
Phosphorylation of Guar Gum/Magnetite/Chitosan Nanocomposites for Uranium (VI) Sorption and Antibacterial Applications

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
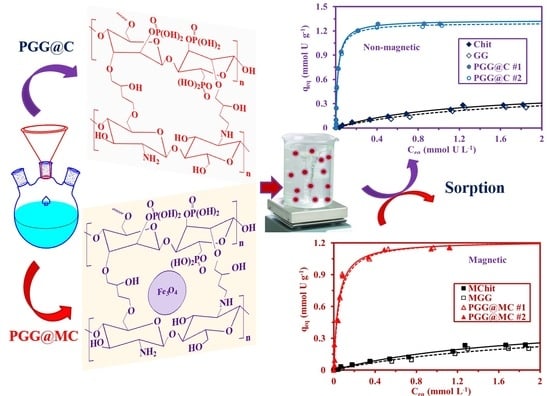
:
1. Introduction
- (a)
- (b)
- (c)
- (d)
- Iminodiacetic acid-bearing groups [24].
2. Materials and Methods
2.1. Materials
2.2. Synthesis Procedures
2.2.1. Synthesis of Magnetite Nanoparticles
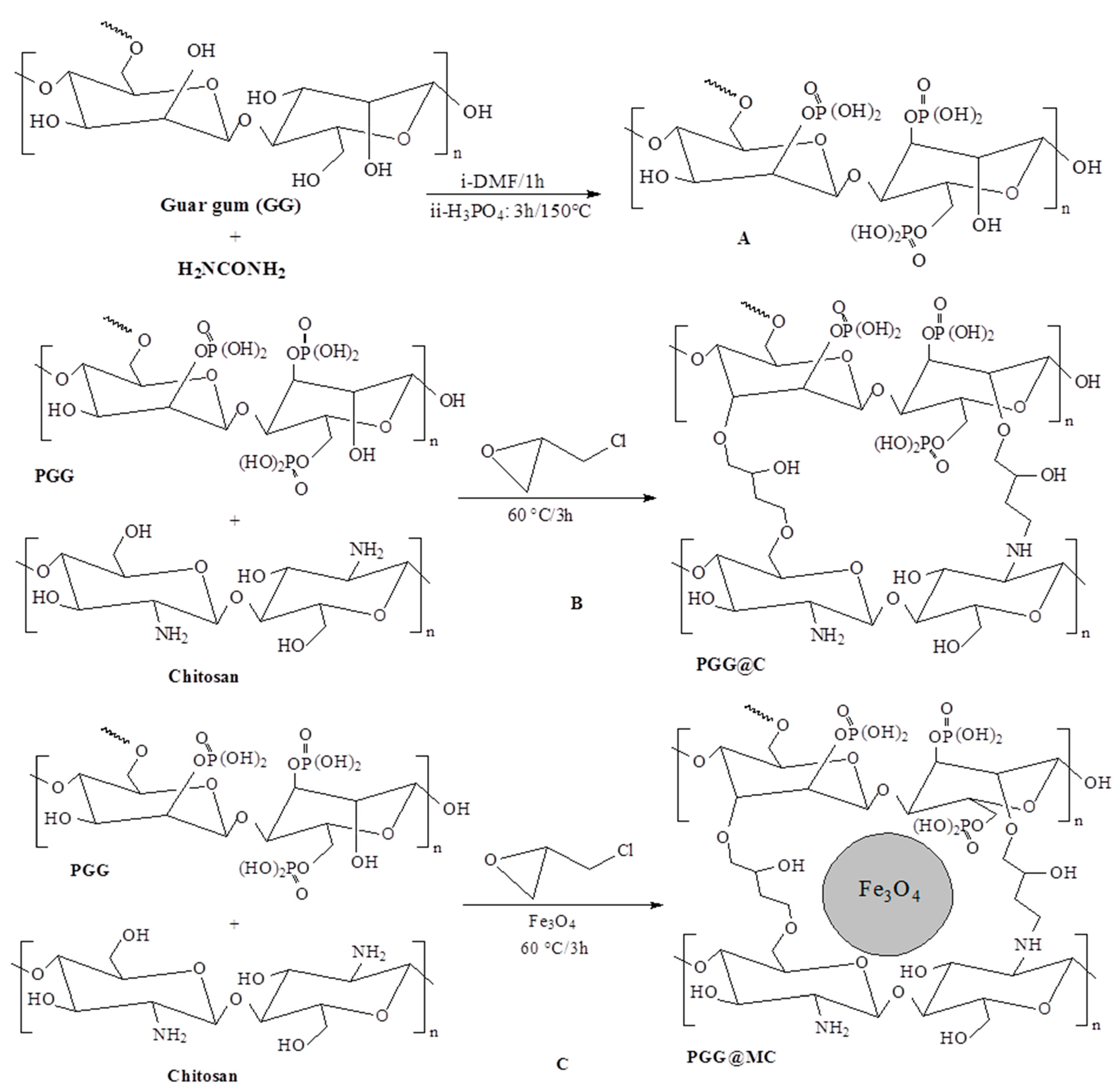
2.2.2. Phosphorylation of Guar Gum
2.2.3. Synthesis of PGG-Chitosan Based Composites
2.2.4. Synthesis of Chit and MChit Composite
2.3. Characterization of Materials
2.4. Sorption Procedures
2.5. Antimicrobial Tests
3. Results and Discussion
