
Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides



Abstract
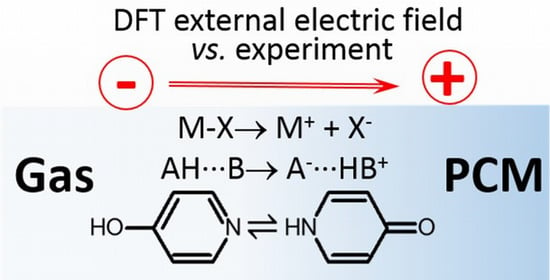
:
1. Introduction
2. Results and Discussion
2.1. Alkali Metal Halides
2.2. H3N···HF Complex
2.3. Pyridone–Hydroxypyridine Equilibria
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Řezáč, J. Non-covalent interactions atlas benchmark data sets: Hydrogen bonding. J. Chem. Theory Comput. 2020, 16, 2355–2368. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.; Matveychuk, Y.; Tsirelson, V. Identification of the tetrel bonds between halide anions and carbon atom of methyl groups using electronic criterion. Molecules 2019, 24, 1083. [Google Scholar] [CrossRef] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. N…C and S…S interactions in complexes, molecules, and transition structures HN(CH)SX:SCO, for X = F, Cl, NC, CCH, H, and CN. Molecules 2019, 24, 3232. [Google Scholar] [CrossRef] [Green Version]
- Sagan, F.; Mitoraj, M.P. Kinetic and Potential energy contributions to a chemical bond from the charge and energy decomposition scheme of extended transition state natural orbitals for chemical valence. J. Phys. Chem. A 2019, 123, 4616–4622. [Google Scholar] [CrossRef]
- Ostras, A.S.; Ivanov, D.M.; Novikov, A.S.; Tolstoy, P.M. Phosphine oxides as spectroscopic halogen bond descriptors: IR and NMR correlations with interatomic distances and complexation energy. Molecules 2020, 25, 1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalczyk, M.; Zierkiewicz, W.; Wysokiński, R.; Scheiner, S. Theoretical studies of IR and NMR spectral changes induced by sigma-hole hydrogen, halogen, chalcogen, pnicogen, and tetrel bonds in a model protein environment. Molecules 2019, 24, 3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panek, J.J.; Jezierska, A. N-oxide Derivatives: Car-Parrinello molecular dynamics and electron localization function study on intrarnolecular hydrogen bonds. J. Phys. Chem. A 2018, 122, 6605–6614. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S. Hydrogen bond and other Lewis acid—Lewis base interactions as preliminary stages of chemical reactions. Molecules 2020, 25, 4668. [Google Scholar] [CrossRef]
- Borštnar, R.; Repič, M.; Kamerlin, S.C.L.; Vianello, R.; Mavri, J. Computational study of the pKa values of potential catalytic residues in the active site of monoamine oxidase, B. J. Chem. Theory Comput. 2012, 8, 3864–3870. [Google Scholar] [CrossRef] [Green Version]
- Lakshmipriya, A.; Chaudhary, M.; Mogurampelly, S.; Klein, M.L.; Suryaprakash, N. Intramolecular hydrogen bonding appetency for conformational penchants in oxalohydrazide fluoro derivatives: NMR, MD, QTAIM, and NCI studies. J. Phys. Chem. A 2018, 122, 2703–2713. [Google Scholar] [CrossRef]
- Bauzá, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the reliability of pure and hybrid DFT methods for the evaluation of halogen, chalcogen, and pnicogen bonds involving anionic and neutral electron donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkorta, I.; Sánchez-Sanz, G.; Elguero, J.; Del Bene, J.E. Influence of hydrogen bonds on the P···P pnicogen bond. J. Chem. Theory Comput. 2012, 8, 2320–2327. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Shikhaliyev, N.G.; Maharramov, A.M.; Bagirova, K.N.; Suleymanova, G.T.; Novikov, A.S.; Khrustalev, V.N.; Tskhovrebov, A.G. Halogenated diazabutadiene dyes: Synthesis, structures, supramolecular features, and theoretical studies. Molecules 2020, 25, 5013. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. intramolecular hydrogen bond patterns and proton dynamics in nitrophthalic acid associates. Molecules 2020, 25, 4720. [Google Scholar] [CrossRef]
