
Compact NMR Spectroscopy for Low-Cost Identification and Quantification of PVC Plasticizers



Abstract
:
1. Introduction
2. Results and Discussions
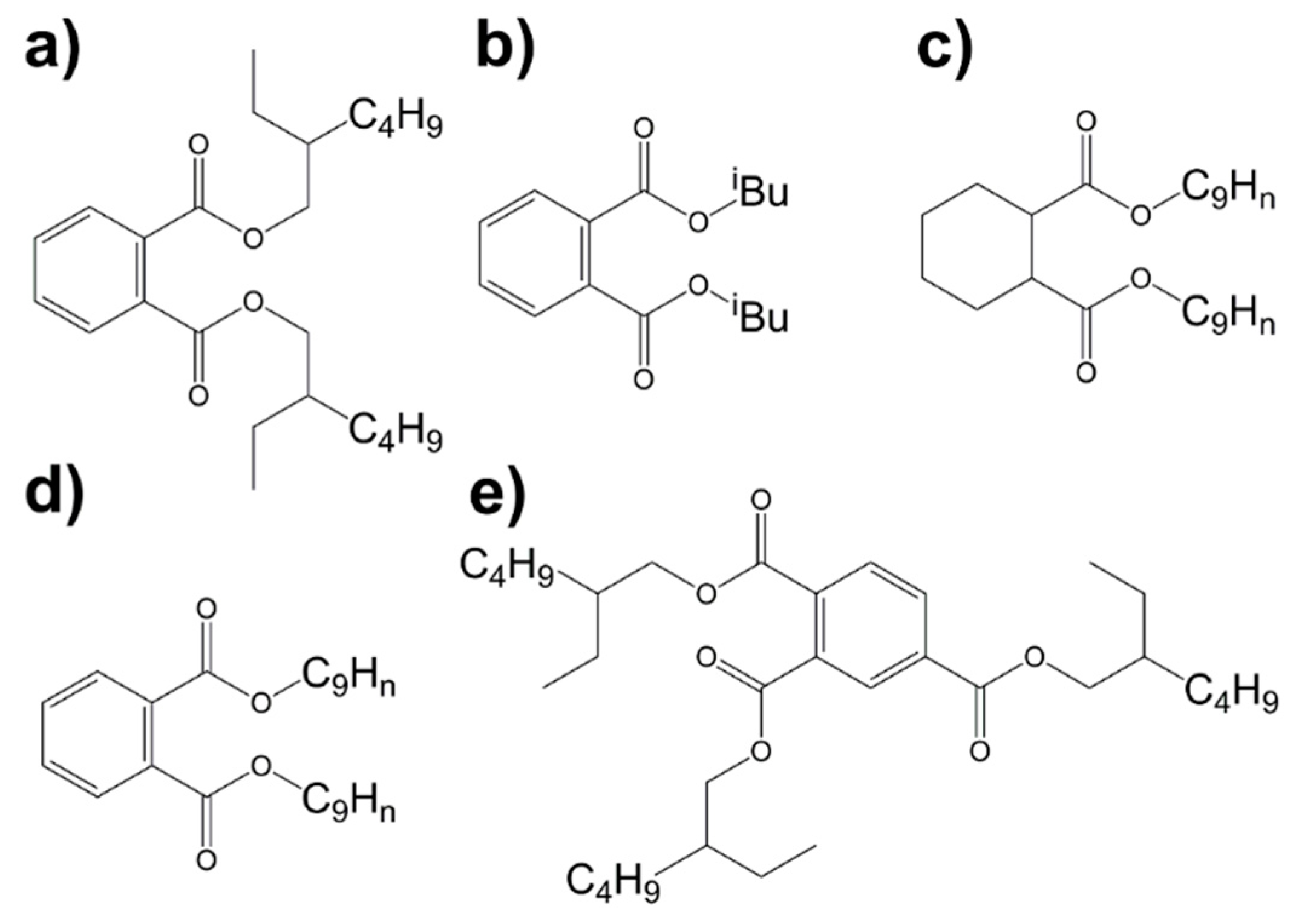
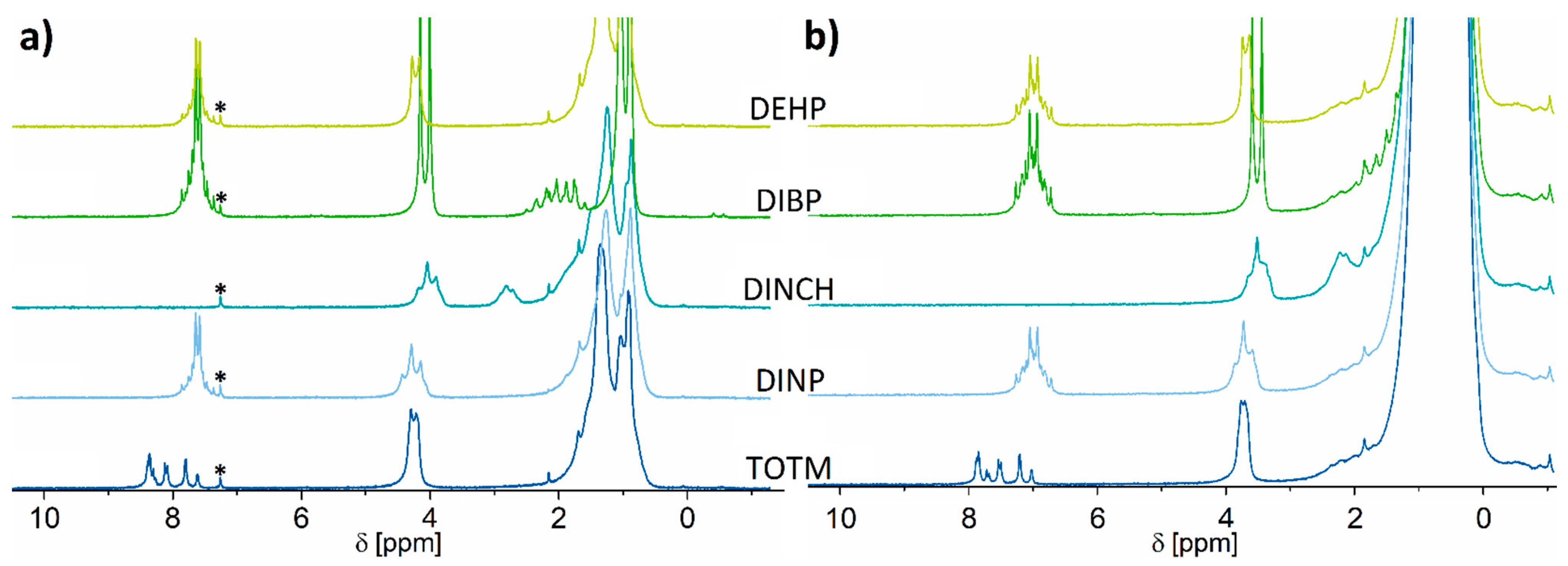
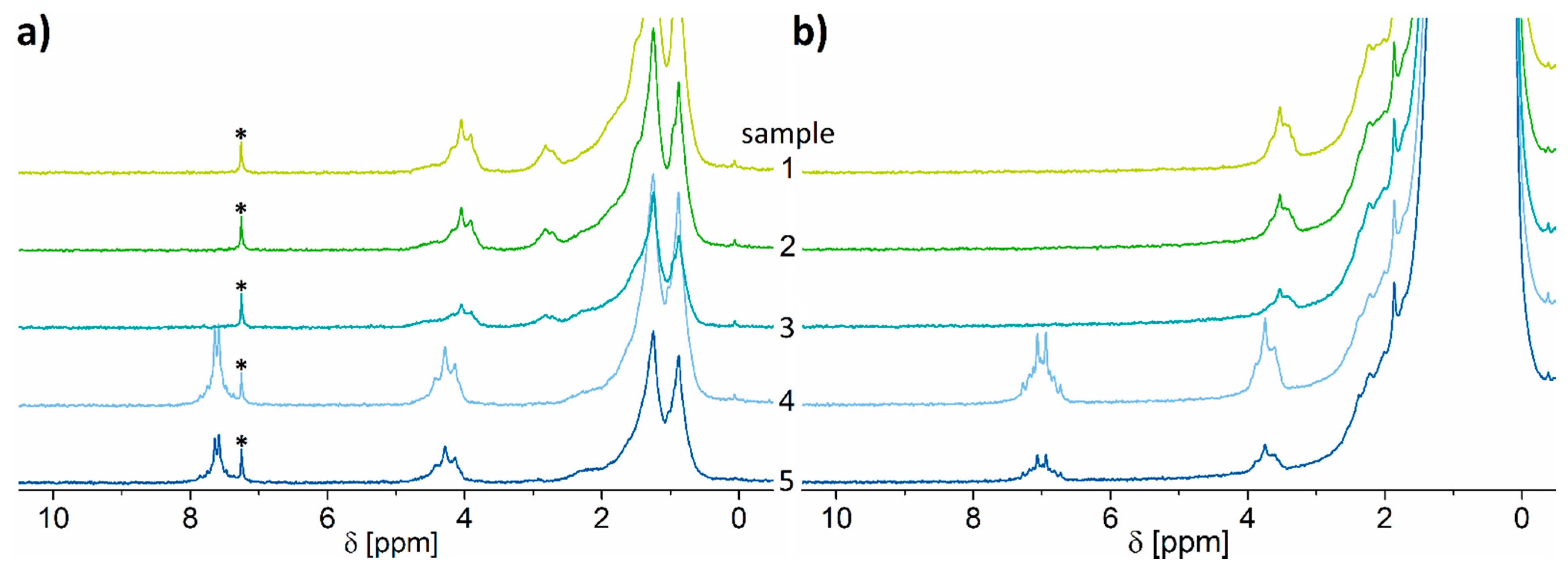
2.1. 1H NMR Spectroscopy
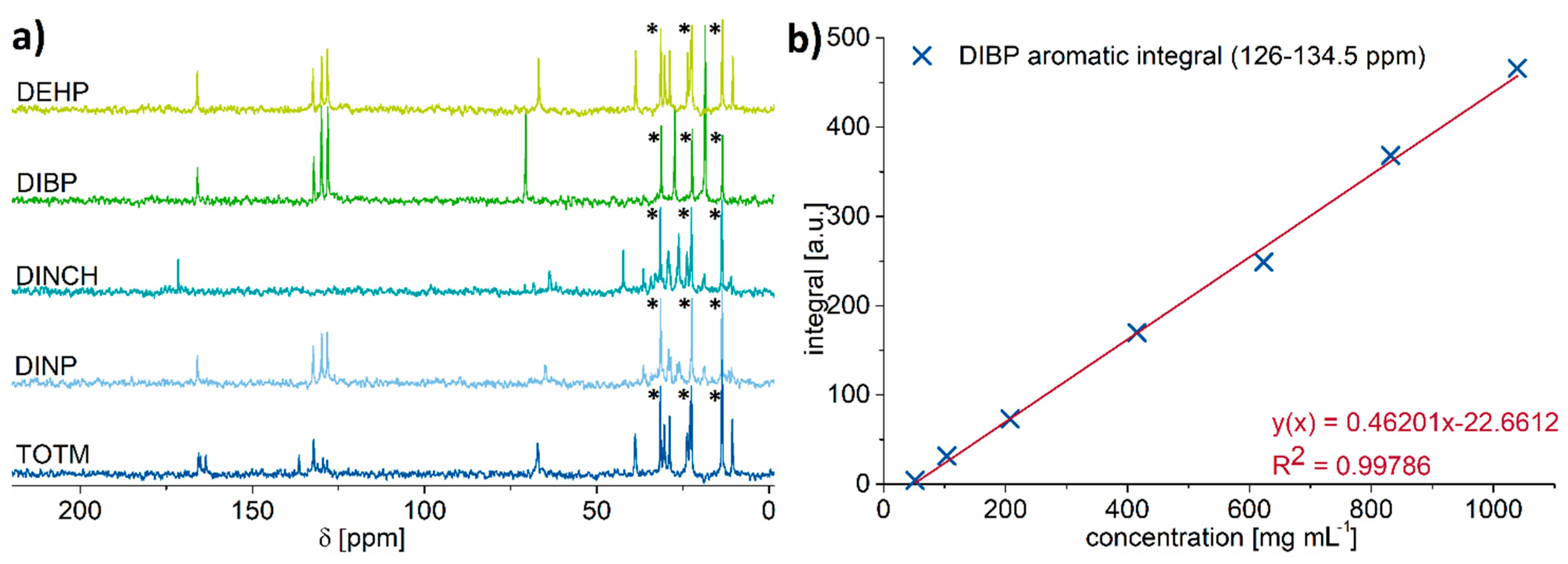
2.2. 13C NMR Spectroscopy
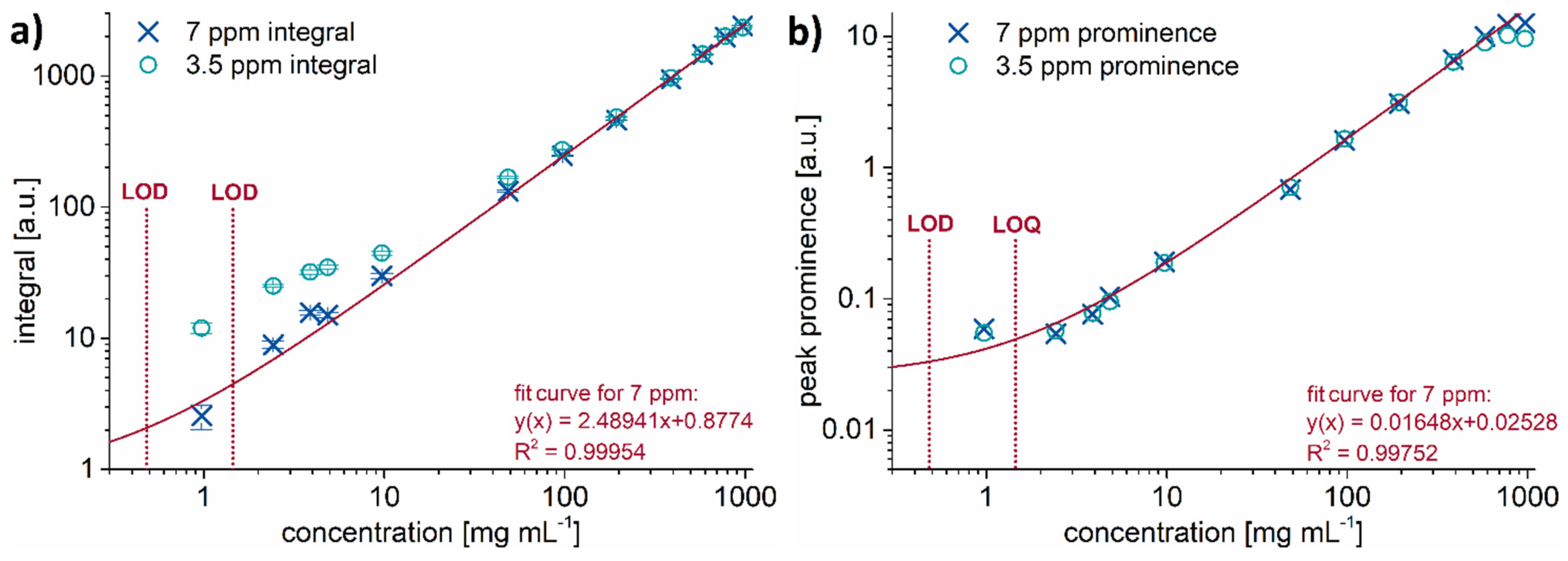
2.3. Test of the Proposed Method
2.4. How to Further Improve the Low-Field NMR Identification and Quantification
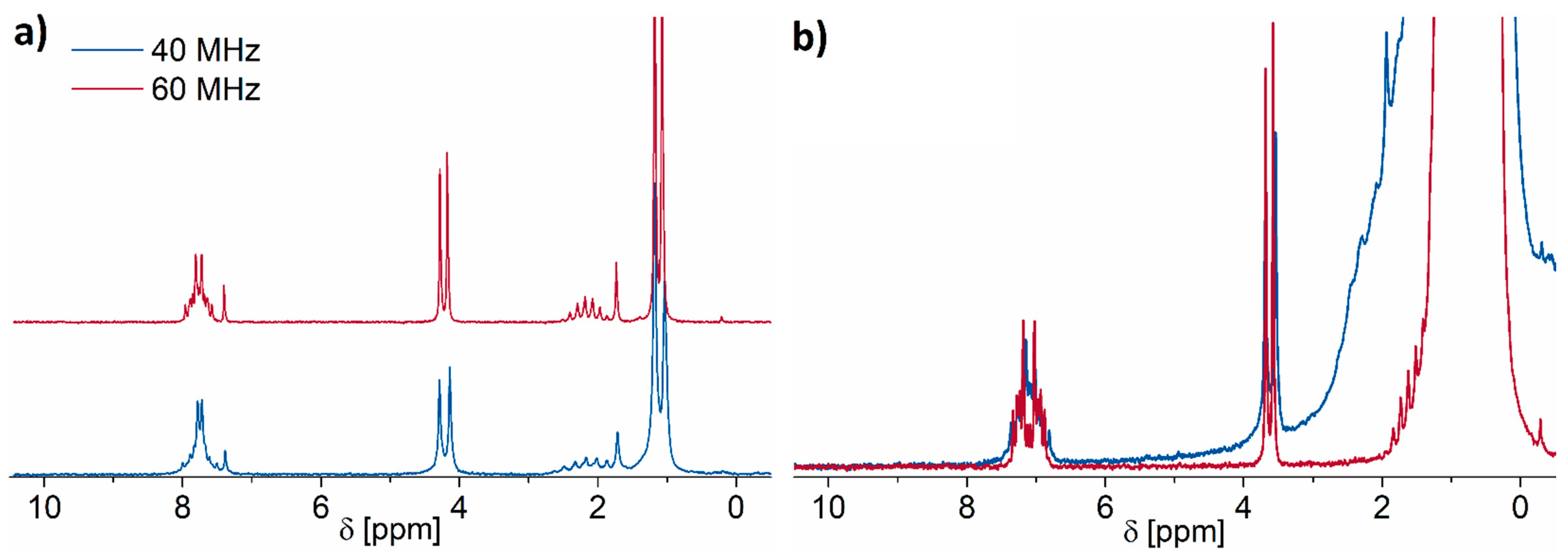
2.5. Low-Field NMR versus Conventional High-Field NMR
3. Materials and Methods
3.1. Samples
3.2. NMR Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Andrady, A.L.; Neal, M.A. Applications and societal benefits of plastics. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Plastic antioxidants market projected to reach US$2.11 billion by 2022. Addit. Polym. 2018. [CrossRef]
- Organic peroxide market forecast to be worth US$1.20 billion by 2025. Addit. Polym. 2018. [CrossRef]