3.1. Characterization of Materials
3.1.1. Morphological Analysis-SEM and TEM
3.1.2. Textural Analysis-BET
3.1.3. Structural Analysis-XRD
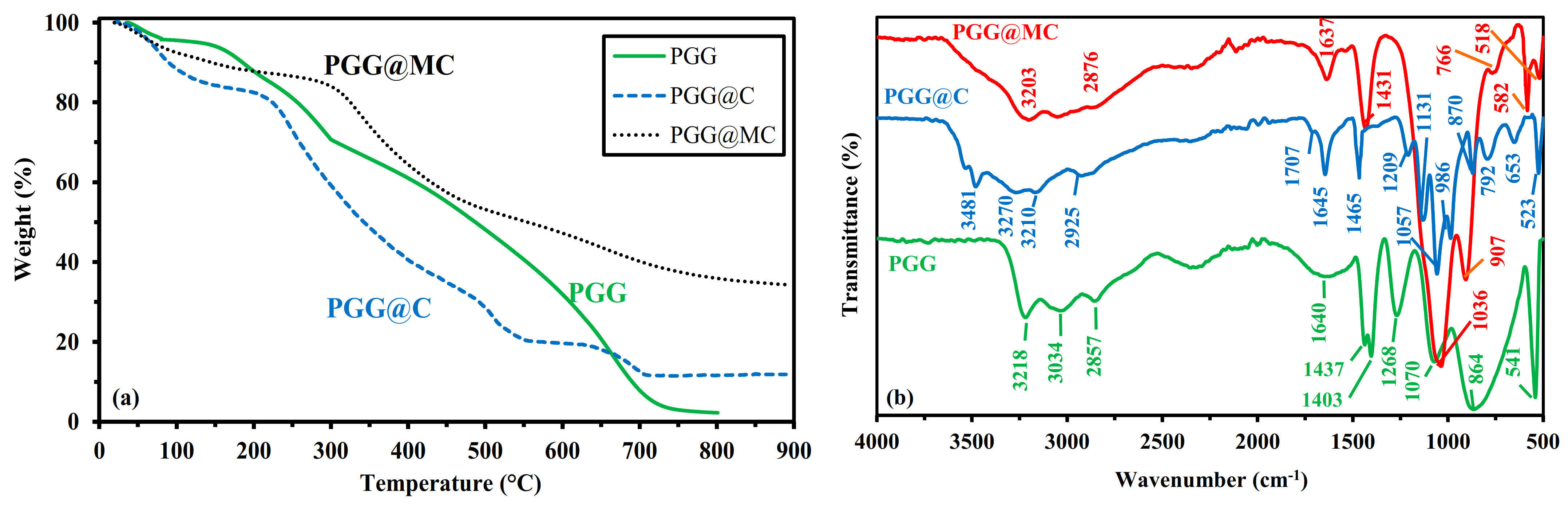
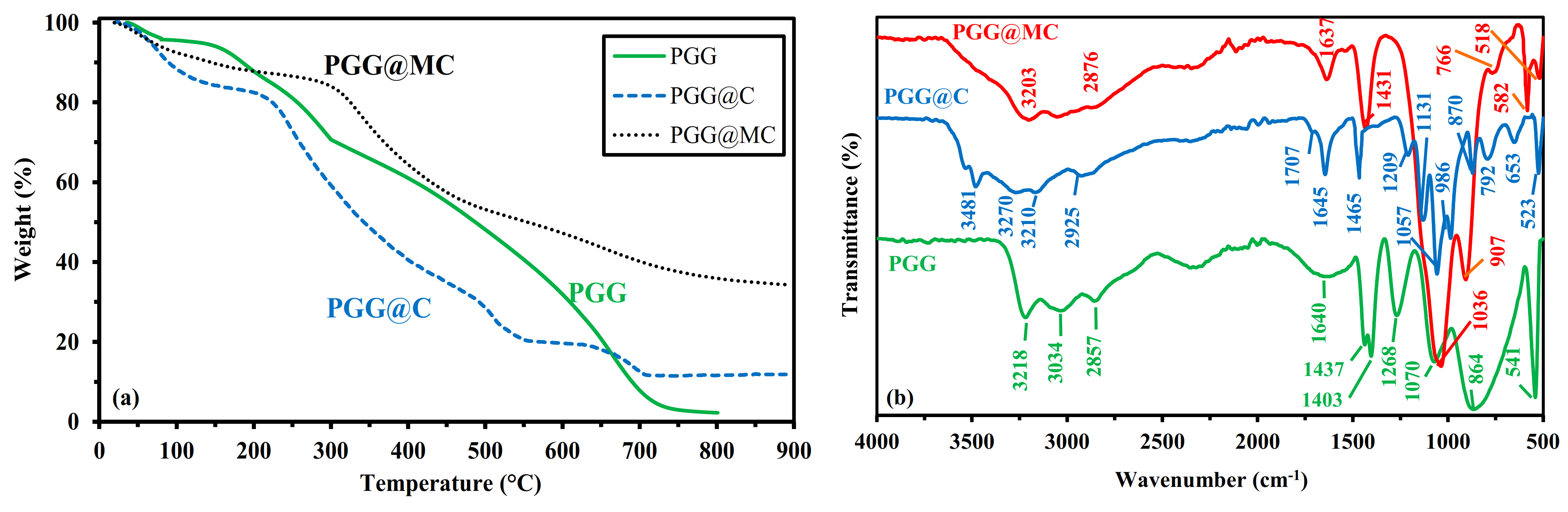
3.1.4. Thermogravimetric Analysis-TGA
- (a)
- Below 151 °C, the weight loss reaches 6%, corresponding to water release and volatile matters.
- (b)
