
Novel Fast Chromatography-Tandem Mass Spectrometric Quantitative Approach for the Determination of Plant-Extracted Phytosterols and Tocopherols


Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development
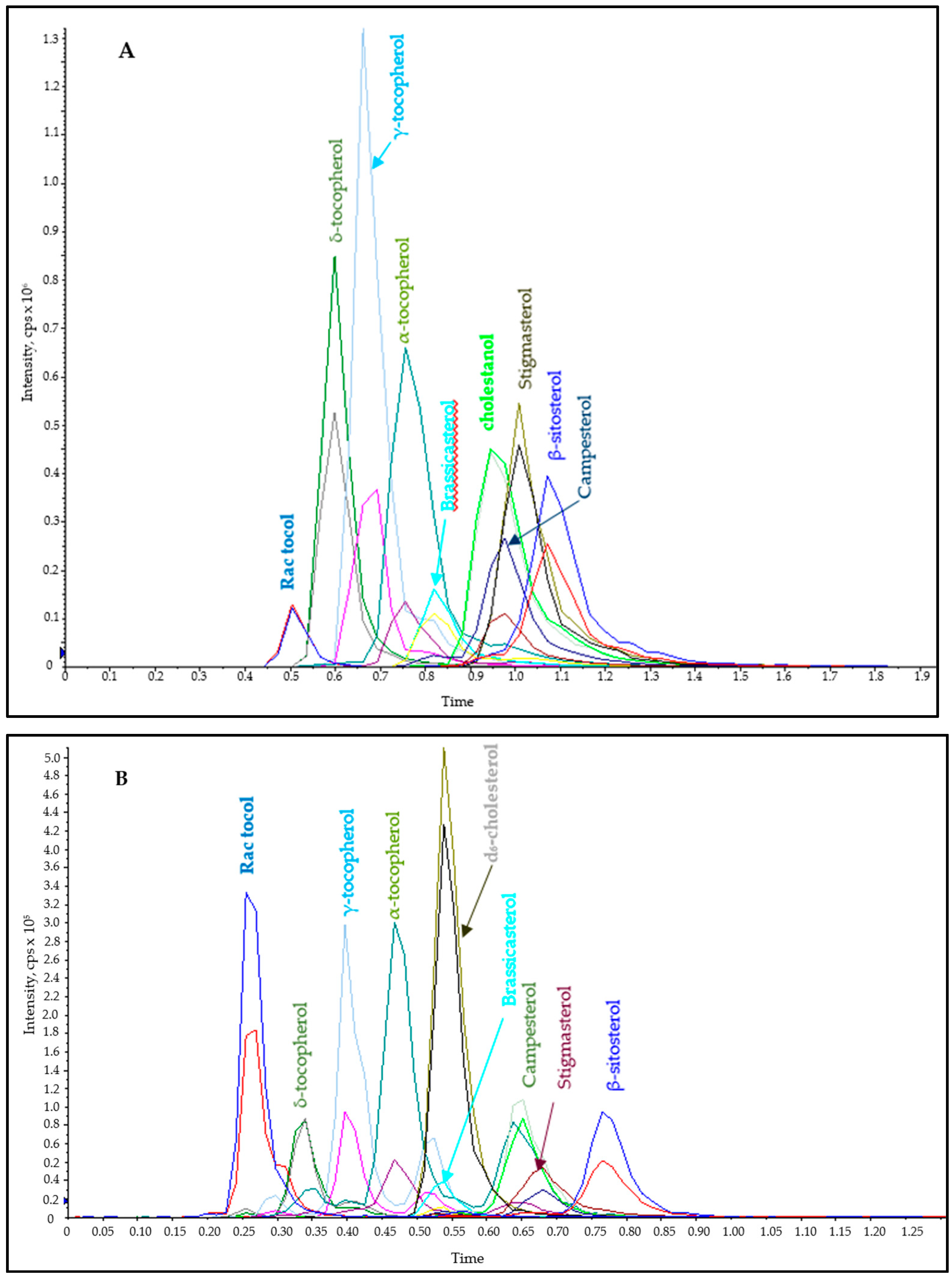
2.1.1. Fast Chromatography (FC)
2.1.2. MS Conditions
2.2. Method Validation
2.2.1. Calibration Range and Sensitivity
2.2.2. Intra and Interday Accuracy and Precision
2.2.3. Recovery and Matrix Effects
2.2.4. Stability
2.3. Application of the Method for the Quantification of Phytosterols and Tocopherols
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Standard Solutions
3.3. Sample Preparation
Unsaponifiable Matter
3.4. HPLC and MS Parameters
3.5. Method Validation
3.5.1. Calibration Curve and Sensitivity
3.5.2. Intraday and Interday Accuracy and Precision
3.5.3. Recovery and Matrix Effect
3.5.4. Stability
4. Application of the Method for the Analysis of Phytosterols and Tocopherols
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Eussen, S.R.; de Jong, N.; Rompelberg, C.J.; Garssen, J.; Verschuren, W.M.; Klungel, O.H. Dose-dependent cholesterol-lowering effects of phytosterol/phytostanol-enriched margarine in statin users and statin non-users under free-living conditions. Public Health Nutr. 2011, 14, 1823–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautwein, E.A.; Demonty, I. Phytosterols: Natural compounds with established and emerging health benefits. Oléagineux Corps gras Lipides 2007, 14, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Frei, B. Reactive oxygen species and antioxidant vitamins: Mechanisms of action. Am. J. Med. 1994, 97, S5–S13. [Google Scholar] [CrossRef]
- Rowlands, J.C.; Hoadley, J.E. Fda perspectives on health claims for food labels. Toxicology 2006, 221, 35–43. [Google Scholar] [CrossRef]
- Commission Regulation (EC), No. 608/2004 concerning the labelling of foods and food ingredients with added phytosterols, phytosterol esters, phytostanols and/or phytostanol (Text with EEA relevance). Off. J. L 2013, 201, 49–50. [Google Scholar]
- Verleyen, T.; Verhé, R.; Garcia, L.; Dewettinck, K.; Huyghebaert, A.; De Greyt, W. Gas chromatographic characterization of vegetable oil deodorization distillate. J. Chromatogr. A 2001, 921, 277–285. [Google Scholar] [CrossRef]
- Xu, B.; You, S.; Zhou, L.; Kang, H.; Luo, D.; Ma, H.; Han, S. Simultaneous determination of free phytosterols and tocopherols in vegetable oils by an improved SPE–GC–FID method. Food Anal. Methods 2020, 13, 358–369. [Google Scholar] [CrossRef]
- Mohamed, H.; Awatif, I. The use of sesame oil unsaponifiable matter as a natural antioxidant. Food Chem. 1998, 62, 269–276. [Google Scholar] [CrossRef]
- Kasim, N.S.; Gunawan, S.; Yuliana, M.; Ju, Y.-H. A simple two-step method for simultaneous isolation of tocopherols and free phytosterols from soybean oil deodorizer distillate with high purity and recovery. Sep. Sci. Technol. 2010, 45, 2437–2446. [Google Scholar] [CrossRef]
