
Assignment of Absolute Configuration of Bromoallenes by Vacuum-Ultraviolet Circular Dichroism (VUVCD)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
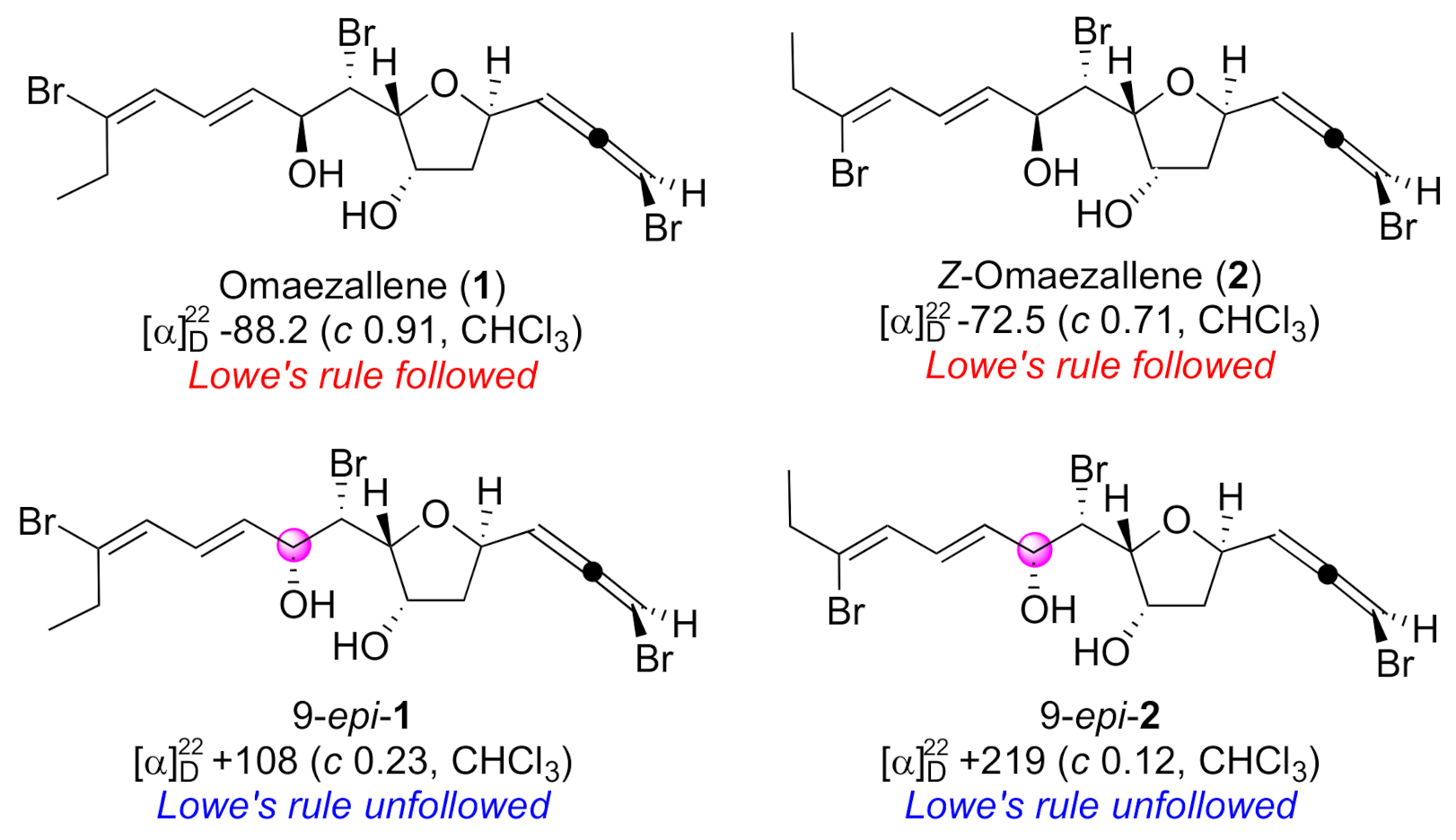
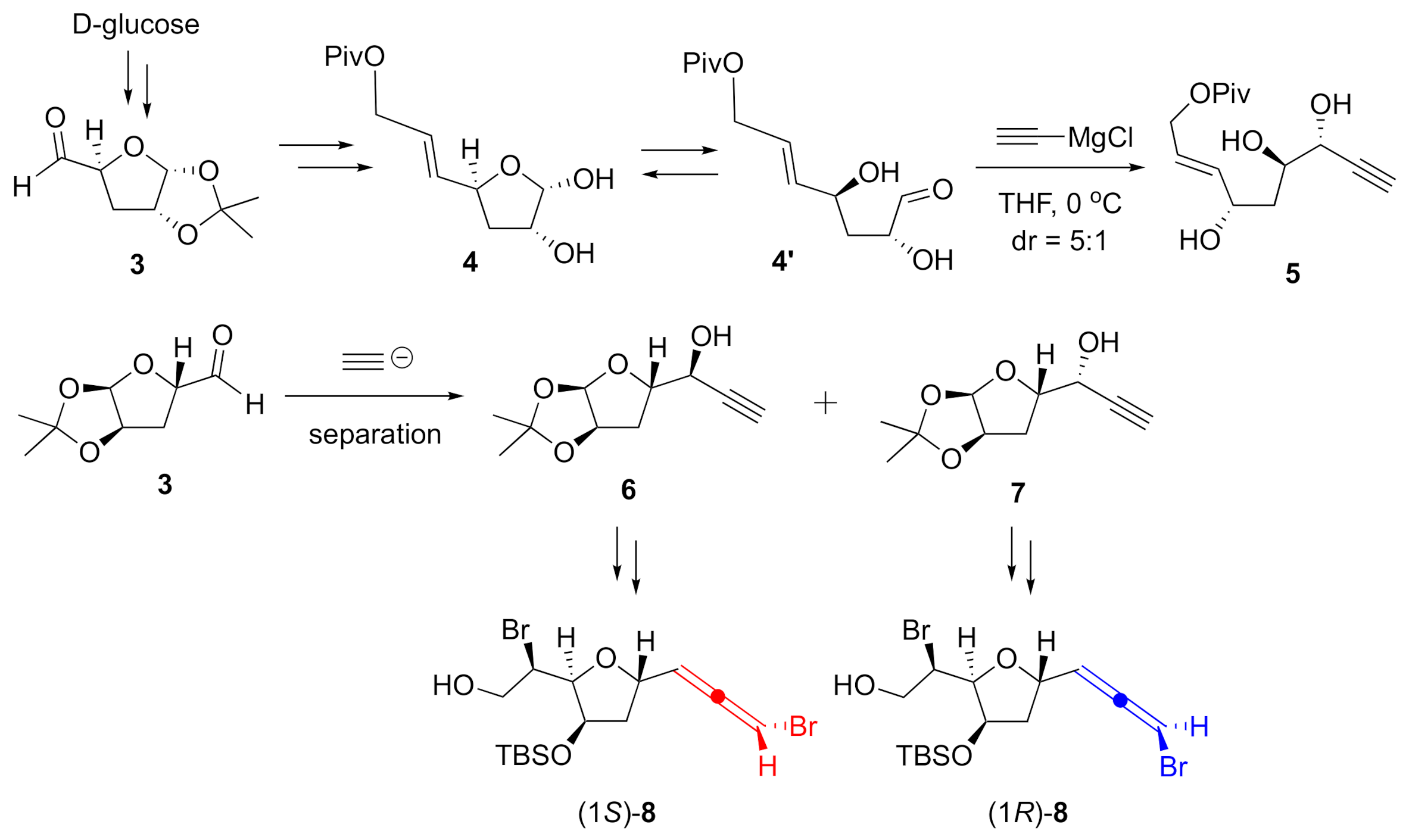
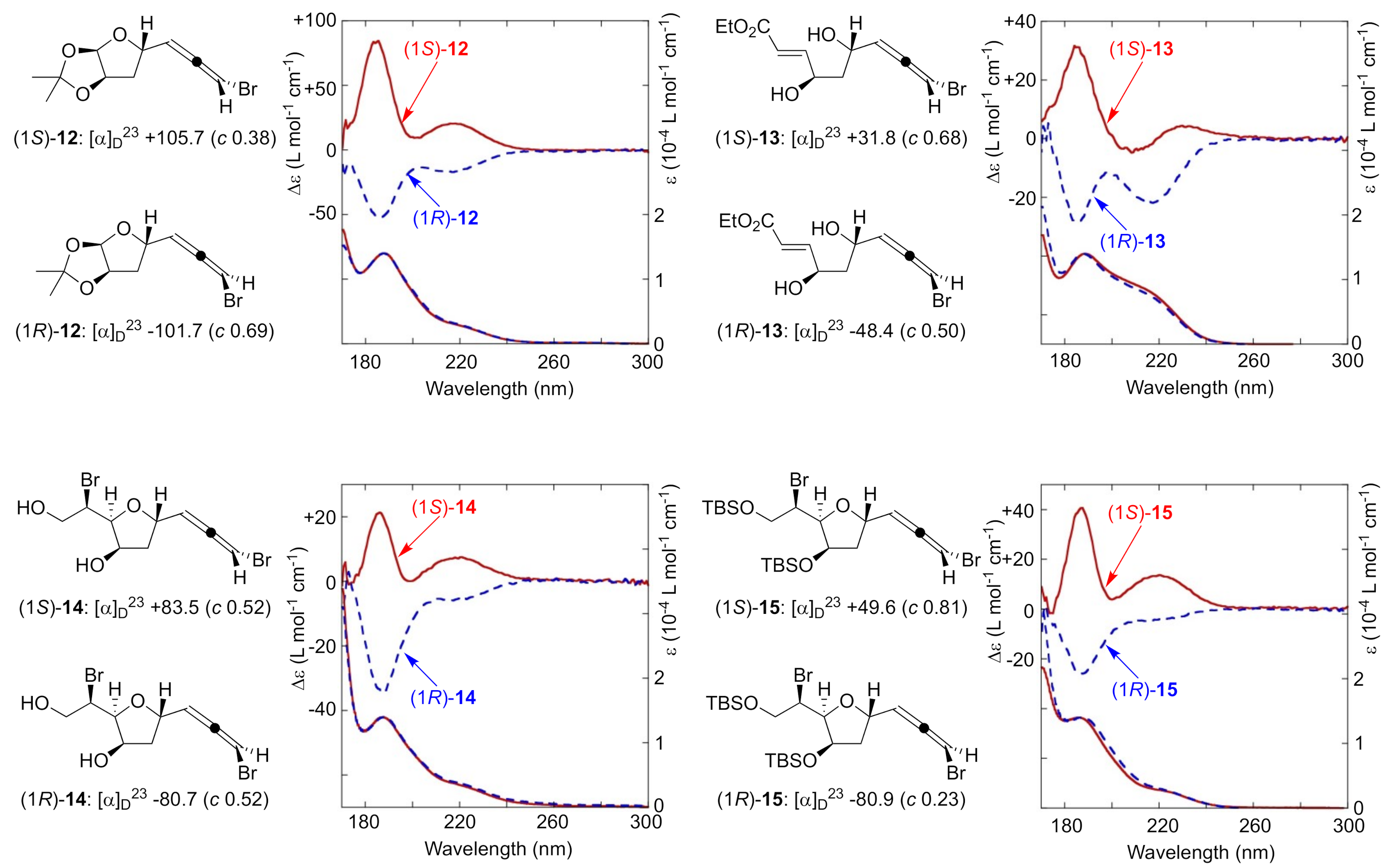
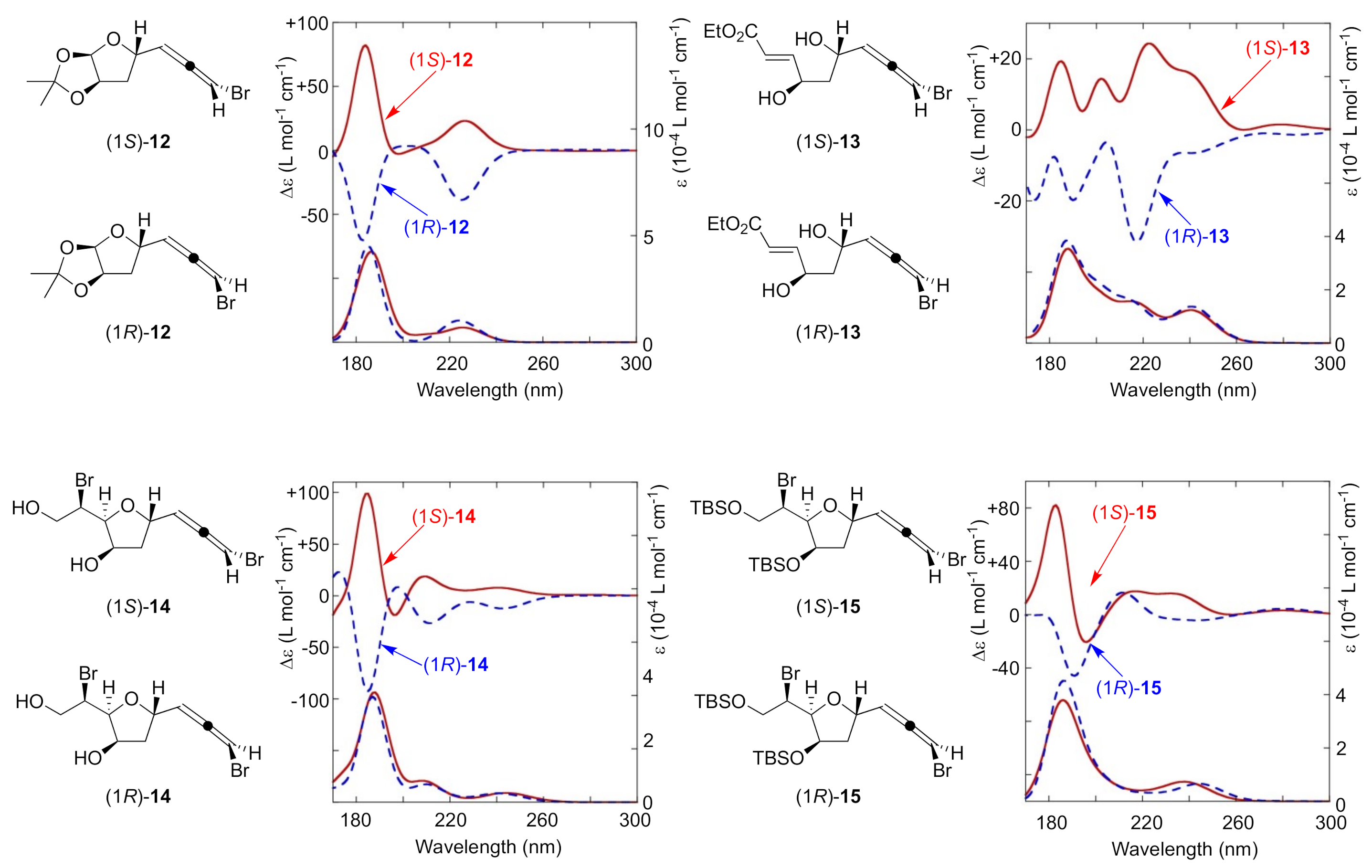
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Syntheses
3.2.1. Propargyl Alcohols 6 and 7
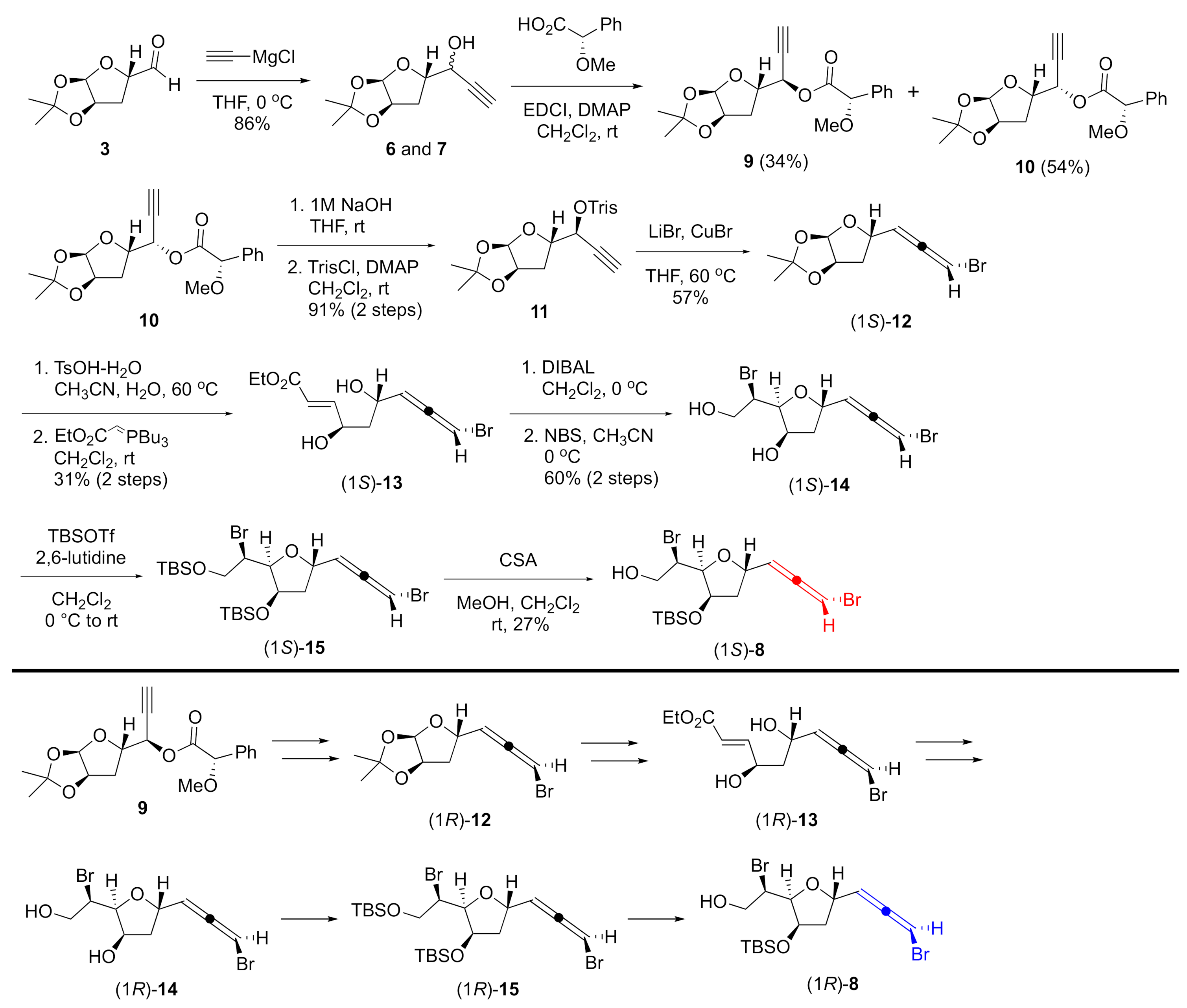
3.2.2. Ester 9 and 10
3.2.3. Sulfonate 11
3.2.4. Bromoallene (1S)-12
3.2.5. Unsaturated Ester (1S)-13
3.2.6. Bromoether (1S)-14
3.2.7. TBS Ether (1S)-15
3.2.8. Alcohol (1S)-8
3.2.9. Sulfonate S1 (the Corresponding Diastereomer of 11)
3.2.10. Bromoallene (1R)-12
3.2.11. Unsaturated Ester (1R)-13
3.2.12. Bromoether (1R)-14
3.2.13. TBS Ether (1R)-15
3.2.14. Alcohol (1R)-8
3.3. CD Calculations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hoffmann-Röder, A.; Krause, Ν. Synthesis and Properties of Allenic Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2004, 43, 1196–1216. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G. The Absolute Configuration of Allenes. Chem. Commun. 1965, 411–413. [Google Scholar] [CrossRef]