- Panek, J.J.; Jezierska, A. Symmetry-adapted perturbation theory analysis of the N···HX hydrogen bonds. J. Phys. Chem. A 2007, 111, 650–655. [Google Scholar] [CrossRef]
- Golubev, N.S.; Melikova, S.M.; Shchepkin, D.N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S. Interpretation of H/D isotope effects on NMR chemical shifts of [FHF]− ion based on calculations of nuclear magnetic shielding tensor surface. Z. Phys. Chem. 2003, 217, 1549–1563. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Afkhami, F.A.; Kennedy, A.R.; Zubkov, F.I.; Zangrando, E.; Kirillov, A.M.; Molins, E.; Mitoraj, M.P.; Safin, D.A. Lead(II) coordination polymers driven by pyridine-hydrazine donors: From anion-guided self-assembly to structural features. Dalton Trans. 2020, 49, 11238–11248. [Google Scholar] [CrossRef] [PubMed]
- Arp, F.F.; Bhuvanesh, N.; Blümel, J. Hydrogen peroxide adducts of triarylphosphine oxides. Dalton Trans. 2019, 48, 14312–14325. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P = O moiety as an ambidextrous hydrogen bond acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Vener, M.V.; Shishkina, A.V.; Rykounov, A.A.; Tsirelson, V.G. Cl··· Cl interactions in molecular crystals: Insights from the theoretical charge density analysis. J. Phys. Chem. A 2013, 117, 8459–8467. [Google Scholar] [CrossRef]
- Ahn, S.H.; Cluff, K.J.; Bhuvanesh, N.; Blümel, J. Hydrogen peroxide and di(hydroperoxy)propane adducts of phosphine oxides as stoichiometric and soluble oxidizing agents. Angew. Chem. Int. Ed. 2015, 54, 13341–13345. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Effect of non-covalent interactions on the 31P chemical shift tensor of phosphine oxides, phosphinic, phosphonic, and phosphoric acids and their complexes with Lead(II). J. Phys. Chem. C 2013, 117, 26689–26702. [Google Scholar] [CrossRef]
- Palusiak, M.; Grabowski, S.J. Do intramolecular halogen bonds exist? Ab initio calculations and crystal structures’ evidences. Struct. Chem. 2008, 19, 5–11. [Google Scholar] [CrossRef]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-bond and Pi-electron delocalization. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef]
- Limbach, H.-H.; Chan-Huot, M.; Sharif, S.; Tolstoy, P.M.; Shenderovich, I.G.; Denisov, G.S. Critical hydrogen bonds and protonation states of pyridoxal 5’-phosphate revealed by NMR. Biochim. Biophys. Acta 2011, 1814, 1426–1437. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Tolstoy, P.M.; Limbach, H.-H.; Shenderovich, I.G. Counteranion-dependent mechanisms of intramolecular proton transfer in aprotic solution. Phys. Chem. Chem. Phys. 2010, 12, 10373–10379. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Rozhkova, Y.A.; Zukal, A.; Čejka, J.; Shenderovich, I.G. Mutable Lewis and Brønsted acidity of aluminated SBA-15 as revealed by NMR of adsorbed pyridine−15N. Langmuir 2011, 27, 12115–12123. [Google Scholar] [CrossRef] [PubMed]
- Gedat, E.; Schreiber, A.; Findenegg, G.H.; Shenderovich, I.; Limbach, H.-H.; Buntkowsky, G. Stray field gradient NMR reveals effects of hydrogen bonding on diffusion coefficients of pyridine in mesoporous silica. Magn. Reson. Chem. 2001, 39, S149–S157. [Google Scholar] [CrossRef]
- Orozco, M.; Luque, F.J. Theoretical methods for the description of the solvent effect in biomolecular systems. Chem. Rev. 2000, 100, 4187–4225. [Google Scholar] [CrossRef]