- Rahman, M.; Brazel, C. The plasticizer market: An assessment of traditional plasticizers and research trends to meet new challenges. Prog. Polym. Sci. 2004, 29, 1223–1248. [Google Scholar] [CrossRef]
- Global demand for plasticizers continues to rise. Addit. Polym. 2017, 10–11. [CrossRef]
- Wilkes, C.E.; Summers, J.W.; Daniels, C.A.; Berard, M.T. PVC Handbook, Hanser, Munich, Cincinnati. In PVC Formulary; Wypych, G., Ed.; ChemTec Publishing: Scarborough, ON, Canada, 2005. [Google Scholar]
- Wypych, G. PVC Formulary, 3rd ed.; ChemTec Publishing: Scarborough, ON, Canada, 2020. [Google Scholar]
- Bernard, L.; Cueff, R.; Bourdeaux, D.; Breysse, C.; Sautou, V. Analysis of plasticizers in poly (vinyl chloride) medical devices for infusion and artificial nutrition: Comparison and optimization of the extraction procedures, a pre-migration test step. Anal. Bioanal. Chem. 2015, 407, 1651–1659. [Google Scholar] [CrossRef]
- Messadi, D.; Vergnaud, J.M. Plasticizer transfer from plasticized PVC into ethanol–water mixtures. J. Appl. Polym. Sci. 1982, 27, 3945–3955. [Google Scholar] [CrossRef]