- Kinnel, R.; Duggan, A.J.; Eisner, T.; Meinwald, J. Panacene: An Aromatic Bromoallene from a Sea Hare (Aplysia brasiliana). Tetrahedron Lett. 1977, 18, 3913–3916. [Google Scholar] [CrossRef]
- Suzuki, M.; Koizumi, K.; Kikuchi, H.; Suzuki, T.; Kurosawa, E. Epilaurallene, a New Nonterpenoid C15-Bromoallene from the Red Alga Laurencia nipponica Yamada. Bull. Chem. Soc. Jpn. 1983, 56, 715–718. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Koizumi, K.; Suzuki, M.; Kurosawa, E. Kumausallene, a New Bromoallene from the Marine Red Alga Laurencia nipponica Yamada. Chem. Lett. 1983, 12, 1639–1642. [Google Scholar] [CrossRef] [Green Version]
- Öztunç, A.; Imre, S.; Wagner, H.; Norte, M.; Fernández, J.J.; González, R. A New Haloether from Laurencia Possessing a Lauroxacyclododecane Ring. Structural and Conformational Studies. Tetrahedron 1991, 47, 2273–2276. [Google Scholar] [CrossRef]
- Suzuki, M.; Kondo, H.; Tanaka, I. The Absolute Stereochemistry of Okamurallene and Its Congeners, Halogenated C15 Nonterpenoids from the Red Alga Laurencia intricate. Chem. Lett. 1991, 20, 33–34. [Google Scholar] [CrossRef]
- Okamoto, Y.; Nitanda, N.; Ojika, M.; Sakagami, Y. Aplysiallene, a New Bromoallene as an Na, K-ATPase Inhibitor from the Sea Hare, Aplysia kurodai. Biosci. Biotechnol. Biochem. 2001, 65, 474–476. [Google Scholar] [CrossRef]