- Benighaus, T.; Thiel, W. A general boundary potential for hybrid QM/MM simulations of solvated biomolecular systems. J. Chem. Theory Comput. 2009, 5, 3114–3128. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, S.; Allolio, C.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H.; Sebastiani, D.; Tolstoy, P.M. Proton transfer in a short hydrogen bond caused by solvation shell fluctuations: An ab initio MD and NMR/UV study of an (OHO)−bonded system. Phys. Chem. Chem. Phys. 2015, 17, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, O. Implicit solvent models in molecular dynamics simulations: A brief overview. Annu. Rep. Comput. Chem. 2008, 4, 125–137. [Google Scholar] [CrossRef]
- Clabaut, P.; Schweitzer, B.; Götz, A.W.; Michel, C.; Steinmann, S.N. Solvation free energies and adsorption energies at the metal/water interface from hybrid quantum-mechanical/molecular mechanics simulations. J. Chem. Theory Comput. 2020, 16, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Vianello, R.; Domene, C.; Mavri, J. The use of multiscale molecular simulations in understanding a relationship between the structure and function of biological systems of the brain: The application to monoamine oxidase enzymes. Front. Neurosci. 2016, 10, 327. [Google Scholar] [CrossRef] [Green Version]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.-H.; Shenderovich, I.G. NMR study of solvation effect on the geometry of proton-bound homodimers of increasing size. J. Phys. Chem. A 2017, 121, 8697–8705. [Google Scholar] [CrossRef] [Green Version]
- Del Bene, J.E.; Bartlett, R.J.; Elguero, J. Interpreting 2hJ(F,N), 1hJ(H,N) and 1J(F,H) in the hydrogen-bonded FH–collidine complex. Magn. Reson. Chem. 2002, 40, 767–771. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Simplified calculation approaches designed to reproduce the geometry of hydrogen bonds in molecular complexes in aprotic solvents. J. Chem. Phys. 2018, 148, 124313. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. Solvent effects on acid-base complexes. What is more important: A macroscopic reaction field or solute-solvent interactions? J. Chem. Phys. 2019, 150, 204505. [Google Scholar] [CrossRef]
- Shenderovich, I.G.; Denisov, G.S. Adduct under field—a qualitative approach to account for solvent effect on hydrogen bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef] [Green Version]
- Dominikowska, J.; Palusiak, M. Tuning aromaticity of para-substituted benzene derivatives with an external electric field. ChemPhysChem 2018, 19, 590–595. [Google Scholar] [CrossRef]
- Liang, H.; Chai, B.; Chen, G.; Chen, W.; Chen, S.; Xiao, H.; Lin, S. Electric field-driven acid-base transformation: Proton transfer from acid (HBr/HF) to base (NH3/H2O). Chem. Res. Chin. Univ. 2015, 31, 418–426. [Google Scholar] [CrossRef]
- Astrakas, L.; Gousias, C.; Tzaphlidou, M. Electric field effects on chignolin conformation. J. Appl. Phys. 2011, 109. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Provasi, P.F.; Pagola, G.I.; Ferraro, M.B. Electric field effects on nuclear magnetic shielding of the 1:1 and 2:1 (homo and heterochiral) complexes of XOOX′ (X, X′ = H, CH3) with lithium cation and their chiral discrimination. J. Chem. Phys. 2011, 135, 104116. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Effect of an external electric field on the dissociation energy and the electron density properties: The case of the hydrogen bonded dimer HF⋯HF. J. Chem. Phys. 2009, 130, 044104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bene, J.E.; Jordan, M.J.T. To what extent do external fields and vibrational and isotopic effects influence NMR coupling constants across hydrogen bonds? Two-bond Cl-N spin-spin coupling constants (2hJCl-N) in model ClH:NH3 complexes. J. Phys. Chem. A 2002, 106, 5385–5392. [Google Scholar] [CrossRef]
