
Insights into the Mechanism and Catalysis of Peptide Thioester Synthesis by Alkylselenols Provide a New Tool for Chemical Protein Synthesis

 ,
,  and
and
Abstract
:
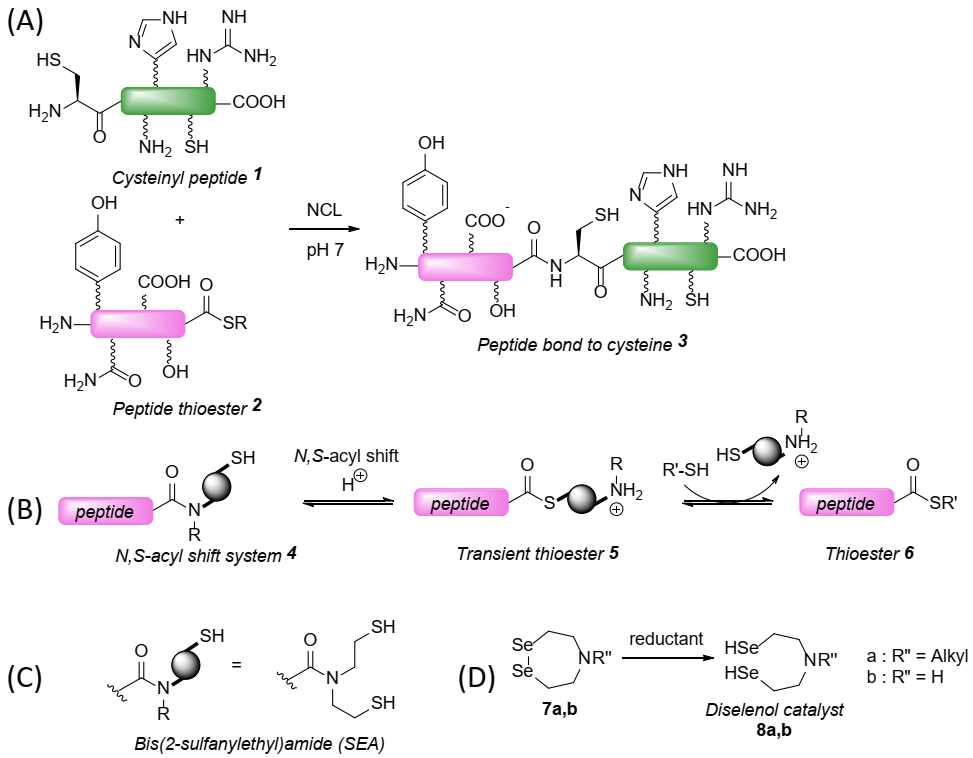
1. Introduction
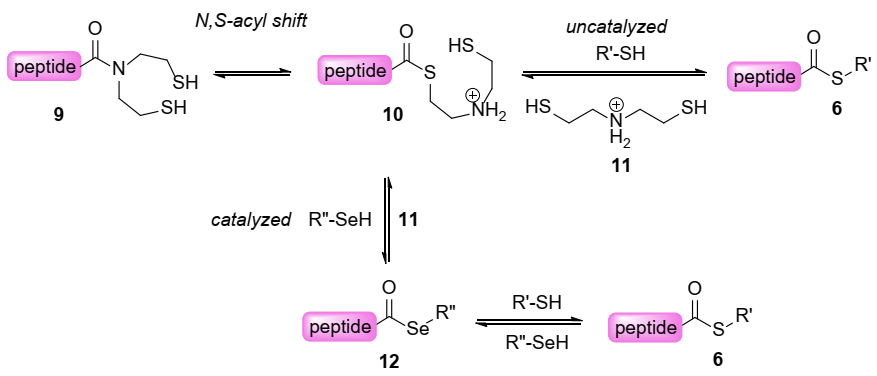
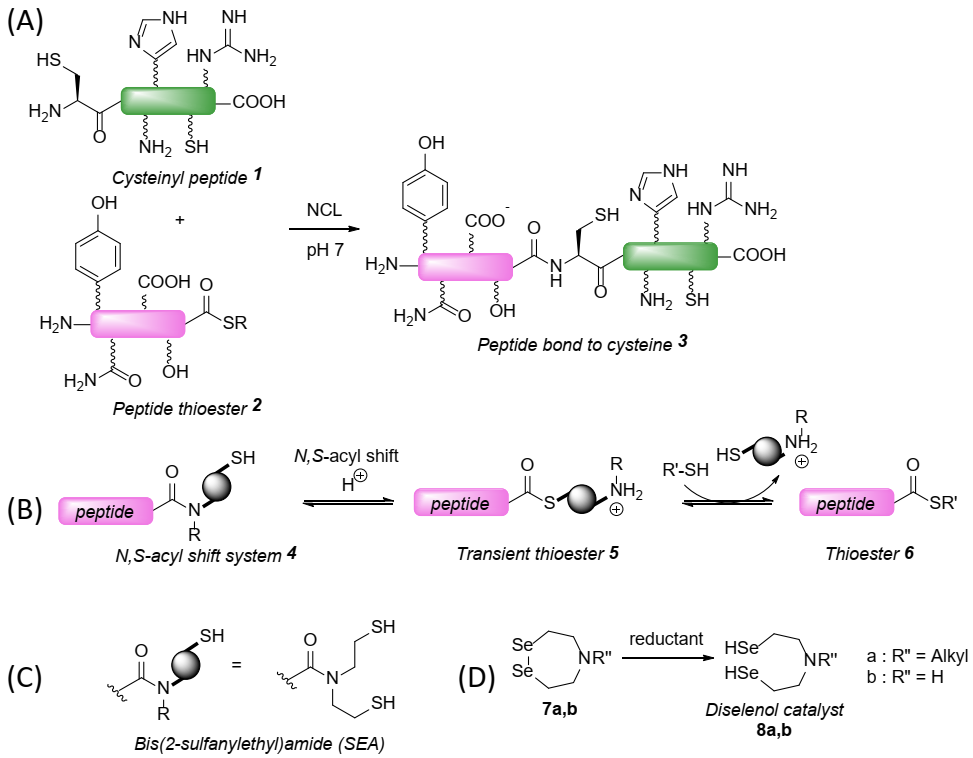
2. Results and Discussion
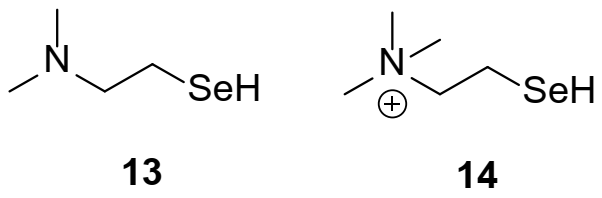
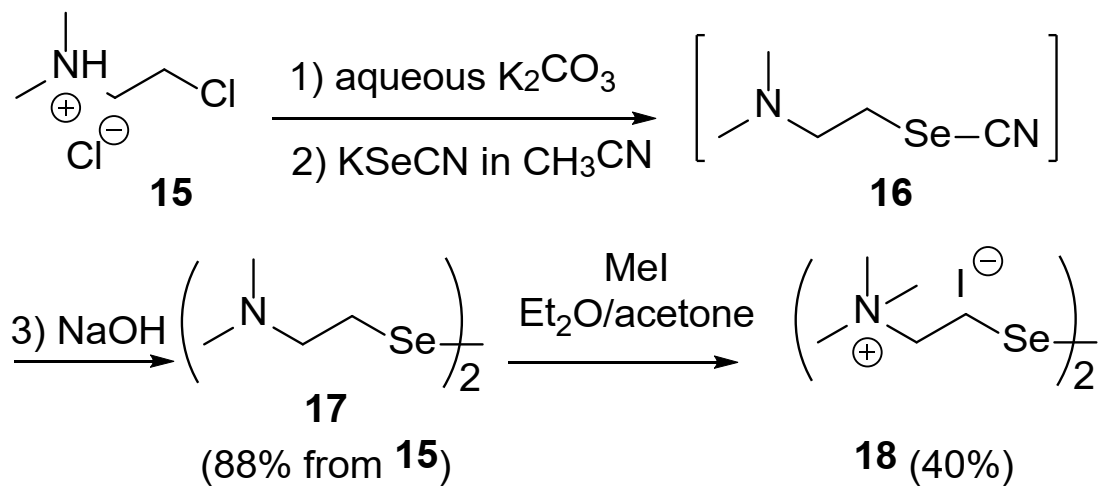
2.1. Catalyst Synthesis
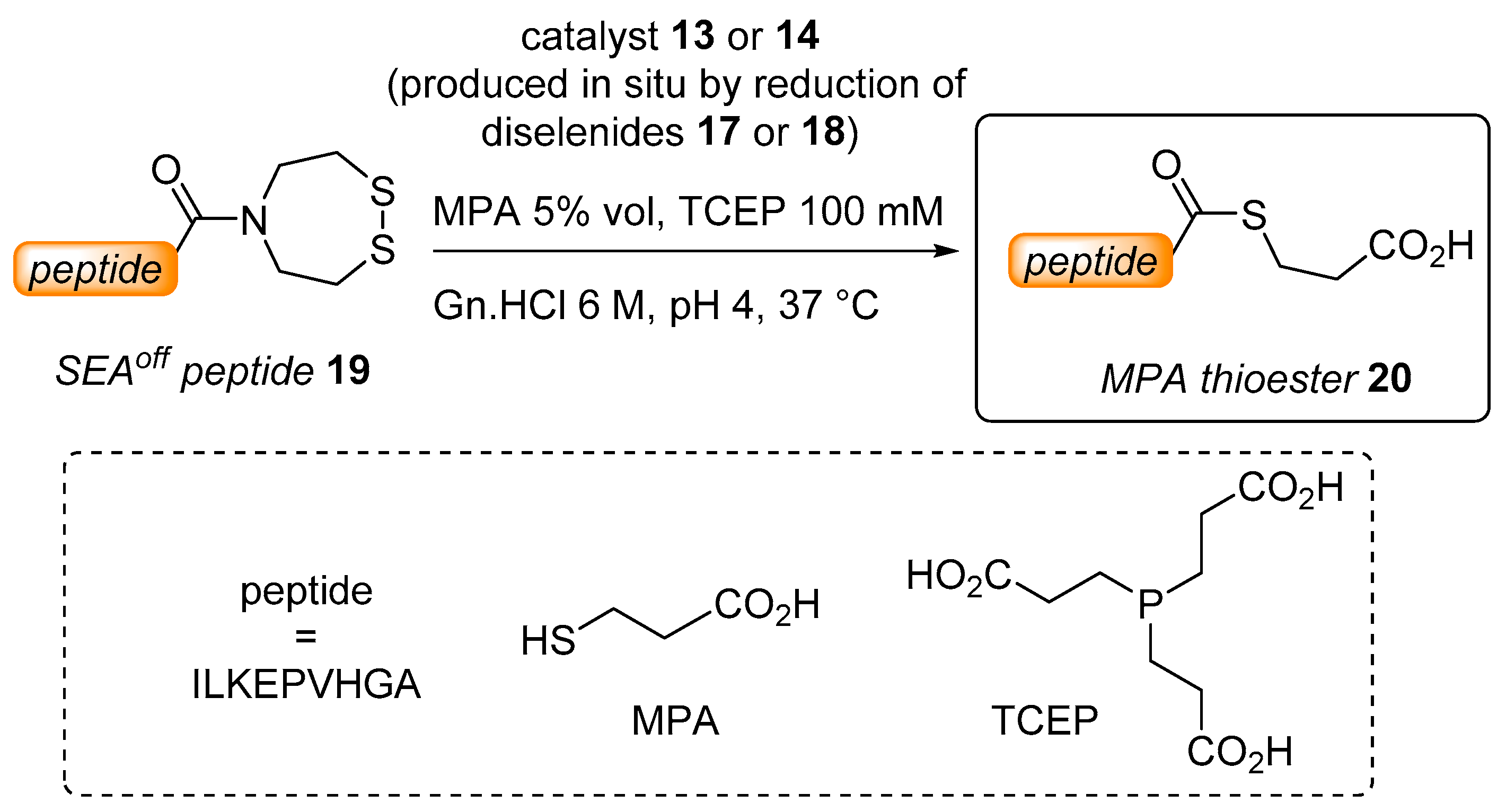
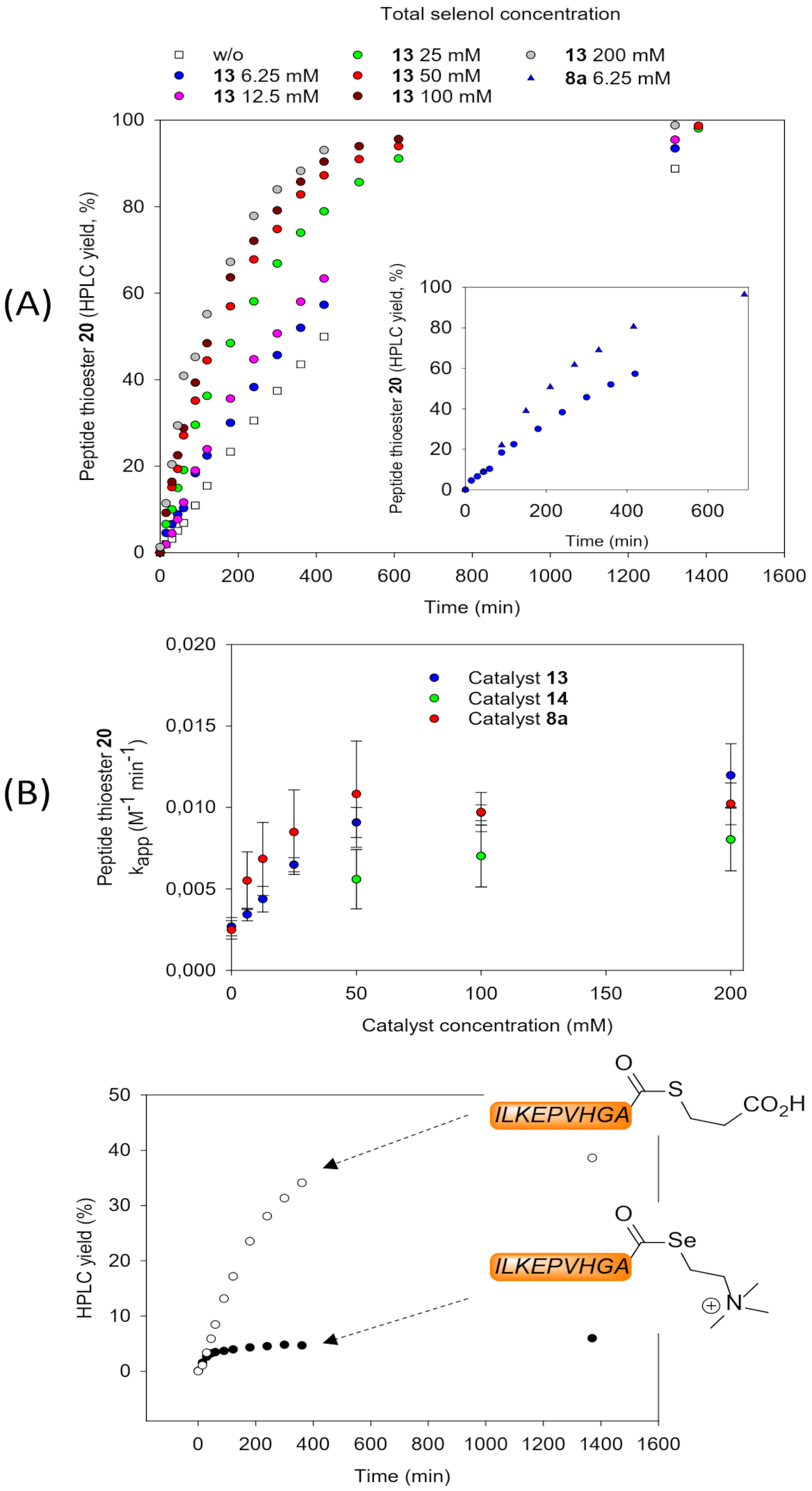
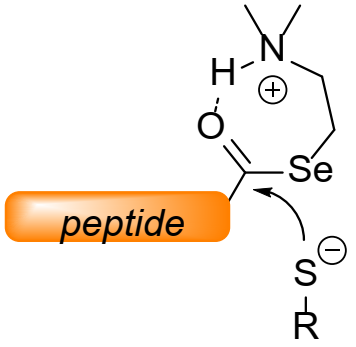
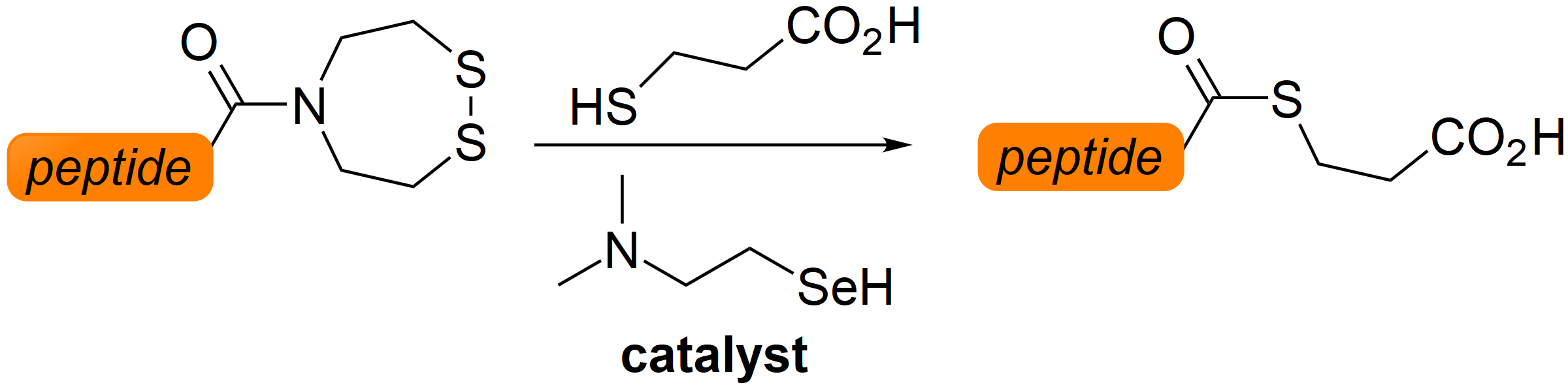
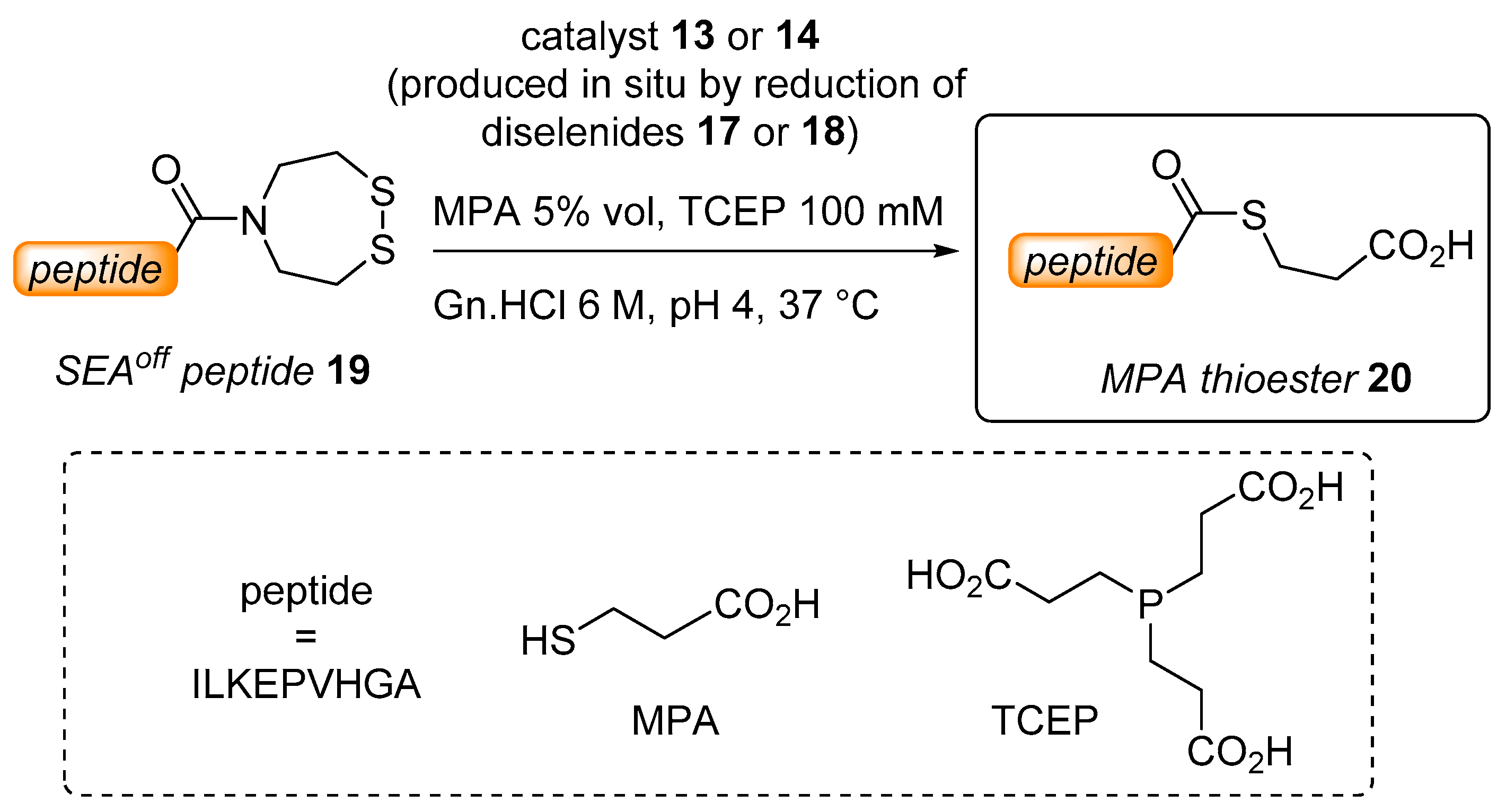
2.2. Kinetic Studies
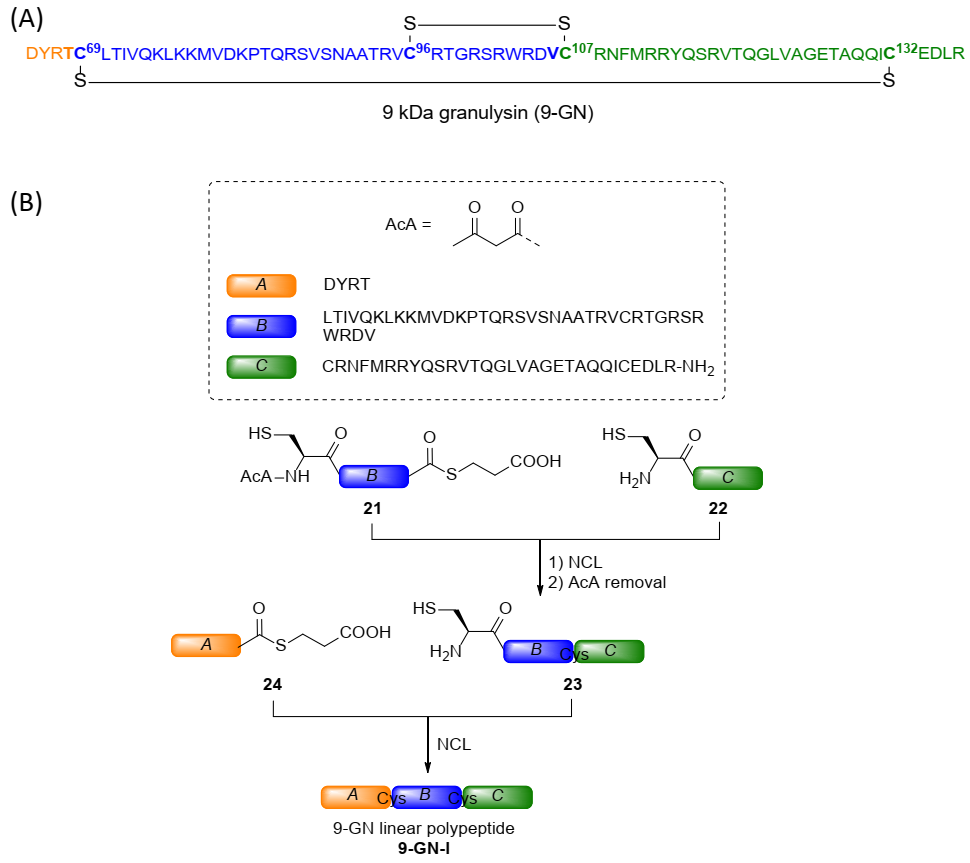
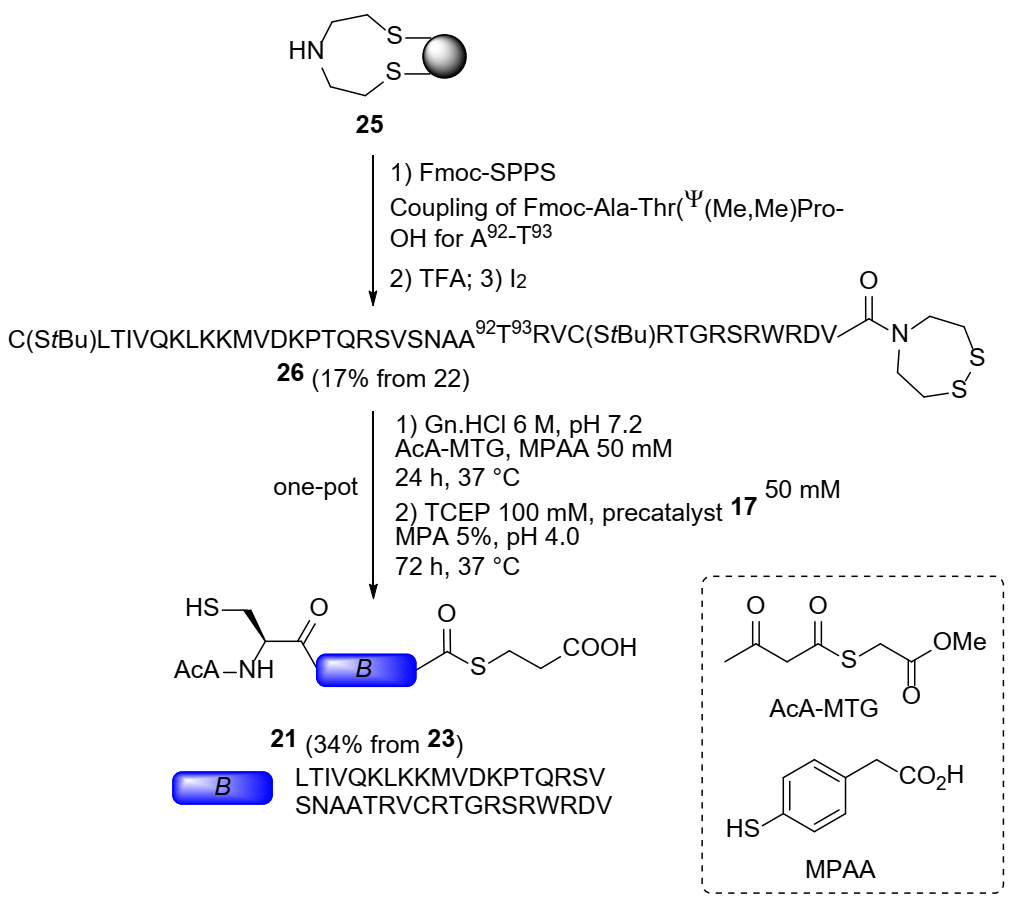
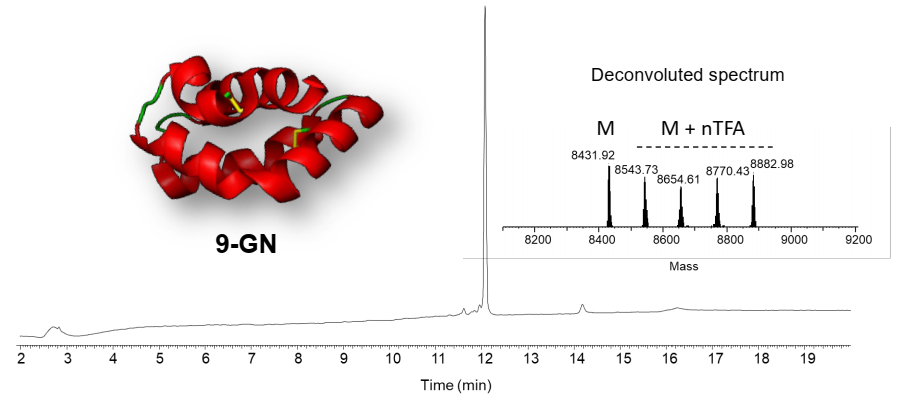
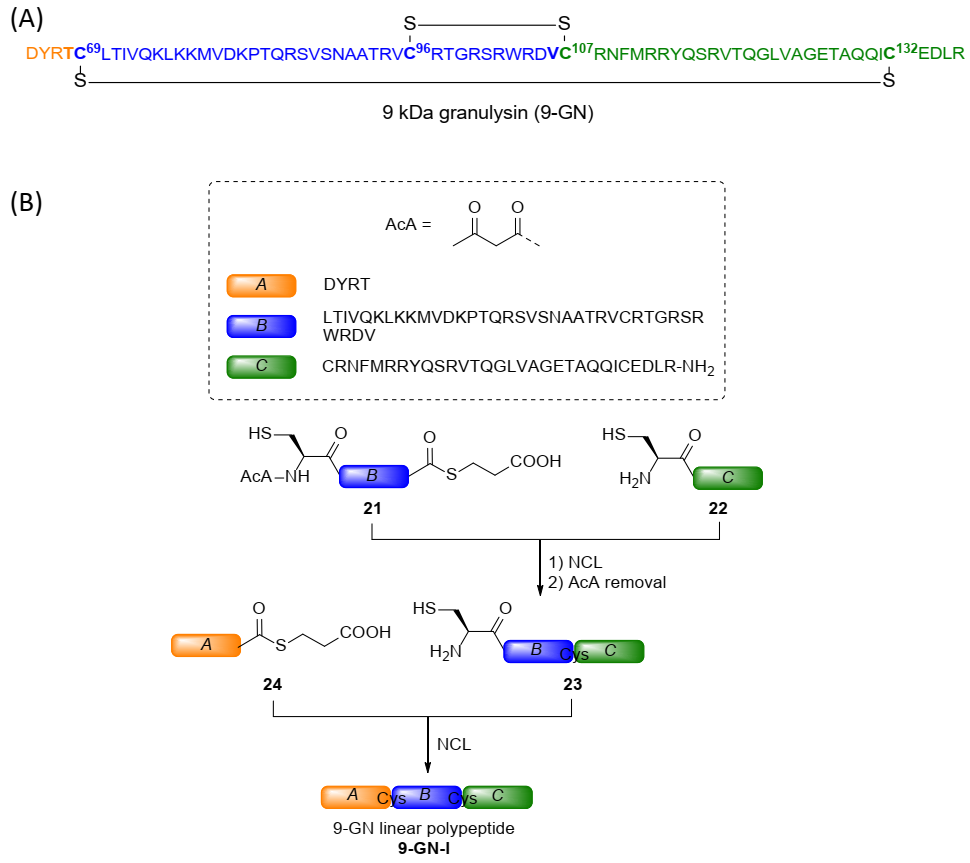
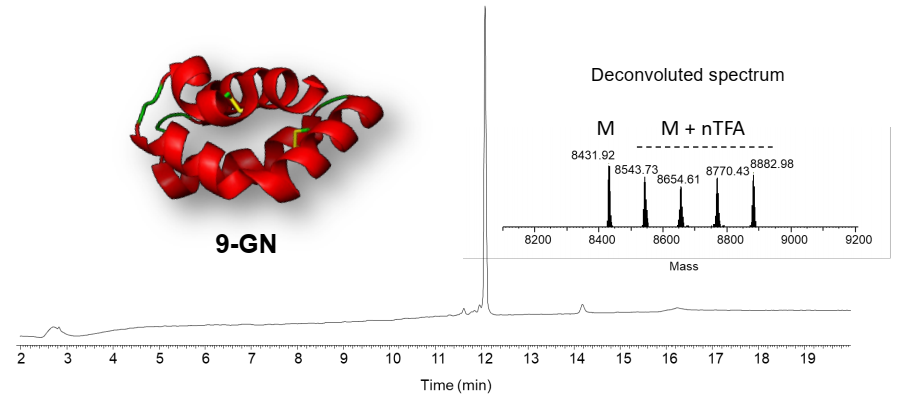
2.3. Total Chemical Synthesis of 9 kDa Granulysin
2.4. Biological Activity of Synthetic 9-GN
3. Materials and Methods
3.1. Synthesis of Diselenide Precatalyst 17
3.2. Synthesis of 9-GN
3.3. Folding
3.4. Chimiotaxis Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agouridas, V.; El Mahdi, O.; Melnyk, O. Chemical Protein Synthesis in Medicinal Chemistry. J. Med. Chem. 2020, 63, 15140–15152. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Raibaut, L.; Ollivier, N.; Melnyk, O. Sequential Native Peptide Ligation Strategies for Total Chemical Protein Synthesis. Chem. Soc. Rev. 2012, 41, 7001–7015. [Google Scholar] [CrossRef] [PubMed]
- Conibear, A.C.; Watson, E.E.; Payne, R.J.; Becker, C.F.W. Native Chemical Ligation in Protein Synthesis and Semi-Synthesis. Chem. Soc. Rev. 2018, 47, 9046–9068. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Sayers, J.; Premdjee, B.; Payne, R.J. Rapid and Efficient Protein Synthesis through Expansion of the Native Chemical Ligation Concept. Nat. Rev. Chem. 2018, 2, 0122. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoet, M.; Monbaliu, J.-C.M.; Melnyk, O. Native Chemical Ligation and Extended Methods. Mechanisms, Catalysis, Scope and Limitations. Chem. Rev. 2019, 12, 7328–7443. [Google Scholar] [CrossRef]