- Messadi, D.; Taverdet, J.L.; Vergnaud, J.M. Plasticizer migration from plasticized poly (vinyl chloride) into liquids. Effect of several parameters on the transfer. Ind. Eng. Chem. Prod. Res. Dev. 1983, 22, 142–146. [Google Scholar] [CrossRef]
- Demir, A.P.T.; Ulutan, S. Migration of phthalate and non-phthalate plasticizers out of plasticized PVC films into air. J. Appl. Polym. Sci. 2012. [Google Scholar] [CrossRef]
- Kovačić, T.; Mrklić, Ž. The kinetic parameters for the evaporation of plasticizers from plasticized poly (vinyl chloride). Thermochim. Acta 2002, 381, 49–60. [Google Scholar] [CrossRef]
- Reddy, N.N.; Mohan, Y.M.; Varaprasad, K.; Ravindra, S.; Vimala, K.; Raju, K.M. Surface treatment of plasticized poly (vinyl chloride) to prevent plasticizer migration. J. Appl. Polym. Sci. 2010, 115, 1589–1597. [Google Scholar] [CrossRef]
- Kastner, J.; Cooper, D.G.; Marić, M.; Dodd, P.; Yargeau, V. Aqueous leaching of di-2-ethylhexyl phthalate and “green” plasticizers from poly (vinyl chloride). Sci. Total. Environ. 2012, 432, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, E.; Gedde, U. Plasticizer migration from PVC cable insulation—The challenges of extrapolation methods. Polym. Degrad. Stab. 2014, 101, 24–31. [Google Scholar] [CrossRef]
- Ekelund, M.; Azhdar, B.; Hedenqvist, M.; Gedde, U. Long-term performance of poly (vinyl chloride) cables, Part 2: Migration of plasticizer. Polym. Degrad. Stab. 2008, 93, 1704–1710. [Google Scholar] [CrossRef]
- Taverdet, J.L.; Vergnaud, J.M. Modelization of matter transfers between plasticized PVC and liquids in case of a maximum for liquid-time curves. J. Appl. Polym. Sci. 1986, 31, 111–122. [Google Scholar] [CrossRef]
- Audouin, L.; Dalle, B.; Metzger, G.; Verdu, J. Thermal Aging of Plasticized PVC. 11. Effect of Plasticizer Loss on Electrical and Mechanical Properties. J. Appl. Polym. Sci. 1992, 45, 2097–2103. [Google Scholar] [CrossRef]
- Ekelund, M.; Edin, H.; Gedde, U. Long-term performance of poly(vinyl chloride) cables. Part 1: Mechanical and electrical performances. Polym. Degrad. Stab. 2007, 92, 617–629. [Google Scholar] [CrossRef]