- Between 151 °C and 288.1 °C, the thermal degradation (about 60.5%) begins with probable dissociation of the linkages between phosphonate groups and GG. Singha et al. [50] reported the main degradation of GG between 230 °C and 320 °C (with a 49% weight loss), while the grafting of other functional groups extended the temperature range up to 400 °C.
- (c)
- Between 288.1 °C and ~600 °C, the degradation continues progressively and almost linearly (weight loss ~38%). This step in the process corresponds to the formation of the char resulting from backbone degradation.
- (d)
- Between ~600 °C and ~720 °C, a steeper step is observed corresponding to 28% weight loss, associated with char degradation.
3.1.5. Chemical Characterization–FTIR Spectroscopy
- (a)
- 1268 cm−1: P=O stretching in phosphonate.
- (b)
- 1070 cm−1: primary –OH stretching vibration, with contribution of P-OH stretching vibration.
- (c)
- 864 cm−1 (broad band): galactose and mannose rink (with possible contributions of (1–4) linkages at ~930 cm−1, and P-O stretching vibration at Wn > 900 cm−1 and P-O-C deformation vibration at Wn < 850 cm−1).
- (d)
- 541 cm−1: P-O-C stretching vibration.
3.1.6. Surface Charge–pHPZC
3.2. Sorption Performances–Synthetic Solutions
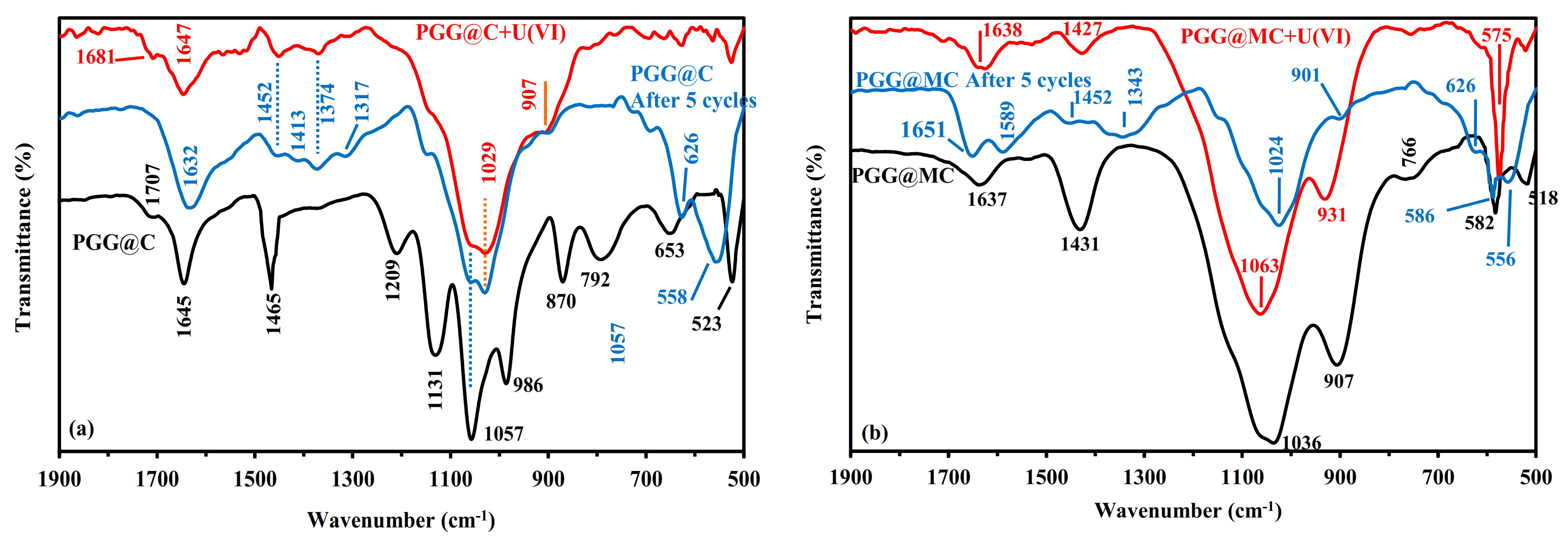
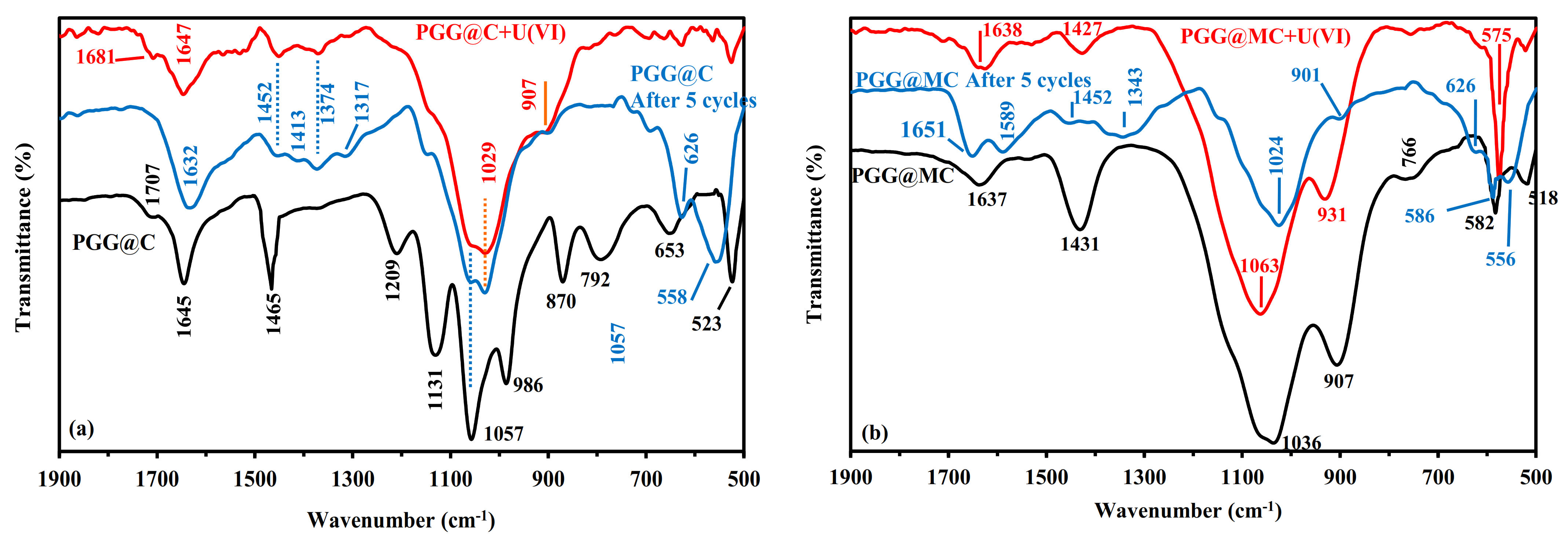
3.2.1. Sorption Mechanisms Correlated with FTIR Analysis
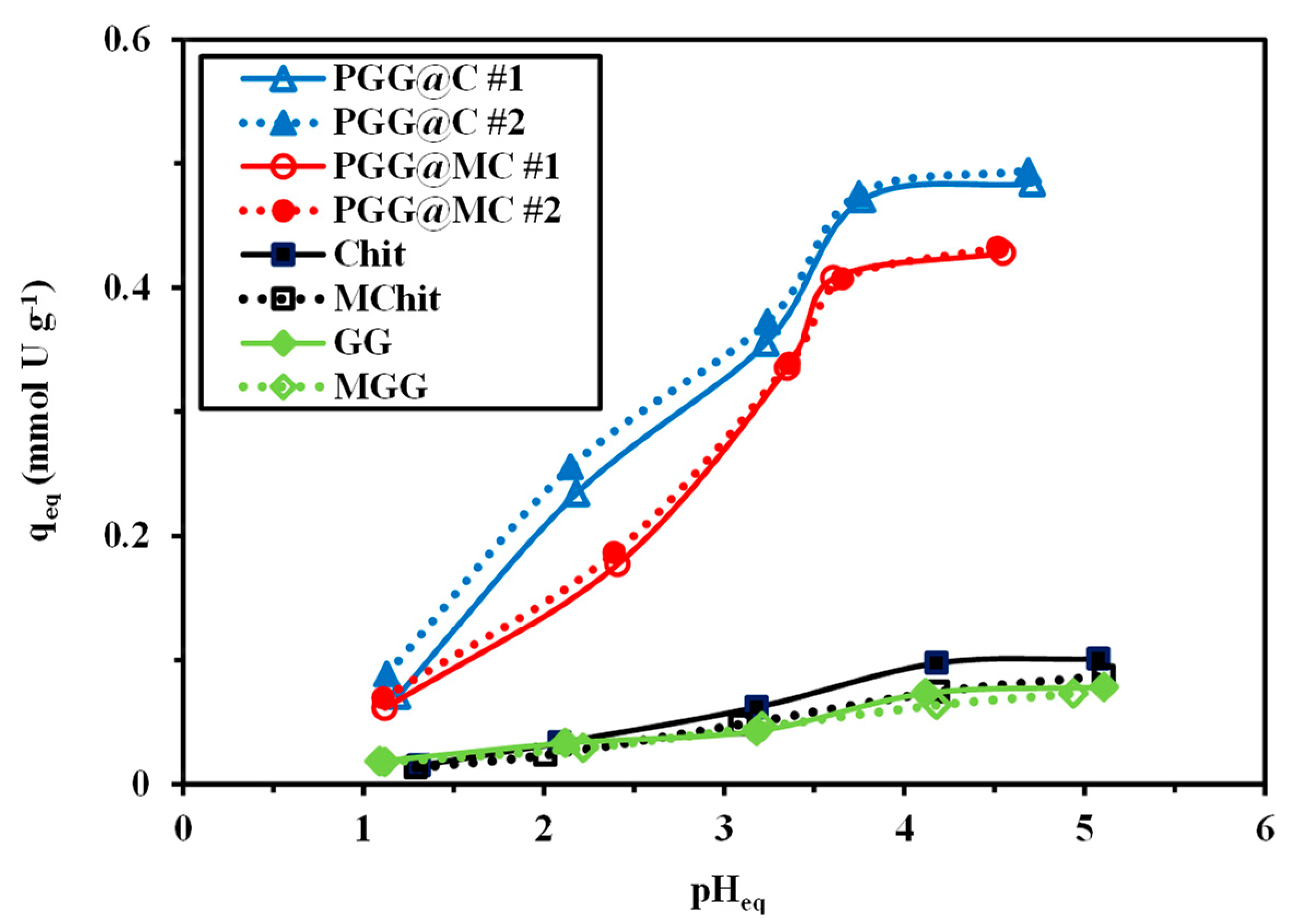