- Wan, Q.; Danishefsky, S.J. Free-Radical-Based, Specific Desulfurization of Cysteine: A Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angew. Chem. Int. Ed. 2007, 46, 9248–9252. [Google Scholar] [CrossRef]
- Yan, L.Z.; Dawson, P.E. Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization. J. Am. Chem. Soc. 2001, 123, 526–533. [Google Scholar] [CrossRef]
- Jin, K.; Li, X. Advances in Native Chemical Ligation–Desulfurization: A Powerful Strategy for Peptide and Protein Synthesis. Chem. A Eur. J. 2018, 24, 17397–17404. [Google Scholar] [CrossRef]
- Jin, K.; Li, T.; Chow, H.Y.; Liu, H.; Li, X. P−B Desulfurization: An Enabling Method for Protein Chemical Synthesis and Site-Specific Deuteration. Angew. Chem. Int. Ed. 2017, 56, 14607–14611. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Cargoët, M.; Melnyk, O. A Statistical View of Protein Chemical Synthesis Using NCL and Extended Methodologies. Bioorg. Med. Chem. 2017, 25, 4938–4945. [Google Scholar] [CrossRef]
- Dawson, P.E.; Churchill, M.J.; Ghadiri, M.R.; Kent, S.B.H. Modulation of Reactivity in Native Chemical Ligation through the Use of Thiol Additives. J. Am. Chem. Soc. 1997, 119, 4325–4329. [Google Scholar] [CrossRef]
- Mitchell, N.J.; Malins, L.R.; Liu, X.; Thompson, R.E.; Chan, B.; Radom, L.; Payne, R.J. Rapid Additive-Free Selenocystine-Selenoester Peptide Ligation. J. Am. Chem. Soc. 2015, 137, 14011–14014. [Google Scholar] [CrossRef]
- Chisholm, T.S.; Clayton, D.; Dowman, L.J.; Sayers, J.; Payne, R.J. Native Chemical Ligation–Photodesulfurization in Flow. J. Am. Chem. Soc. 2018, 29, 9020–9024. [Google Scholar] [CrossRef]