- Raja Rajan, R.; Gopala Krishna, A. A simple method for purification of deodorizer distillate from indian rice (oryza sativa) bran oil and preparation of phytosterols. Grasas y Aceites 2014, 65, e050. [Google Scholar]
- Schott, H.-F.; Krautbauer, S.; Höring, M.; Liebisch, G.; Matysik, S. A validated, fast method for quantification of sterols and gut microbiome derived 5α/β-stanols in human feces by isotope dilution LC–high-resolution MS. Anal. Chem. 2018, 90, 8487–8494. [Google Scholar] [CrossRef] [PubMed]
- Holder, C.; Adams, A.; McGahee, E.; Xia, B.; Blount, B.C.; Wang, L. High-throughput and sensitive analysis of free and total 8-isoprostane in urine with isotope-dilution liquid chromatography-tandem mass spectrometry. ACS Omega 2020, 5, 10919–10926. [Google Scholar] [CrossRef] [PubMed]
- Beldean-Galea, M.S.; Horga, C.; Coman, M.V. Separation and determination of tocopherols in vegetable oils by solid phase extraction on porous polymers spe cartridges and capillary gas chromatography analysis. Cent. Eur. J. Chem. 2010, 8, 1110–1116. [Google Scholar] [CrossRef]
- dos Santos, M.; Roehrs, M.; de Pereira, C.; Freitag, B.; de Bairros, A. Analysis of phytosterols in plants and derived products by gas chromatography—A short critical review. Austin Chromatogr. 2014, 1, 1–4. [Google Scholar]
- Pyka, A.; Sliwiok, J. Chromatographic separation of tocopherols. J. Chromatogr. A 2001, 935, 71–76. [Google Scholar] [CrossRef]
- Lechner, M.; Reiter, B.; Lorbeer, E. Determination of tocopherols and sterols in vegetable oils by solid-phase extraction and subsequent capillary gas chromatographic analysis. J. Chromatogr. A 1999, 857, 231–238. [Google Scholar] [CrossRef]
- Fernández-Cuesta, Á.; Aguirre-González, M.R.; Ruiz-Méndez, M.V.; Velasco, L. Validation of a method for the analysis of phytosterols in sunflower seeds. Eur. J. Lipid Sci. Technol. 2012, 114, 325–331. [Google Scholar] [CrossRef]
- Du, M.; Ahn, D. Simultaneous analysis of tocopherols, cholesterol, and phytosterols using gas chromatography. J. Food Sci. 2002, 67, 1696–1700. [Google Scholar] [CrossRef]
- García-Llatas, G.; Vidal, C.; Cilla, A.; Barberá, R.; Lagarda, M.J. Simultaneous quantification of serum phytosterols and cholesterol precursors using a simple gas chromatographic method. Eur. J. Lipid Sci. Technol. 2012, 114, 520–526. [Google Scholar] [CrossRef]
- Santos, R.; Limas, E.; Sousa, M.; da Conceição Castilho, M.; Ramos, F.; da Silveira, M.I.N. Optimization of analytical procedures for GC–MS determination of phytosterols and phytostanols in enriched milk and yoghurt. Food Chem. 2007, 102, 113–117. [Google Scholar] [CrossRef]
