 0.453 | 0.152 | −0.205 | 0.488 | −0.109 | 0.698 |
| MNDO_LUMO | −0.001 | 0.05 | −0.134 | 0.014 | −0.007 | 0.004 |
| SlogP_VSA5 | 0.253 | 0.268 | −0.131 | 0.292 | −0.012 | 0.412 |
| vsurf_CP | −0.263 | −0.4 | −0.107 | −0.203 | 0.086 | −0.211 |
| vsurf_CW1 | −0.308 | 0.247 | 0.23 | −0.373 | −0.038 | −0.591 |
| vsurf_DD13 | 0.415 | 0.053 | −0.163 | 0.499 | −0.008 | 0.47 |
| vsurf_IW7 | −0.07 | −0.185 | 0.102 | −0.08 | 1 | −0.074 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Yang, Z.; Gao, Y.; Li, C. Fast Identification of Adverse Drug Reactions (ADRs) of Digestive and Nervous Systems of Organic Drugs by In Silico Models. Molecules 2021, 26, 930. https://doi.org/10.3390/molecules26040930
Chen M, Yang Z, Gao Y, Li C. Fast Identification of Adverse Drug Reactions (ADRs) of Digestive and Nervous Systems of Organic Drugs by In Silico Models. Molecules. 2021; 26(4):930. https://doi.org/10.3390/molecules26040930
Chicago/Turabian StyleChen, Meimei, Zhaoyang Yang, Yuxing Gao, and Candong Li. 2021. "Fast Identification of Adverse Drug Reactions (ADRs) of Digestive and Nervous Systems of Organic Drugs by In Silico Models" Molecules 26, no. 4: 930. https://doi.org/10.3390/molecules26040930
APA StyleChen, M., Yang, Z., Gao, Y., & Li, C. (2021). Fast Identification of Adverse Drug Reactions (ADRs) of Digestive and Nervous Systems of Organic Drugs by In Silico Models. Molecules, 26(4), 930. https://doi.org/10.3390/molecules26040930