- Ollivier, N.; Toupy, T.; Hartkoorn, R.C.; Desmet, R.; Monbaliu, J.-C.M.; Melnyk, O. Accelerated Microfluidic Native Chemical Ligation at Difficult Amino Acids toward Cyclic Peptides. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durek, T.; Alewood, P.F. Preformed Selenoesters Enable Rapid Native Chemical Ligation at Intractable Sites. Angew. Chem. Int. Ed. 2011, 50, 12042–12045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Tang, S.; Huang, Y.-C.; Liu, L. Development of New Thioester Equivalents for Protein Chemical Synthesis. Acc. Chem. Res. 2013, 46, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, N.; Behr, J.B.; El-Mahdi, O.; Blanpain, A.; Melnyk, O. Fmoc Solid-Phase Synthesis of Peptide Thioesters Using an Intramolecular N,S-Acyl Shift. Org. Lett. 2005, 7, 2647–2650. [Google Scholar] [CrossRef]
- Ohta, Y.; Itoh, S.; Shigenaga, A.; Shintaku, S.; Fujii, N.; Otaka, A. Cysteine-Derived S-Protected Oxazolidinones: Potential Chemical Devices for the Preparation of Peptide Thioesters. Org. Lett. 2006, 8, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Onuma, Y.; Akimoto, Y.; Nakahara, Y.; Nakahara, Y. N-Alkyl Cysteine-Assisted Thioesterification of Peptides. Tetrahedron Lett. 2007, 48, 25–28. [Google Scholar] [CrossRef]
- Kawakami, T.; Aimoto, S. The Use of a Cysteinyl Prolyl Ester (CPE) Autoactivating Unit in Peptide Ligation Reactions. Tetrahedron 2009, 65, 3871–3877. [Google Scholar] [CrossRef]
- Kang, J.; Richardson, J.P.; Macmillan, D. 3-Mercaptopropionic Acid-Mediated Synthesis of Peptide and Protein Thioesters. Chem. Commun. 2009, 4, 407–409. [Google Scholar] [CrossRef] [Green Version]
- Ollivier, N.; Dheur, J.; Mhidia, R.; Blanpain, A.; Melnyk, O. Bis(2-Sulfanylethyl)Amino Native Peptide Ligation. Org. Lett. 2010, 12, 5238–5241. [Google Scholar] [CrossRef]
- Macmillan, D.; De Cecco, M.; Reynolds, N.L.; Santos, L.F.A.; Barran, P.E.; Dorin, J.R. Synthesis of Cyclic Peptides through an Intramolecular Amide Bond Rearrangement. ChemBioChem 2011, 12, 2133–2136. [Google Scholar] [CrossRef]
- Hou, W.; Zhang, X.; Li, F.; Liu, C.F. Peptidyl N,N-Bis(2-Mercaptoethyl)-Amides as Thioester Precursors for Native Chemical Ligation. Org. Lett. 2011, 13, 386–389. [Google Scholar] [CrossRef]