- Ahmida, H.M.; Bertucci, P.; Franzò, L.; Massoud, R.; Cortese, C.; Lala, A.; Federici, G. Simultaneous determination of plasmatic phytosterols and cholesterol precursors using gas chromatography–mass spectrometry (GC–MS) with selective ion monitoring (sim). J. Chromatogr. B 2006, 842, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Bartosińska, E.; Buszewska-Forajta, M.; Siluk, D. GC–MS and LC–MS approaches for determination of tocopherols and tocotrienols in biological and food matrices. J. Pharm. Biomed. Anal. 2016, 127, 156–169. [Google Scholar] [CrossRef]
- Zuba, D.; Adamowicz, P. Distinction of constitutional isomers of mephedrone by chromatographic and spectrometric methods. Aust. J. Forensic Sci. 2017, 49, 637–649. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, S.; Li, G.; Zhang, X.; Yang, J.; Lai, X.; Yang, G. Simultaneous analysis of tocopherols, tocotrienols, phospholipids, γ-oryzanols and β-carotene in rice by ultra-high performance liquid chromatography coupled to a linear ion trap-orbitrap mass spectrometer. Anal. Methods 2016, 8, 5628–5637. [Google Scholar] [CrossRef]
- Carretero, A.S.; Carrasco-Pancorbo, A.; Cortacero, S.; Gori, A.; Cerretani, L.; Fernández-Gutiérrez, A. A simplified method for HPLC-MS analysis of sterols in vegetable oil. Eur. J. Lipid Sci. Technol. 2008, 110, 1142–1149. [Google Scholar] [CrossRef]
- Flakelar, C.L.; Prenzler, P.D.; Luckett, D.J.; Howitt, J.A.; Doran, G. A rapid method for the simultaneous quantification of the major tocopherols, carotenoids, free and esterified sterols in canola (brassica napus) oil using normal phase liquid chromatography. Food Chem. 2017, 214, 147–155. [Google Scholar] [CrossRef]
- Cañabate-Díaz, B.; Carretero, A.S.; Fernández-Gutiérrez, A.; Vega, A.B.; Frenich, A.G.; Vidal, J.L.M.; Martos, J.D. Separation and determination of sterols in olive oil by HPLC-MS. Food Chem. 2007, 102, 593–598. [Google Scholar] [CrossRef]
- Baila-Rueda, L.; Cenarro, A.; Cofán, M.; Orera, I.; Barcelo-Batllori, S.; Pocoví, M.; Ros, E.; Civeira, F.; Nerín, C.; Domeño, C. Simultaneous determination of oxysterols, phytosterols and cholesterol precursors by high performance liquid chromatography tandem mass spectrometry in human serum. Anal. Methods 2013, 5, 2249–2257. [Google Scholar] [CrossRef]
- Lanina, S.A.; Toledo, P.; Sampels, S.; Kamal-Eldin, A.; Jastrebova, J.A. Comparison of reversed-phase liquid chromatography–mass spectrometry with electrospray and atmospheric pressure chemical ionization for analysis of dietary tocopherols. J. Chromatogr. A 2007, 1157, 159–170. [Google Scholar] [CrossRef]
- Huang, L.; Zhong, T.; Chen, T.; Ye, Z.; Chen, G. Identification of β-sitosterol, stigmasterol and ergosterin in a. Roxburghii using supercritical fluid extraction followed by liquid chromatography/atmospheric pressure chemical ionization ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 3024–3032. [Google Scholar] [CrossRef]
- Varga, M.; Bartók, T.; Mesterházy, Á. Determination of ergosterol in fusarium-infected wheat by liquid chromatography–atmospheric pressure photoionization mass spectrometry. J. Chromatogr. A 2006, 1103, 278–283. [Google Scholar] [CrossRef]