- Bevitt, J.; Chapman, K.; Crittenden, D.; Jordan, M.J.T.; Del Bene, J.E. An ab initio study of anharmonicity and field effects in hydrogen-bonded complexes of the deuterated analogues of HCl and HBr with NH3 and N(CH3). J. Phys. Chem. A 2001, 105, 3371–3378. [Google Scholar] [CrossRef]
- Jordan, M.J.T.; Del Bene, J.E. Unraveling environmental effects on hydrogen-bonded complexes: Matrix effects on the structures and proton-stretching frequencies of hydrogen-halide complexes with ammonia and trimethylamine. J. Am. Chem. Soc. 2000, 122, 2101–2115. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Chipman, D.M. Reaction field effects on nitrogen shielding. J. Chem. Phys. 1999, 110, 1611. [Google Scholar] [CrossRef]
- Ramos, M.; Alkorta, I.; Elguero, J.; Golubev, N.S.; Denisov, G.S.; Benedict, H.; Limbach, H.-H. Theoretical study of the influence of electric fields on hydrogen-bonded acid−base complexes. J. Phys. Chem. A 1997, 101, 9791–9800. [Google Scholar] [CrossRef]
- Suydam, I.T.; Snow, C.D.; Pande, V.S.; Boxer, S.G. Electric fields at the active site of an enzyme: Direct comparison of experiment with theory. Science 2006, 313, 200–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenderovich, I.G. Electric field effect on 31P NMR magnetic shielding. J. Chem. Phys. 2020, 153, 184501. [Google Scholar] [CrossRef]
- Frank, J.; Katritzky, A.R. Tautomeric pyridines. Part XV. Pyridone–hydroxypyridine equilibria in solvents of differing polarity. J. Chem. Soc. Perkin Trans. 1976, 2, 1428–1431. [Google Scholar] [CrossRef]
- Wang, Z.; Danovich, D.; Ramanan, R.; Shaik, S. Oriented-external electric fields create absolute enantioselectivity in Diels–Alder reactions: Importance of the molecular dipole moment. J. Am. Chem. Soc. 2018, 140, 13350–13359. [Google Scholar] [CrossRef]
- Chranina, O.V.; Czerniakowski, F.P.; Denisov, G.S. UV-Vis electrochromism due to proton-transfer. J. Mol. Struct. 1988, 177, 309–315. [Google Scholar] [CrossRef]
- Bergmann, K.; Eigen, M.; de Maeyer, L. Dielektrische absorption als folge chemischer relaxation. Ber. Bunsenges. Phys. Chem. 1963, 67, 819–826. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Covalent bond lengthening in hydroxyl groups involved in three-center and in cooperative hydrogen bonds. Analysis of low-temperature neutron diffraction data. J. Am. Chem. Soc. 1992, 114, 7123–7126. [Google Scholar] [CrossRef]
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and spectral properties of the protonated homodimer of pyridine in the liquid and solid states. A combined NMR, X-ray diffraction and inelastic neutron scattering study. J. Phys. Chem. A 2011, 115, 8041–8048. [Google Scholar] [CrossRef]
- Grabowski, S.J. The bond valence model in analysing H-bonds of crystal structures. J. Mol. Struct. 2000, 552, 153–157. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. A Systematic comparison of second-order polarization propagator approximation (SOPPA) and equation-of-motion coupled cluster singles and doubles (EOM-CCSD) spin-spin coupling constants for selected singly bonded molecules, and the hydrides NH3, H2O, and HF and their protonated and deprotonated ions and hydrogen-bonded complexes. J. Chem. Theory Comput. 2008, 4, 967–973. [Google Scholar] [CrossRef]
- Biczysko, M.; Latajka, Z. The influence of water molecules on the proton position in H3N ‒ HX (X = F, Cl, Br) complexes. Chem. Phys. Lett. 1999, 313, 366–373. [Google Scholar] [CrossRef]
- Tsuchida, N.; Yamabe, S. Reaction paths of tautomerization between hydroxypyridines and pyridones. J. Phys. Chem. A 2005, 109, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Katritzky, A.R.; Denisko, O.V. Prototropic tautomerism of heterocycles: Heteroaromatic tautomerism-General overview and methodology. Adv. Heterocycl. Chem. 2000, 76, 1–84. [Google Scholar] [CrossRef]