- Sato, K.; Shigenaga, A.; Tsuji, K.; Tsuda, S.; Sumikawa, Y.; Sakamoto, K.; Otaka, A. N-sulfanylethylanilide Peptide as a Crypto-Thioester Peptide. ChemBioChem 2011, 12, 1840–1844. [Google Scholar] [CrossRef]
- Adams, A.L.; Macmillan, D. Investigation of Peptide Thioester Formation via N→Se Acyl Transfer. J. Pept. Sci. 2013, 19, 65–73. [Google Scholar] [CrossRef]
- Taichi, M.; Hemu, X.; Qiu, Y.; Tam, J.P. A Thioethylalkylamido (TEA) Thioester Surrogate in the Synthesis of a Cyclic Peptide via a Tandem Acyl Shift. Org. Lett. 2013, 15, 2620–2623. [Google Scholar] [CrossRef] [PubMed]
- Ruff, Y.; Garavini, V.; Giuseppone, N. Reversible Native Chemical Ligation: A Facile Access to Dynamic Covalent Peptides. J. Am. Chem. Soc. 2014, 136, 6333–6339. [Google Scholar] [CrossRef]
- Burlina, F.; Papageorgiou, G.; Morris, C.; White, P.D.; Offer, J. In Situ Thioester Formation for Protein Ligation Using a-Methylcysteine. Chem. Sci. 2014, 5, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Asahina, Y.; Nabeshima, K.; Hojo, H. Peptidyl N-Alkylcysteine as a Peptide Thioester Surrogate in the Native Chemical Ligation. Tetrahedron Lett. 2015, 56, 1370–1373. [Google Scholar] [CrossRef]
- Eto, M.; Naruse, N.; Morimoto, K.; Yamaoka, K.; Sato, K.; Tsuji, K.; Inokuma, T.; Shigenaga, A.; Otaka, A. Development of an Anilide-Type Scaffold for the Thioester Precursor N-Sulfanylethylcoumarinyl Amide. Org. Lett. 2016, 18, 4416–4419. [Google Scholar] [CrossRef]
- Terrier, V.P.; Adihou, H.; Arnould, M.; Delmas, A.F.; Aucagne, V. A Straightforward Method for Automated Fmoc-Based Synthesis of Bio-Inspired Peptide Crypto-Thioesters. Chem. Sci. 2016, 7, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, S.; Mochizuki, M.; Sakamoto, K.; Denda, M.; Nishio, H.; Otaka, A.; Yoshiya, T. N-Sulfanylethylaminooxybutyramide (SEAoxy): A Crypto-Thioester Compatible with Fmoc Solid-Phase Peptide Synthesis. Org. Lett. 2016, 18, 5940–5943. [Google Scholar] [CrossRef]