- Suzuki, M.; Takahashi, Y.; Mitome, Y.; Itoh, T.; Abe, T.; Masuda, M. Brominated Metabolites from an Okinawan Laurencia intricate. Phytochemistry 2002, 60, 861–867. [Google Scholar] [CrossRef]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Norte, M.; Souto, M.L. New Bicyclotridecane C15 Nonterpenoid Bromoallenes from Laurencia marilzae. Org. Lett. 2011, 13, 2690–2693. [Google Scholar] [CrossRef]
- Kokkotou, K.; Ioannou, E.; Nomikou, M.; Pitterl, F.; Vonaparti, A.; Siapi, E.; Zervou, M.; Roussis, V. An Integrated Approach Using UHPLC–PDA–HRMS and 2D HSQC NMR for the Metabolic Profiling of the Red Alga Laurencia: Dereplication and Tracing of Natural Products. Phytochemistry 2014, 108, 208–219. [Google Scholar] [CrossRef]
- Sutour, S.; Therrien, B.; von Reuss, S.H.; Tomi, F. Halogenated C15 Acetogenin Analogues of Obtusallene III from a Laurenciella sp. Collected in Corsica. J. Nat. Prod. 2018, 81, 279–285. [Google Scholar] [CrossRef]
- Umezawa, T.; Oguri, Y.; Matsuura, H.; Yamazaki, S.; Suzuki, M.; Yoshimura, E.; Furuta, T.; Nogata, Y.; Serisawa, Y.; Abe, T.; et al. Omaezallene from Red Alga Laurencia sp.: Structure Elucidation, Total Synthesis, and Antifouling Activity. Angew. Chem. Int. Ed. 2014, 53, 3909–3912. [Google Scholar] [CrossRef] [PubMed]