- Jakubowicz, I.; Yarahmadi, N.; Gevert, T. Effects of accelerated and natural ageing on plasticized PVC. Polym. Degrad. Stab. 1999, 66, 415–421. [Google Scholar] [CrossRef]
- Yu, Q.; Selvadurai, A. Mechanical behaviour of a plasticized PVC subjected to ethanol exposure. Polym. Degrad. Stab. 2005, 89, 109–124. [Google Scholar] [CrossRef]
- Latini, G.; Ferri, M.; Chiellini, F. Materials Degradation in PVC Medical Devices, DEHP Leaching and Neonatal Outcomes. Curr. Med. Chem. 2010, 17, 2979–2989. [Google Scholar] [CrossRef]
- Mersiowsky, I. Long-term fate of PVC products and their additives in landfills. Prog. Polym. Sci. 2002, 27, 2227–2277. [Google Scholar] [CrossRef]
- Abb, M.; Heinrich, T.; Sorkau, E.; Lorenz, W. Phthalates in house dust. Environ. Int. 2009, 35, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Yost, E.E.; Euling, S.Y.; Weaver, J.A.; Beverly, B.E.; Keshava, N.; Mudipalli, A.; Arzuaga, X.; Blessinger, T.; Dishaw, L.; Hotchkiss, A.; et al. Hazards of diisobutyl phthalate (DIBP) exposure: A systematic review of animal toxicology studies. Environ. Int. 2019, 125, 579–594. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Implementing Decision (EU) 2017/1210 of 4 July 2017 on the Identification Of Bis(2-Ethylhexyl) Phthalate (DEHP), Dibutyl Phthalate (DBP), Benzyl Butyl Phthalate (BBP) and Diisobutyl Phthalate (DIBP) as Substances of very High Concern According to Article 57(F) of Regulation (EC) No 1907/2006 of the European Parliament and of the Council (Notified Under Document C(2017) 4462) (Text with EEA Relevance), OPOCE, Brussels EU/2017/4462; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar]
- Sazan, P.; Karin, A.; Orna, C.; Bert-Ove, L.; Ana, P.P.; Stefania, V. European Union Summary Risk Assessment Report-Bis (2-ethylhexyl) Phthalate (DEHP); European Chemicals Bureau: Ispra, Italy, 2008.
- Allanou, R.; Munn, S.J.; Hansen, B.G. European Union Risk Assessment Report Dibutyl Phthalate; European Chemicals Bureau: Ispra, Italy, 2009.