- Lauridsen, C.; Leonard, S.; Griffin, D.; Liebler, D.; McClure, T.; Traber, M. Quantitative analysis by liquid chromatography–tandem mass spectrometry of deuterium-labeled and unlabeled vitamin e in biological samples. Anal. Biochem. 2001, 289, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kalman, A.; Mujahid, C.; Mottier, P.; Heudi, O. Determination of α-tocopherol in infant foods by liquid chromatography combined with atmospheric pressure chemical ionisation mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, W.; Carrasco-Pancorbo, A.; Segura-Carretero, A.; Fernandez-Gutierrez, A.; Zarrouk, M. Exploratory characterization of the unsaponifiable fraction of tunisian virgin olive oils by a global approach with HPLC-APCI-IT MS/MS analysis. J. Agric. Food Chem. 2010, 58, 6418–6426. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.F.; Makahleh, A.; Saad, B.; Ibrahim, M.N.M.; Rahim, A.A.; Brosse, N. Uplc method for the determination of vitamin e homologues and derivatives in vegetable oils, margarines and supplement capsules using pentafluorophenyl column. Talanta 2014, 130, 299–306. [Google Scholar] [CrossRef]
- Grebenstein, N.; Frank, J. Rapid baseline-separation of all eight tocopherols and tocotrienols by reversed-phase liquid-chromatography with a solid-core pentafluorophenyl column and their sensitive quantification in plasma and liver. J. Chromatogr. A 2012, 1243, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Decloedt, A.; Van Landschoot, A.; Watson, H.; Vanderputten, D.; Vanhaecke, L. Plant-based beverages as good sources of free and glycosidic plant sterols. Nutrients 2018, 10, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataglion, G.A.; Meurer, E.; de Albergaria-Barbosa, A.C.R.; Bícego, M.C.; Weber, R.R.; Eberlin, M.N. Determination of geochemically important sterols and triterpenols in sediments using ultrahigh-performance liquid chromatography tandem mass spectrometry (UHPLC–MS/MS). Anal. Chem. 2015, 87, 7771–7778. [Google Scholar] [CrossRef]
- Poudel, A.; Gachumi, G.; Badea, I.; Bashi, Z.D.; El-Aneed, A. The simultaneous quantification of phytosterols and tocopherols in liposomal formulations using validated atmospheric pressure chemical ionization-liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 2020, 183, 113104. [Google Scholar] [CrossRef]
- Jiang, K.; Gachumi, G.; Poudel, A.; Shurmer, B.; Bashi, Z.; El-Aneed, A. The establishment of tandem mass spectrometric fingerprints of phytosterols and tocopherols and the development of targeted profiling strategies in vegetable oils. J. Am. Soc. Mass Spectrom. 2019, 30, 1700–1712. [Google Scholar] [CrossRef]
- Mohammed-Saeid, W.; Michel, D.; Badea, I.; El-Aneed, A. Rapid and simple flow injection analysis tandem mass spectrometric method for the quantification of melphalan in a lipid-based drug delivery system. Rapid Commun. Mass Spectrom. 2017, 31, 1481–1490. [Google Scholar] [CrossRef]