- Sohn, T.; Kim, D.; Paton, R.S. Substrate Controlled Asymmetric Total Syntheses of Microcladallenes A, B, and C Based on the Proposed Structures. Chem. Eur. J. 2015, 21, 15988–15997. [Google Scholar] [CrossRef] [PubMed]
- Ojima, N.; Sakai, K.; Fukazawa, T.; Gekko, K. Vacuum-Ultraviolet Circular Dichroism Spectrophotometer Using Synchrotron Radiation: Optical System and Off-line Performance. Chem. Lett. 2000, 29, 832–833. [Google Scholar] [CrossRef]
- Ojima, N.; Sakai, K.; Matsuo, K.; Matsui, T.; Fukazawa, T.; Namatame, H.; Taniguchi, M.; Gekko, K. Vacuum-Ultraviolet Circular Dichroism Spectrophotometer Using Synchrotron Radiation: Optical System and On-line Performance. Chem. Lett. 2001, 30, 522–523. [Google Scholar] [CrossRef]
- Dickerson, H.; Ferber, S.; Richardson, F.S. Molecular Orbital Calculations on the Optical Rotatory Properties of Chiral Allene Systems. Theor. Chim. Acta 1976, 42, 333–344. [Google Scholar] [CrossRef]
- Matsuo, K.; Gekko, K. Construction of a Synchrotron-Radiation Vacuum-Ultraviolet Circular-Dichroism Spectrophotometer and Its Application to the Structural Analysis of Biomolecules. Bull. Chem. Soc. Jpn. 2013, 86, 675–689. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Gekko, K. Vacuum-Ultraviolet Circular Dichroism Study of Saccharides by Synchrotron Radiation Spectrophotometry. Carbohydr. Res. 2004, 339, 591–597. [Google Scholar] [CrossRef]