- Navarro, R.; Perrino, M.P.; Tardajos, M.G.; Reinecke, H. Phthalate Plasticizers Covalently Bound to PVC: Plasticization with Suppressed Migration. Macromolecules 2010, 43, 2377–2381. [Google Scholar] [CrossRef]
- Greco, A.; Brunetti, D.; Renna, G.; Mele, G.; Maffezzoli, A. Plasticizer for poly(vinyl chloride) from cardanol as a renewable resource material. Polym. Degrad. Stab. 2010, 95, 2169–2174. [Google Scholar] [CrossRef]
- Chiellini, F.; Ferri, M.; Morelli, A.; Dipaola, L.; Latini, G. Perspectives on alternatives to phthalate plasticized poly(vinyl chloride) in medical devices applications. Prog. Polym. Sci. 2013, 38, 1067–1108. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.T.; Giovanoulis, G.; Cousins, A.P.; Magnér, J.; Cousins, I.T.; de Wit, C.A. Human exposure, hazard and risk of alternative plasticizers to phthalate esters. Sci. Total. Environ. 2016, 541, 451–467. [Google Scholar] [CrossRef]
- Weiss, J.M.; Gustafsson, Å.; Gerde, P.; Bergman, Å.; Lindh, C.H.; Krais, A.M. Daily intake of phthalates, MEHP, and DINCH by ingestion and inhalation. Chemosphere 2018, 208, 40–49. [Google Scholar] [CrossRef]
- Wypych, G. (Ed.) 15 Specialized Analytical Methods in Plasticizer Testing. In Handbook of Plasticizers, 3rd ed.; ChemTec Publishing: Scarborough, ON, Canada, 2017; pp. 661–669. [Google Scholar]
- Marcilla, A.; Garcia, S.; Garcia-Quesada, J. Migrability of PVC plasticizers. Polym. Test. 2008, 27, 221–233. [Google Scholar] [CrossRef]
- Gimeno, P.; Thomas, S.; Bousquet, C.; Maggio, A.-F.; Civade, C.; Brenier, C.; Bonnet, P.-A. Identification and quantification of 14 phthalates and 5 non-phthalate plasticizers in PVC medical devices by GC–MS. J. Chromatogr. B 2014, 949–950, 99–108. [Google Scholar] [CrossRef]
- Bernard, L.; Bourdeaux, D.; Pereira, B.; Azaroual, N.; Barthelemy, C.; Breysse, C.; Chennell, P.; Cueff, R.; Dine, T.; Eljezi, T.; et al. Analysis of plasticizers in PVC medical devices: Performance comparison of eight analytical methods. Talanta 2017, 162, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Barendswaard, W.; Litvinov, V.M.; Souren, F.; Scherrenberg, R.L.; Gondard, C.; Colemonts, C. Crystallinity and Microstructure of Plasticized Poly (vinyl chloride). A 13C and 1H Solid State NMR Study. Macromolecules 1999, 32, 167–180. [Google Scholar] [CrossRef]
- Giachet, M.T.; Schilling, M.R.; McCormick, K.; Mazurek, J.; Richardson, E.; Khanjian, H.; Learner, T. Assessment of the composition and condition of animation cels made from cellulose acetate. Polym. Degrad. Stab. 2014, 107, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Adams, A.; Kwamen, R.; Woldt, B.; Grass, M. Nondestructive Quantification of Local Plasticizer Concentration in PVC by (1)H NMR Relaxometry. Macromol. Rapid Commun. 2015, 36, 2171–2175. [Google Scholar] [CrossRef]
- Sommer, S.; Koch, M.; Adams, A. Terahertz Time-Domain Spectroscopy of Plasticized Poly (vinyl chloride). Anal. Chem. 2018, 90, 2409–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genay, S.; Feutry, F.; Masse, M.; Barthelemy, C.; Sautou, V.; Odou, P.; Decaudin, B.; Azaroual, N. Armed Study Group. Identification and quantification by (1)H nuclear magnetic resonance spectroscopy of seven plasticizers in PVC medical devices. Anal. Bioanal. Chem. 2017, 409, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.; Gladden, L.F.; Chandrasekera, T.C.; Fordham, E. Low-Field Permanent Magnets for Industrial Process and Quality Control. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 76, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Blümich, B.; Singh, K. Desktop NMR and Its Applications from Materials Science to Organic Chemistry. Angew. Chem. Int. Ed. 2018, 57, 6996–7010. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.; Limer, E.; Watson, A.; Defernez, M.; Williamson, D.; Kemsley, E.K. 60MHz 1H NMR spectroscopy for the analysis of edible oils. TrAC Trends Anal. Chem. 2014, 57, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Dalitz, F.; Cudaj, M.; Maiwald, M.; Guthausen, G. Process and reaction monitoring by low-field NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 52–70. [Google Scholar] [CrossRef]
- Adams, A. Analysis of solid technical polymers by compact NMR. TrAC Trends Anal. Chem. 2016, 83, 107–119. [Google Scholar] [CrossRef]