- Michel, D.; Gaunt, M.C.; Arnason, T.; El-Aneed, A. Development and validation of fast and simple flow injection analysis–tandem mass spectrometry (FIA–MS/MS) for the determination of metformin in dog serum. J. Pharm. Biomed. Anal. 2015, 107, 229–235. [Google Scholar] [CrossRef]
- Nanita, S.C.; Kaldon, L.G. Emerging flow injection mass spectrometry methods for high-throughput quantitative analysis. Anal. Bioanal. Chem. 2016, 408, 23–33. [Google Scholar] [CrossRef]
- Gachumi, G.N.; Purves, R.W.; Hopf, C.; El-Aneed, A. Fast quantification without conventional chromatography, the growing power of mass spectrometry. Anal. Chem. 2020, 92, 8628–8637. [Google Scholar] [CrossRef]
- Vogeser, M.; Seger, C. Pitfalls associated with the use of liquid chromatography–tandem mass spectrometry in the clinical laboratory. Clin. Chem. 2010, 56, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Nischwitz, V.; Pergantis, S.A. Optimisation of an hplc selected reaction monitoring electrospray tandem mass spectrometry method for the detection of 50 arsenic species. J. Anal. At. Spectrom. 2006, 21, 1277–1286. [Google Scholar] [CrossRef]
- Song, F. “Cross-talk” in scheduled multiple reaction monitoring caused by in-source fragmentation in herbicide screening with liquid chromatography electrospray tandem mass spectrometry. J. Agric. Food Chem. 2011, 59, 4361–4364. [Google Scholar] [CrossRef] [PubMed]
- Bedner, M.; Schantz, M.M.; Sander, L.C.; Sharpless, K.E. Development of liquid chromatographic methods for the determination of phytosterols in standard reference materials containing saw palmetto. J. Chromatogr. A 2008, 1192, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N. A method for simultaneous analysis of phytosterols and phytosterol esters in tobacco leaves using non aqueous reversed phase chromatography and atmospheric pressure chemical ionization mass spectrometry detector. J. Chromatogr. A 2014, 1340, 99–108. [Google Scholar] [CrossRef]
- Sánchez-Machado, D.; López-Hernández, J.; Paseiro-Losada, P.; López-Cervantes, J. An HPLC method for the quantification of sterols in edible seaweeds. Biomed. Chromatogr. 2004, 18, 183–190. [Google Scholar] [CrossRef]
- Rozenberg, R.; Ruibal-Mendieta, N.L.; Petitjean, G.; Cani, P.; Delacroix, D.L.; Delzenne, N.M.; Meurens, M.; Quetin-Leclercq, J.; Habib-Jiwan, J.-L. Phytosterol analysis and characterization in spelt (Triticum aestivum ssp. spelta l.) and wheat (T. aestivum L.) lipids by LC/APCI-MS. J. Cereal Sci. 2003, 38, 189–197. [Google Scholar] [CrossRef]
- Buse, J.; Purves, R.W.; Verrall, R.E.; Badea, I.; Zhang, H.; Mulligan, C.C.; Peru, K.M.; Bailey, J.; Headley, J.V.; El-Aneed, A. The development and assessment of high-throughput mass spectrometry-based methods for the quantification of a nanoparticle drug delivery agent in cellular lysate. J. Mass Spectrom. 2014, 49, 1171–1180. [Google Scholar] [CrossRef]