- Matsuo, K.; Matsushima, Y.; Fukuyama, T.; Senba, S.; Gekko, K. Vacuum-Ultraviolet Circular Dichroism of Amino Acids as Revealed by Synchrotron Radiation Spectrophotometer. Chem. Lett. 2002, 31, 826–827. [Google Scholar] [CrossRef]
- Matsuo, K.; Yonehara, R.; Gekko, K. Secondary-Structure Analysis of Proteins by Vacuum-Ultraviolet Circular Dichroism Spectroscopy. J. Biochem. 2004, 135, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Yonehara, R.; Gekko, K. Improved Estimation of the Secondary Structures of Proteins by Vacuum-Ultraviolet Circular Dichroism Spectroscopy. J. Biochem. 2005, 138, 79–88. [Google Scholar] [CrossRef]
- Egron, D.; Durand, T.; Roland, A.; Videl, J.-P.; Rossi, J.-C. C-Furanoside Synthesis via Intramolecular Cyclization. Synlett 1999, 435–437. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s Method. The Absolute Configurations of Marine Terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Elsevier, C.L.; Vermeer, P.; Gedanken, A.; Runge, W. Synthesis and Absolute Configurations of Halogenoallenes. J. Org. Chem. 1985, 50, 364–367. [Google Scholar] [CrossRef]