- Rao, C.; Liu, C.F. Peptide Weinreb Amide Derivatives as Thioester Precursors for Native Chemical Ligation. Org. Biomol. Chem. 2017, 15, 2491–2496. [Google Scholar] [CrossRef]
- Shelton, P.M.; Weller, C.E.; Chatterjee, C. A Facile N-Mercaptoethoxyglycinamide (MEGA) Linker Approach to Peptide Thioesterification and Cyclization. J. Am. Chem. Soc. 2017, 139, 3946–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollivier, N.; Desmet, R.; Drobecq, H.; Blanpain, A.; Boll, E.; Leclercq, B.; Mougel, A.; Vicogne, J.; Melnyk, O. A Simple and Traceless Solid Phase Method Simplifies the Assembly of Large Peptides and the Access to Challenging Proteins. Chem. Sci. 2017, 8, 5362–5370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raibaut, L.; Cargoët, M.; Ollivier, N.; Chang, Y.M.; Drobecq, H.; Boll, E.; Desmet, R.; Monbaliu, J.-C.M.; Melnyk, O. Accelerating Chemoselective Peptide Bond Formation Using Bis(2-Selenylethyl)Amido Peptide Selenoester Surrogates. Chem. Sci. 2016, 7, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchenna, J.; Sénéchal, M.; Drobecq, H.; Vicogne, J.; Melnyk, O. Total Chemical Synthesis of All SUMO-2/3 Dimer Combinations. Bioconjug. Chem. 2019, 30, 2967–2973. [Google Scholar] [CrossRef] [PubMed]
- Abboud, S.A.; Cisse, E.h.; Doudeau, M.; Bénédetti, H.; Aucagne, V. A Straightforward Methodology to Overcome Solubility Challenges for N-Terminal Cysteinyl Peptide Segments Used in Native Chemical Ligation. Chem. Sci. 2021. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, K.; Naruse, N.; Kitakaze, K.; Inokuma, T.; Hirokawa, T.; Shigenaga, A.; Itoh, K.; Otaka, A. Tailored Synthesis of 162-Residue S-Monoglycosylated GM2-Activator Protein (GM2AP) Analogues That Allows Facile Access to a Protein Library. Chembiochem 2016, 17, 1986–1992. [Google Scholar] [CrossRef]
- Sato, K.; Shigenaga, A.; Kitakaze, K.; Sakamoto, K.; Tsuji, D.; Itoh, K.; Otaka, A. Chemical Synthesis of Biologically Active Monoglycosylated GM2-Activator Protein Analogue Using N-Sulfanylethylanilide Peptide. Angew. Chem. Int. Ed. 2013, 52, 7855–7859. [Google Scholar] [CrossRef]