- Samanidou, V.F. Basic LC method development and optimization. Anal. Sep. Sci. 2015, 1, 25–42. [Google Scholar]
- Song, W.; Pabbisetty, D.; Groeber, E.A.; Steenwyk, R.C.; Fast, D.M. Comparison of fused-core and conventional particle size columns by LC–MS/MS and UV: Application to pharmacokinetic study. J. Pharm. Biomed. Anal. 2009, 50, 491–500. [Google Scholar] [CrossRef]
- Guideline, I.H.T. Validation of Analytical Procedures: Text and Methodology Q2 (R1); International Conference on Harmonization: Geneva, Switzerland, 2005; pp. 11–12. [Google Scholar]
- Guideline, I.H. M10 Bioanalytical Method Validation; Center for Drug Evaluation and Research: Rockville, MD, USA, 2019. [Google Scholar]
- Khamis, M.M.; Adamko, D.J.; El-Aneed, A. Strategies and challenges in method development and validation for the absolute quantification of endogenous biomarker metabolites using liquid chromatography-tandem mass spectrometry. Mass Spectrom. Rev. 2019, 40, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Poudel, A.; Gachumi, G.; Wasan, K.M.; Dallal Bashi, Z.; El-Aneed, A.; Badea, I. Development and characterization of liposomal formulations containing phytosterols extracted from canola oil deodorizer distillate along with tocopherols as food additives. Pharmaceutics 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornsteiner, M.; Wagner, K.-H.; Elmadfa, I. Tocopherols and total phenolics in 10 different nut types. Food Chem. 2006, 98, 381–387. [Google Scholar] [CrossRef]
- Zarrouk, W.; Carrasco-Pancorbo, A.; Zarrouk, M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Multi-component analysis (sterols, tocopherols and triterpenic dialcohols) of the unsaponifiable fraction of vegetable oils by liquid chromatography–atmospheric pressure chemical ionization–ion trap mass spectrometry. Talanta 2009, 80, 924–934. [Google Scholar] [CrossRef]
- Ghazani, S.M.; García-Llatas, G.; Marangoni, A.G. Micronutrient content of cold-pressed, hot-pressed, solvent extracted and rbd canola oil: Implications for nutrition and quality. Eur. J. Lipid Sci. Technol. 2014, 116, 380–387. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | Molecular Weight | a [M + H − H2O]+ m/z | [M + H − 4H]+ m/z | [M + H − 2H]+ m/z |
|---|---|---|---|---|
| Stigmasterol | 412.7 |  | 409 |  |
| Brassicasterol | 398.7 | 381 |  |  |
| β-sitosterol | 414.7 |  |  | 413 |
| Campesterol | 400.7 | 383 |  | 399 |
| Cholestanol (IS) | 388.7 | 371 | 385 | 387 |
| d6-cholesterol (IS) | 392.7 | 375 | 389 | 391 |
| Compound | Retention Time (min) | Precursor Ion (m/z) | MRM Transitions (m/z) | CE (V) | |
|---|---|---|---|---|---|
| Phytosterols | Brassicasterol | 0.50 | 381.4 | 297.4 | 22 30 |
| 147.1 | |||||
| Campesterol | 0.61 | 383.4 | 161.1 | 30 30 | |
| 147.1 | |||||
| Stigmasterol | 0.63 | 395.4 | 297.4 | 23 26 | |
| 83.2 | |||||