- Grese, T.A.; Hutchinson, K.D.; Overman, L.E. General Approach to Halogenated Tetrahydrofuran Natural Products from Red Algae of the Genus Laurencia. Total Synthesis of (±)-Kumausallene and (±)-1-epi-Kumausallene. J. Org. Chem. 1993, 58, 2466–2477. [Google Scholar] [CrossRef]
- Harcken, C.; Martin, S.F. Improved E-Selectivity in the Wittig Reaction of Stabilized Ylides with α-Alkoxyaldehydes and Sugar Lactols. Org. Lett. 2001, 3, 3591–3593. [Google Scholar] [CrossRef]
- Saito, S.; Moriwake, Y.; Moriwake, T. Synthesis of Optically Pure 2,3,4,5-Substituted Tetrahydrofurans from a Bis-Allylic Framework via an Osmylation-Haloetherification Sequence. Synlett 1990, 523–524. [Google Scholar] [CrossRef]
- Saito, S.; Harunari, T.; Shimamura, N.; Asahara, M.; Moriwake, T. Protected Vicinal Diol Controller in Diastereoselective Reduction of β-Oxo Esters. Synlett 1992, 325–327. [Google Scholar] [CrossRef]
- Murata, Y.; Kamino, T.; Aoki, T.; Hosokawa, S.; Kobayashi, S. Highly Efficient Total Synthesis of (+)-Citreoviral. Angew. Chem. Int. Ed. 2004, 43, 3175–3177. [Google Scholar] [CrossRef]
- Houk, K.N.; Moses, S.R.; Wu, Y.D.; Rondan, N.G.; Jäger, V.; Schohe, R.; Fronczek, F.R. Stereoselective Nitrile Oxide Cycloadditions to Chiral Allyl Ethers and Alcohols. The “Inside Alkoxy” Effect. J. Am. Chem. Soc. 1984, 106, 3880–3882. [Google Scholar] [CrossRef]