- Sun, H.; Brik, A. The Journey for the Total Chemical Synthesis of a 53 kDa Protein. Acc. Chem. Res. 2019, 52, 3361–3371. [Google Scholar] [CrossRef]
- Kumar, K.S.; Bavikar, S.N.; Spasser, L.; Moyal, T.; Ohayon, S.; Brik, A. Total Chemical Synthesis of a 304 Amino Acid K48-Linked Tetraubiquitin Protein. Angew. Chem. Int. Ed. 2011, 50, 6137–6141. [Google Scholar] [CrossRef] [PubMed]
- Pira, S.L.; El Mahdi, O.; Raibaut, L.; Drobecq, H.; Dheur, J.; Boll, E.; Melnyk, O. Insight into the SEA Amide Thioester Equilibrium. Application to the Synthesis of Thioesters at Neutral pH. Org. Biomol. Chem. 2016, 14, 7211–7216. [Google Scholar] [CrossRef]
- Hupe, D.J.; Jencks, W.P. Nonlinear Structure-Reactivity Correlations. Acyl Transfer between Sulfur and Oxygen Nucleophiles. J. Am. Chem. Soc. 1977, 99, 451–464. [Google Scholar] [CrossRef]
- Cargoët, M.; Diemer, V.; Snella, B.; Desmet, R.; Blanpain, A.; Drobecq, H.; Agouridas, V.; Melnyk, O. Catalysis of Thiol-Thioester Exchange by Water-Soluble Alkyldiselenols Applied to the Synthesis of Peptide Thioesters and SEA-Mediated Ligation. J. Org. Chem. 2018, 83, 12584–12594. [Google Scholar] [CrossRef]
- Shefter, E.; Kennard, O. Crystal and Molecular Structure of Acetylselenocholine Iodide. Science 1966, 153, 1389. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.-H.; Mautner, H.G. Analogs of Neuroeffectors. V. Neighboring-Group Effects in the Reactions of Esters, Thiolesters, and Selenolesters. The Hydrolysis and Aminolysis of Benzoylcholine, Benzoylthiolcholine, Benzoylselenolcholine, and of their Dimethylamino Analogs. J. Org. Chem. 1966, 31, 308–312. [Google Scholar] [CrossRef]
- Günther, W.H.H.; Mautner, H.G. Analogs of Parasympathetic Neuroeffectors. I. Acetylselenocholine, Selenocholine, and Related Compounds. J. Med. Chem. 1964, 7, 229–232. [Google Scholar]