- Kwamen, R.; Blumich, B.; Adams, A. Estimation of Self-Diffusion Coefficients of Small Penetrants in Semicrystalline Polymers Using Single-Sided NMR. Macromol. Rapid Commun. 2012, 33, 943–947. [Google Scholar] [CrossRef]
- Duffy, J.; Urbas, A.; Niemitz, M.; Lippa, K.; Marginean, I. Differentiation of fentanyl analogues by low-field NMR spectroscopy. Anal. Chim. Acta 2019, 1049, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.; Piechatzek, A.; Schmitt, G.; Siegmund, G. Single-sided Nuclear Magnetic Resonance for condition monitoring of cross-linked polyethylene exposed to aggressive media. Anal. Chim. Acta 2015, 887, 163–171. [Google Scholar] [CrossRef]
- Adams, A. Non-destructive analysis of polymers and polymer-based materials by compact NMR. Magn. Reson. Imaging 2019, 56, 119–125. [Google Scholar] [CrossRef]
- Höpfner, J.; Ratzsch, K.-F.; Botha, C.; Wilhelm, M. Medium Resolution 1 H-NMR at 62 MHz as a New Chemically Sensitive Online Detector for Size-Exclusion Chromatography (SEC-NMR). Macromol. Rapid Commun. 2018, 39, e1700766. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Blümich, B. Compact low-field NMR spectroscopy and chemometrics: A tool box for quality control of raw rubber. Polymer 2018, 141, 154–165. [Google Scholar] [CrossRef]
- Grootveld, M.; Percival, B.; Gibson, M.; Osman, Y.; Edgar, M.; Molinari, M.; Mather, M.L.; Casanova, F.; Wilson, P.B. Progress in low-field benchtop NMR spectroscopy in chemical and biochemical analysis. Anal. Chim. Acta 2019, 1067, 11–30. [Google Scholar] [CrossRef] [Green Version]
- Chakrapani, S.B.; Minkler, M.J.; Beckingham, B.S. Low-field 1H-NMR spectroscopy for compositional analysis of multicomponent polymer systems. Anal. Chim. Acta 2019, 144, 1679–1686. [Google Scholar] [CrossRef]
- Garg, B.; Bisht, T.; Ling, Y.-C. Sulfonated graphene as highly efficient and reusable acid carbocatalyst for the synthesis of ester plasticizers. RSC Adv. 2014, 4, 57297–57307. [Google Scholar] [CrossRef]
- Bernard, L.; Décaudin, B.; Lecoeur, M.; Richard, D.; Bourdeaux, D.; Cueff, R.; Sautou, V. Analytical methods for the determination of DEHP plasticizer alternatives present in medical devices: A review. Talanta 2014, 129, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- European Commission. REGULATION (EC) No 1907/2006 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), Establishing a European Chemicals Agency, Amending Directive 1999/45/EC and Repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC, OPOCE; European Commission: Brussels, Belgium, 2006. [Google Scholar]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DINP | DIBP | DEHP | TOTM | DINCH | |
|---|---|---|---|---|---|
| LOD [mg mL−1] | 0.48 | 0.42 | 0.57 | 1.52 | 0.63 |
| LOQ [mg mL−1] | 1.45 | 1.25 | 1.70 | 4.58 | 1.90 |
| LOD [wt% in PVC] | 0.96 | 0.83 | 1.13 | 3.05 | 1.27 |
| LOQ [wt% in PVC] | 2.89 | 2.49 | 3.39 | 9.15 | 3.80 |
| Sample | Identified Plasticizer Type | Determined Plasticizer Content [wt.%] from CDCl3 Extraction | Determined Plasticizer Content [wt.%] from n-hexane Extraction | ||
|---|---|---|---|---|---|
| 1 | DINCH | 38.49 | ±1.93 | 42.69 | ±1.64 |
| 2 | DINCH | 31.68 | ±1.83 | 34.14 | ±1.19 |
| 3 | DINCH | 15.92 | ±3.63 | 17.97 | ±2.60 |
| 4 | DINP | 40.94 | ±0.10 | 43.26 | ±3.98 |
| 5 | DINP | 23.85 | ±0.95 | 22.64 | ±1.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duchowny, A.; Adams, A. Compact NMR Spectroscopy for Low-Cost Identification and Quantification of PVC Plasticizers. Molecules 2021, 26, 1221. https://doi.org/10.3390/molecules26051221
Duchowny A, Adams A. Compact NMR Spectroscopy for Low-Cost Identification and Quantification of PVC Plasticizers. Molecules. 2021; 26(5):1221. https://doi.org/10.3390/molecules26051221
Chicago/Turabian StyleDuchowny, Anton, and Alina Adams. 2021. "Compact NMR Spectroscopy for Low-Cost Identification and Quantification of PVC Plasticizers" Molecules 26, no. 5: 1221. https://doi.org/10.3390/molecules26051221
APA StyleDuchowny, A., & Adams, A. (2021). Compact NMR Spectroscopy for Low-Cost Identification and Quantification of PVC Plasticizers. Molecules, 26(5), 1221. https://doi.org/10.3390/molecules26051221