- Bandur, N.G.; Brückner, D.; Hoffmann, R.W.; Koert, U. Total Synthesis of Jimenezin via an Intramolecular Allylboration. Org. Lett. 2006, 8, 3829–3831. [Google Scholar] [CrossRef]
- Elsevier, C.L.; Vermeer, P.; Gedanken, A.; Runge, W. Excited States of the Allene Chromophore: Photoelectron, Circular Dichroism, and Absorption Spectroscopy of Alkyl- and Halogenoallenes. J. Am. Chem. Soc. 1985, 107, 2537–2547. [Google Scholar] [CrossRef]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The Assignment of Absolute Stereostructures through Quantum Chemical Circular Dichroism Calculations. Eur. J. Org. Chem. 2009, 2717–2727. [Google Scholar] [CrossRef]
- Takekawa, H.; Tanaka, K.; Fukushi, E.; Matsuo, K.; Nehira, T.; Hashimoto, M. Roussoellols A and B, Tetracyclic Fusicoccanes from Roussoella hysterioides. J. Nat. Prod. 2013, 76, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nehira, T.; Matsuo, K.; Kawabata, T.; Kobashigawa, Y.; Morioka, H.; Losung, F.; Mangindaan, R.E.P.; de Voogd, N.J.; Yokosawa, H.; et al. Isolation from the Marine Sponge Niphates olemda and Determination of its Absolute Configuration by an ECD Analysis. Tetrahedron 2015, 71, 6956–6960. [Google Scholar] [CrossRef]