- Krensky, A.M.; Clayberger, C. Biology and Clinical Relevance of Granulysin. Tissue Antigens 2009, 73, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Stenger, S.; Hanson, D.A.; Teitelbaum, R.; Dewan, P.; Niazi, K.R.; Froelich, C.J.; Ganz, T.; Thoma-Uszynski, S.; Melian, A.; Bogdan, C.; et al. An Antimicrobial Activity of Cytolytic T Cells Mediated by Granulysin. Science 1998, 282, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, S.V.; Hanson, D.A.; Carr, B.A.; Goralski, T.J.; Krensky, A.M. Processing, Subcellular Localization, and Function of 519 (Granulysin), a Human Late T cell Activation Molecule with Homology to Small, Lytic, Granule Proteins. J. Immunol. 1997, 158, 2680–2688. [Google Scholar]
- Krief, A.; Dumont, W.; Delmotte, C. Reaction of Organic Selenocyanates with Hydroxides: The One-Pot Synthesis of Dialkyl Diselenides from Alkyl Bromides. Angew. Chem. Int. Ed. 2000, 39, 1669–1672. [Google Scholar] [CrossRef]
- Pena, S.V.; Krensky, A.M. Granulysin, a New Human Cytolytic Granule-Associated Protein with Possible Involvement in Cell-Mediated Cytotoxicity. Semin. Immunol. 1997, 9, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.; Chen, S.; Li, Q.; Lyu, S.C.; Clayberger, C.; Krensky, A.M. Granulysin, a Cytolytic Molecule, Is Also a Chemoattractant and Proinflammatory Activator. J. Immunol. 2005, 174, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Boll, E.; Ebran, J.P.; Drobecq, H.; El-Mahdi, O.; Raibaut, L.; Ollivier, N.; Melnyk, O. Access to Large Cyclic Peptides by a One-Pot Two-Peptide Segment Ligation/Cyclization Process. Org. Lett. 2015, 17, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.; Mutter, M. Serine Derived Oxazolidines as Secondary Structure Disrupting, Solubilizing Building Blocks in Peptide Synthesis. Tetrahedron Lett. 1992, 33, 1589–1592. [Google Scholar] [CrossRef]
- Anderson, D.H.; Sawaya, M.R.; Cascio, D.; Ernst, W.; Modlin, R.; Krensky, A.; Eisenberg, D. Granulysin Crystal Structure and a Structure-Derived Lytic Mechanism. J. Mol. Biol. 2003, 325, 355–365. [Google Scholar] [CrossRef]