| β-Sitosterol | 0.73 | 397.4 | 161.2 | 27 28 | |
| 134.9 | |||||
| Tocopherols | Delta Tocopherol (δ) | 0.32 | 402.4 | 177 | 32 38 |
| 137 | |||||
| Gamma Tocopherol (γ) | 0.38 | 416 | 151.1 | 37 34 | |
| 191.2 | |||||
| Alpha Tocopherol (α) | 0.44 | 430 | 165.3 | 38 36 | |
| 205.2 | |||||
| Internal Standards | D6-Cholesterol | 0.51 | 375.4 | 152 | 30 28 |
| 167 | |||||
| Cholestanol | 0.95 | 371.4 | 95 | 32 30 | |
| 109 | |||||
| Rac Tocol | 0.26 | 388.4 | 163.2 | 30 | |
| 122 | 40 | ||||
| Compound a | Concentration (µg/mL) | Intra-day | Inter-day 1 | Inter-day 2 | Inter-day 3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision (%RSD) | Accuracy (%) | Precision (%RSD) | Accuracy (%) | Precision (%RSD) | Accuracy (%) | Precision (%RSD) | |||
| δ-Tocopherol | LLOQC | 0.25 | 97.83 | 6.06 | 92.86 | 3.96 | 95.05 | 5.42 | 96.85 | 3.45 |
| LQC | 0.75 | 94.19 | 4.73 | 90.96 | 4.59 | 96.08 | 0.85 | 102.59 | 5.90 | |
| MQC | 5.5 | 101.08 | 1.82 | 94.46 | 3.86 | 96.59 | 2.51 | 95.70 | 1.13 | |
| HQC | 8 | 98.16 | 5.37 | 96.08 | 3.14 | 99.42 | 2.63 | 99.23 | 3.64 | |
| β/γ-Tocopherol | LLOQC | 0.25 | 107.29 | 2.93 | 88.74 | 0.72 | 94.80 | 3.68 | 100.43 | 3.22 |
| LQC | 0.75 | 96.04 | 2.62 | 90.64 | 4.39 | 94.34 | 1.70 | 94.20 | 3.50 | |
| MQC | 5.5 | 100.00 | 4.97 | 90.30 | 4.32 | 96.68 | 3.86 | 95.67 | 2.35 | |
| HQC | 8 | 98.01 | 2.57 | 90.49 | 3.62 | 99.49 | 1.46 | 100.35 | 2.77 | |
| α-Tocopherol | LLOQC | 0.25 | 118.38 | 0.49 | 109.49 | 2.32 | 118.32 | 2.29 | 115.59 | 1.70 |
| LQC | 0.75 | 94.22 | 3.34 | 89.46 | 3.51 | 91.76 | 1.65 | 94.58 | 4.65 | |
| MQC | 5.5 | 104.32 | 2.78 | 92.00 | 2.82 | 95.14 | 2.86 | 99.50 | 1.92 | |
| HQC | 8 | 102.69 | 4.83 | 96.42 | 2.02 | 102.74 | 3.43 | 103.81 | 2.86 | |
| Brassicasterol | LLOQC | 0.05 | 93.91 | 9.66 | 99.01 | 9.66 | 109.43 | 6.81 | 117.57 | 3.43 |
| LQC | 0.15 | 97.14 | 7.50 | 98.98 | 7.50 | 102.26 | 1.07 | 96.47 | 2.70 | |
| MQC | 5.5 | 96.75 | 1.80 | 96.65 | 1.80 | 97.06 | 3.97 | 95.72 | 0.72 | |
| HQC | 8 | 95.91 | 3.00 | 101.17 | 3.00 | 99.55 | 1.83 | 101.44 | 3.73 | |
| Campesterol | LLOQC | 0.05 | 99.39 | 0.67 | 114.21 | 5.57 | 104.15 | 2.88 | 108.77 | 1.37 |
| LQC | 0.15 | 101.60 | 3.01 | 106.84 | 3.52 | 10.3.9 | 0.77 | 102.95 | 2.93 | |
| MQC | 5.5 | 101.77 | 1.68 | 102.33 | 1.37 | 103.91 | 2.95 | 97.43 | 4.10 | |
| HQC | 8 | 97.57 | 1.24 | 102.37 | 5.16 | 101.16 | 3.35 | 98.53 | 2.80 | |
| Stigmasterol | LLOQC | 0.05 | 94.56 | 0.96 | 113.98 | 8.81 | 116.74 | 3.80 | 118.44 | 4.26 |
| LQC | 0.15 | 94.00 | 3.38 | 103.28 | 6.18 | 103.93 | 1.25 | 102.48 | 2.52 | |
| MQC | 5.5 | 96.72 | 2.47 | 95.68 | 2.42 | 98.97 | 1.82 | 95.05 | 4.09 | |
| HQC | 8 | 94.75 | 2.83 | 97.21 | 6.27 | 98.19 | 4.36 | 95.32 | 3.82 | |
| β-Sitosterol | LLOQC | 0.05 | 99.28 | 2.86 | 108.12 | 3.23 | 103.45 | 5.96 | 110.09 | 3.55 |
| LQC | 0.15 | 100.18 | 2.65 | 108.53 | 4.56 | 107.89 | 2.88 | 102.19 | 2.36 | |