- Luque, F.J.; Zhang, Y.; Alemán, C.; Bachs, M.; Gao, J.; Orozco, M. Solvent effects in chloroform solution: Parametrization of the MST/SCRF continuum model. J. Phys. Chem. 1996, 100, 4269–4276. [Google Scholar] [CrossRef]
- Gao, J.; Shao, L. Polarization effects on the tautomeric equilibria of 2- and 4-hydroxypyridine in aqueous and organic solution. J. Phys. Chem. 1994, 98, 13772–13779. [Google Scholar] [CrossRef]
- Andreeva, D.V.; Ip, B.; Gurinov, A.A.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G.; Limbach, H.-H. Geometrical features of hydrogen bonded complexes involving sterically hindered pyridines. J. Phys. Chem. A 2006, 110, 10872–10879. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G. The partner does matter: The structure of heteroaggregates of acridine orange in water. Molecules 2019, 24, 2816. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. A new basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Winget, P.; Dolney, D.M.; Giesen, D.J.; Cramer, C.J.; Truhlar, D.G. Minnesota Solvent Descriptor Database. Available online: http://comp.chem.umn.edu/solvation/mnsddb.pdf (accessed on 26 February 2021).
- Onsager, L. Electric moments of molecules in liquids. J. Am. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar] [CrossRef]
- Sellner, B.; Valiev, M.; Kathmann, S.M. Charge and electric field fluctuations in aqueous NaCl electrolytes. J. Phys. Chem. B 2013, 117, 10869–10882. [Google Scholar] [CrossRef]
- Nardo, V.M.; Cassone, G.; Ponterio, R.C.; Saija, F.; Sponer, J.; Tommasini, M.; Trusso, S. Electric-field-induced effects on the dipole moment and vibrational modes of the centrosymmetric indigo molecule. J. Phys. Chem. A 2020, 124, 10856–10869. [Google Scholar] [CrossRef]
- Cassone, G.; Sponer, J.; Trusso, S.; Saija, F. Ab initio spectroscopy of water under electric fields. Phys. Chem. Chem. Phys. 2019, 21, 21205–21212. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G. Nuclear quantum effects largely influence molecular dissociation and proton transfer in liquid water under an electric field. J. Phys. Chem. Lett. 2020, 11, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G.; Sofia, A.; Rinaldi, G.; Sponer, J. Catalyst-free hydrogen synthesis from liquid ethanol: An ab initio molecular dynamics study. J. Phys. Chem. C 2019, 123, 9202–9208. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | E(NH)–E(OH), a.u. |
|---|---|
| Exp. in cyclohexane [57] | 0.0020 |
| Exp. in chloroform [57] | −0.0021 |
| Gas-phase DFT | 0.0036 |
| PCM cyclohexane | −0.0007 |
| PCM chloroform | −0.0043 |
| SMD cyclohexane | −0.0017 |
| SMD chloroform | −0.0053 |
| Field 0.0007 a.u. | 0.0019 |
| Field 0.0023 a.u. | −0.0019 |
| PCM cyclohexane and Field 0.0030 a.u. on the OH-form | 0.0021 |
| PCM chloroform and Field 0.0023 a.u. on the OH-form | −0.0021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shenderovich, I.G.; Denisov, G.S. Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides. Molecules 2021, 26, 1283. https://doi.org/10.3390/molecules26051283
Shenderovich IG, Denisov GS. Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides. Molecules. 2021; 26(5):1283. https://doi.org/10.3390/molecules26051283
Chicago/Turabian StyleShenderovich, Ilya G., and Gleb S. Denisov. 2021. "Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides" Molecules 26, no. 5: 1283. https://doi.org/10.3390/molecules26051283
APA StyleShenderovich, I. G., & Denisov, G. S. (2021). Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides. Molecules, 26(5), 1283. https://doi.org/10.3390/molecules26051283