- Pichavant, M.; Taront, S.; Jeannin, P.; Breuilh, L.; Charbonnier, A.S.; Spriet, C.; Fourneau, C.; Corvaia, N.; Heliot, L.; Brichet, A.; et al. Impact of Bronchial Epithelium on Dendritic Cell Migration and Function: Modulation by the Bacterial Motif KpOmpA. J. Immunol. 2006, 177, 5912–5919. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Total Selenol Concentration 8a, 13, 14 (mM) | Catalyst t1/2 (h) | ||
|---|---|---|---|---|
| 8a c | 13 | 14 d | ||
| 1 | 200 | 1.97 | 1.68 | 2.51 |
| 2 | 100 | 2.03 | 2.08 | 2.87 |
| 3 | 50 | 1.95 | 2.22 | 3.60 |
| 4 | 25 | 2.35 | 3.10 | nd |
| 5 | 12.5 | 3.00 | 4.60 | nd |
| 6 | 6.25 | 3.35 | 5.87 | nd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerdraon, F.; Bogard, G.; Snella, B.; Drobecq, H.; Pichavant, M.; Agouridas, V.; Melnyk, O. Insights into the Mechanism and Catalysis of Peptide Thioester Synthesis by Alkylselenols Provide a New Tool for Chemical Protein Synthesis. Molecules 2021, 26, 1386. https://doi.org/10.3390/molecules26051386
Kerdraon F, Bogard G, Snella B, Drobecq H, Pichavant M, Agouridas V, Melnyk O. Insights into the Mechanism and Catalysis of Peptide Thioester Synthesis by Alkylselenols Provide a New Tool for Chemical Protein Synthesis. Molecules. 2021; 26(5):1386. https://doi.org/10.3390/molecules26051386
Chicago/Turabian StyleKerdraon, Florent, Gemma Bogard, Benoît Snella, Hervé Drobecq, Muriel Pichavant, Vangelis Agouridas, and Oleg Melnyk. 2021. "Insights into the Mechanism and Catalysis of Peptide Thioester Synthesis by Alkylselenols Provide a New Tool for Chemical Protein Synthesis" Molecules 26, no. 5: 1386. https://doi.org/10.3390/molecules26051386
APA StyleKerdraon, F., Bogard, G., Snella, B., Drobecq, H., Pichavant, M., Agouridas, V., & Melnyk, O. (2021). Insights into the Mechanism and Catalysis of Peptide Thioester Synthesis by Alkylselenols Provide a New Tool for Chemical Protein Synthesis. Molecules, 26(5), 1386. https://doi.org/10.3390/molecules26051386