| MQC | 5.5 | 102.31 | 0.87 | 99.72 | 1.73 | 103.56 | 1.66 | 97.56 | 3.81 | |
| HQC | 8 | 98.92 | 1.82 | 100.75 | 4.24 | 103.31 | 2.94 | 99.01 | 2.95 | |
| Compoundb | Concentration (µg/mL) | Intra-Day | Inter-Day 1 | Inter-Day 2 | Inter-Day 3 | |||||
| Accuracy (%) | Precision (%RSD) | Accuracy (%) | Precision (%RSD) | Accuracy (%) | Precision (%RSD) | Accuracy (%) | Precision (%RSD) | |||
| δ-Tocopherol | LLOQC | 0.25 | 87.4 | 1.08 | 113.57 | 3.84 | 106.18 | 5.49 | 117.17 | 2.23 |
| LQC | 0.75 | 96.4 | 1.62 | 103.04 | 3.82 | 96.81 | 0.63 | 108.50 | 4.29 | |
| MQC | 5.5 | 94.1 | 1.97 | 97.06 | 1.70 | 106.0 | 1.33 | 97.76 | 5.06 | |
| HQC | 8 | 99.9 | 3.25 | 96.59 | 3.91 | 91.20 | 1.40 | 91.99 | 2.67 | |
| Stigmasterol | MQC | 5.5 | 93.3 | 3.63 | 95.95 | 0.05 | 98.38 | 4.59 | 99.32 | 7.63 |
| HQC | 8 | 96.5 | 2.42 | 97.11 | 1.94 | 103.02 | 3.91 | 103.37 | 0.87 | |
| Concentration (µg/mL) | Delta Tocopherol | Stigmasterol | ||
|---|---|---|---|---|
| Matrix Effect | %Recovery | Matrix Effect | %Recovery | |
| 0.75 | 108.22 ± 0.96 | 104.60 ± 4.05 | - | - |
| 5.5 | - | 99.94 ± 1.68 | - | 95.49 ± 0.06 |
| 8 | 93.96 ± 2.1 | 98.76 ± 3.72 | 106.74 ± 5.8 | 96.89 ± 2.03 |
| Hot Pressed CODD | Hot Pressed CODD | Cold Pressed CODD | |
|---|---|---|---|
| Batch 1 | Batch 2 | ||
| Gamma tocopherol | 2.20 ± 0.02 | 1.26 ± 0.14 | 2.47 ± 0.17 |
| Alpha tocopherol | 0.90 ± 0.01 | 0.63 ± 0.01 | 1.43 ± 0.01 |
| Delta tocopherol | nd | nd | nd |
| Brassicasterol | 7.59 ± 0.13 | 7.10 ± 0.14 | 6.57 ± 0.21 |
| Campesterol | 11.24 ± 0.51 | 9.75 ± 0.46 | 11.93 ± 0.27 |
| Stigmasterol | 1.08 ± 0.10 | 1.00 ± 0.03 | 1.13 ± 0.18 |
| β-Sitosterol | 14.73 ± 0.31 | 12.03 ± 0.46 | 15.68 ± 0.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gachumi, G.; Demelenne, A.; Poudel, A.; Dallal Bashi, Z.; El-Aneed, A. Novel Fast Chromatography-Tandem Mass Spectrometric Quantitative Approach for the Determination of Plant-Extracted Phytosterols and Tocopherols. Molecules 2021, 26, 1402. https://doi.org/10.3390/molecules26051402
Gachumi G, Demelenne A, Poudel A, Dallal Bashi Z, El-Aneed A. Novel Fast Chromatography-Tandem Mass Spectrometric Quantitative Approach for the Determination of Plant-Extracted Phytosterols and Tocopherols. Molecules. 2021; 26(5):1402. https://doi.org/10.3390/molecules26051402
Chicago/Turabian StyleGachumi, George, Alice Demelenne, Asmita Poudel, Zafer Dallal Bashi, and Anas El-Aneed. 2021. "Novel Fast Chromatography-Tandem Mass Spectrometric Quantitative Approach for the Determination of Plant-Extracted Phytosterols and Tocopherols" Molecules 26, no. 5: 1402. https://doi.org/10.3390/molecules26051402
APA StyleGachumi, G., Demelenne, A., Poudel, A., Dallal Bashi, Z., & El-Aneed, A. (2021). Novel Fast Chromatography-Tandem Mass Spectrometric Quantitative Approach for the Determination of Plant-Extracted Phytosterols and Tocopherols. Molecules, 26(5), 1402. https://doi.org/10.3390/molecules26051402