- Goto, H.; Obata, N.; Nakayama, N.; Ohta, K. CONFLEX 8; CONFLEX Corporation: Tokyo, Japan, 2017. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Goto, H.; Osawa, E. Corner Flapping: A Simple and Fast Algorithm for Exhaustive Generation of Ring Conformations. J. Am. Chem. Soc. 1989, 111, 8950–8951. [Google Scholar] [CrossRef]
- Goto, H.; Osawa, E. An Efficient Algorithm for Searching Low-energy Conformers of Cyclic and Acyclic Molecules. J. Chem. Soc. Perkin Trans. 2 1993, 187–198. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umezawa, T.; Mizutani, N.; Matsuo, K.; Tokunaga, Y.; Matsuda, F.; Nehira, T. Assignment of Absolute Configuration of Bromoallenes by Vacuum-Ultraviolet Circular Dichroism (VUVCD). Molecules 2021, 26, 1296. https://doi.org/10.3390/molecules26051296
Umezawa T, Mizutani N, Matsuo K, Tokunaga Y, Matsuda F, Nehira T. Assignment of Absolute Configuration of Bromoallenes by Vacuum-Ultraviolet Circular Dichroism (VUVCD). Molecules. 2021; 26(5):1296. https://doi.org/10.3390/molecules26051296
Chicago/Turabian StyleUmezawa, Taiki, Nakaba Mizutani, Koichi Matsuo, Yuugo Tokunaga, Fuyuhiko Matsuda, and Tatsuo Nehira. 2021. "Assignment of Absolute Configuration of Bromoallenes by Vacuum-Ultraviolet Circular Dichroism (VUVCD)" Molecules 26, no. 5: 1296. https://doi.org/10.3390/molecules26051296