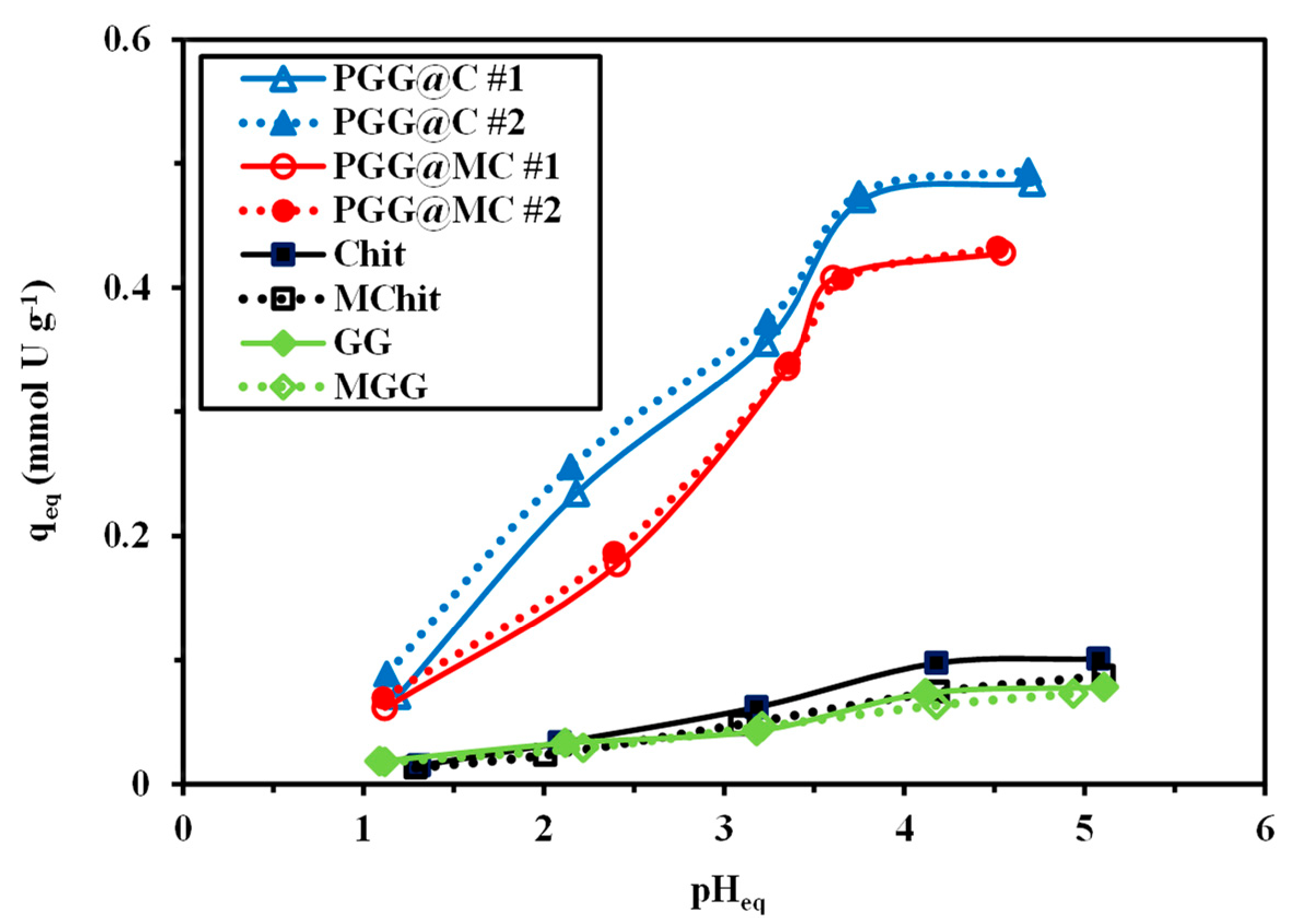
3.2.2. PH Effect
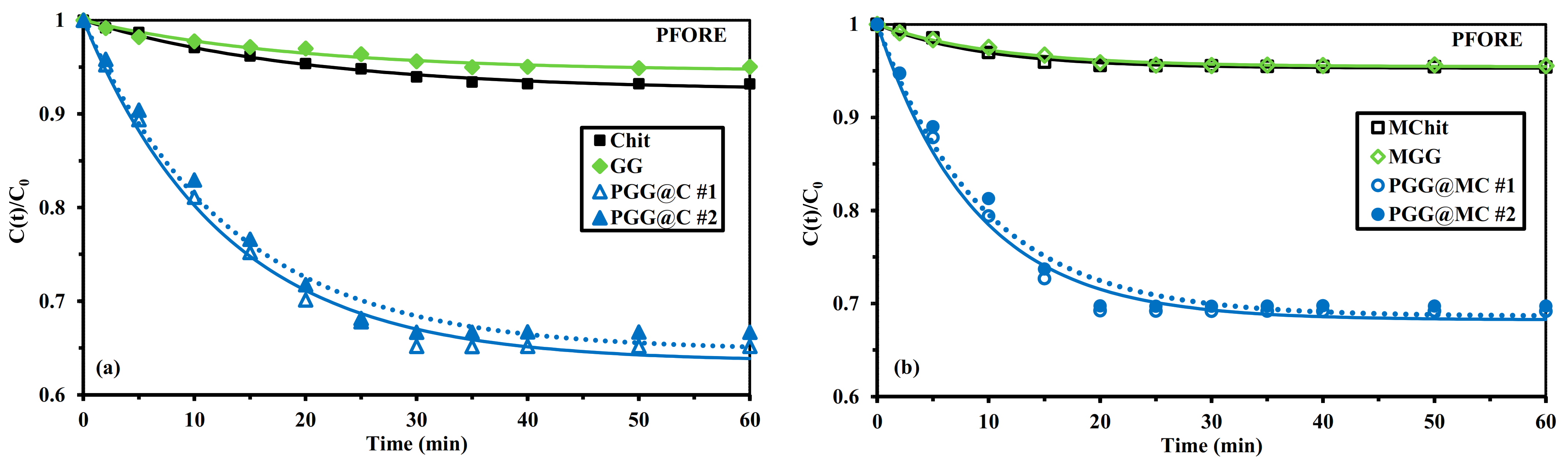
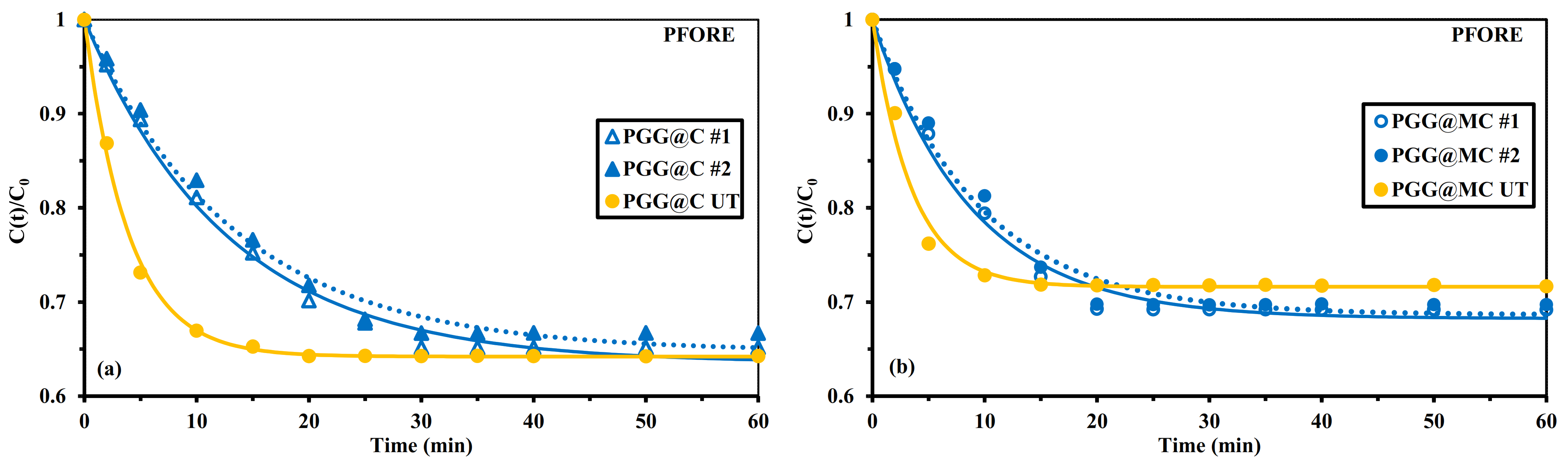
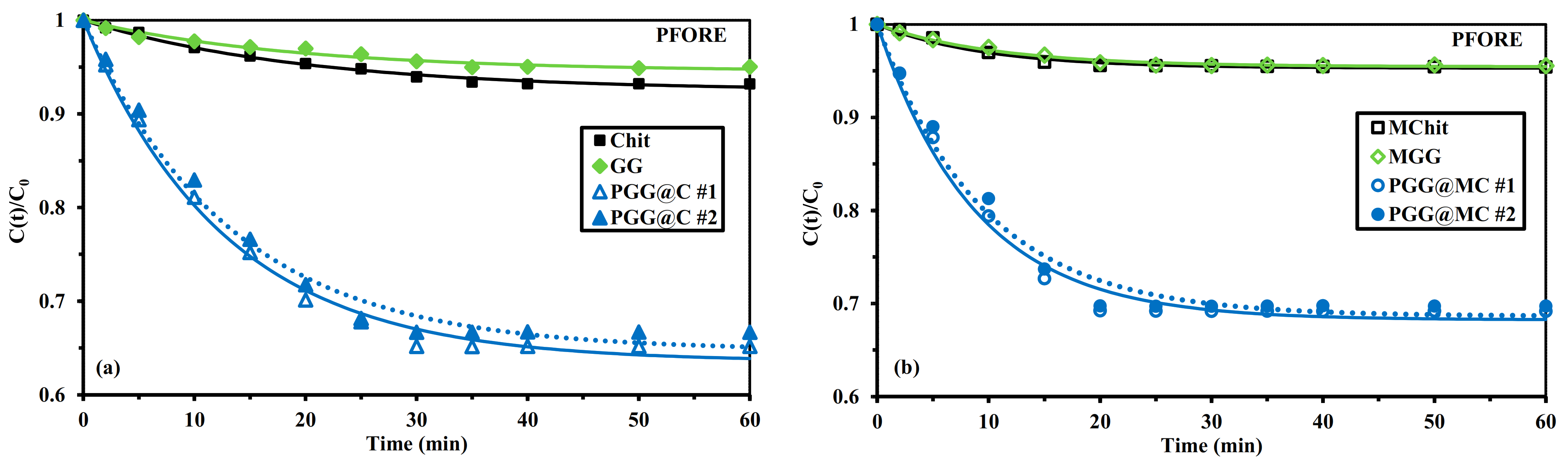
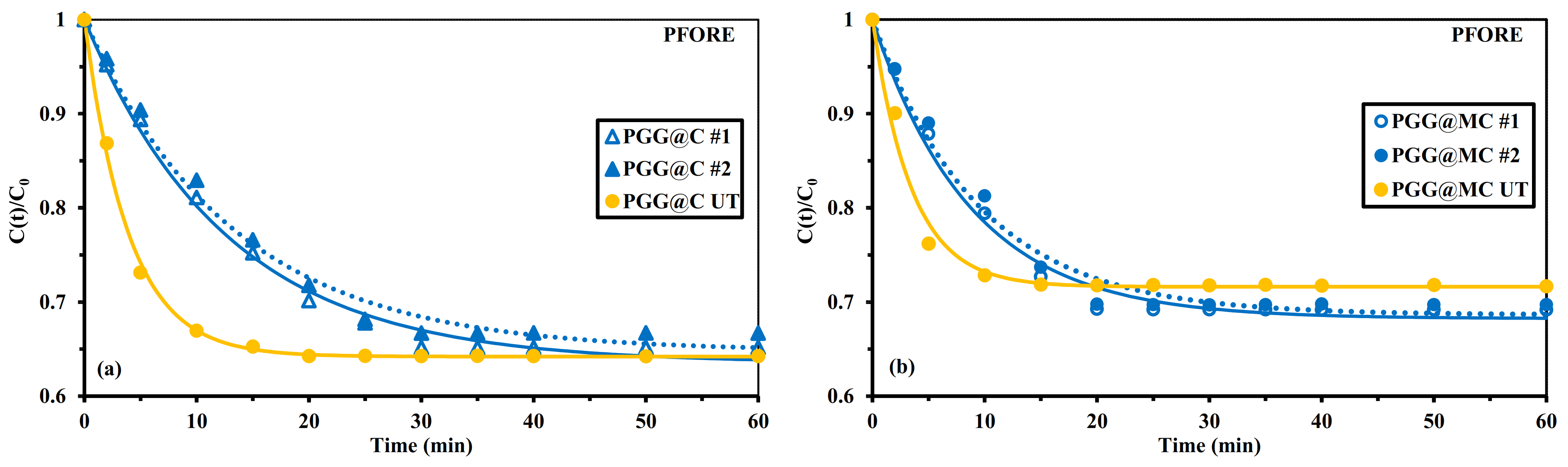
3.2.3. Uptake Kinetics
Mechanical Agitation
Sonication-Assisted Sorption
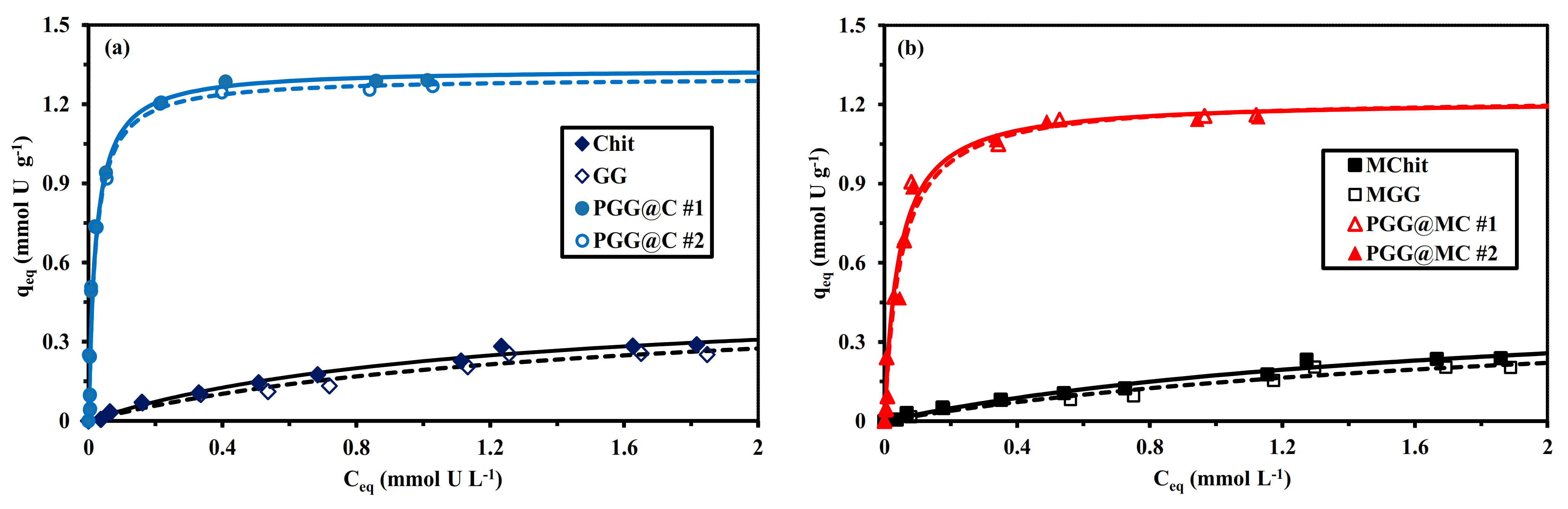
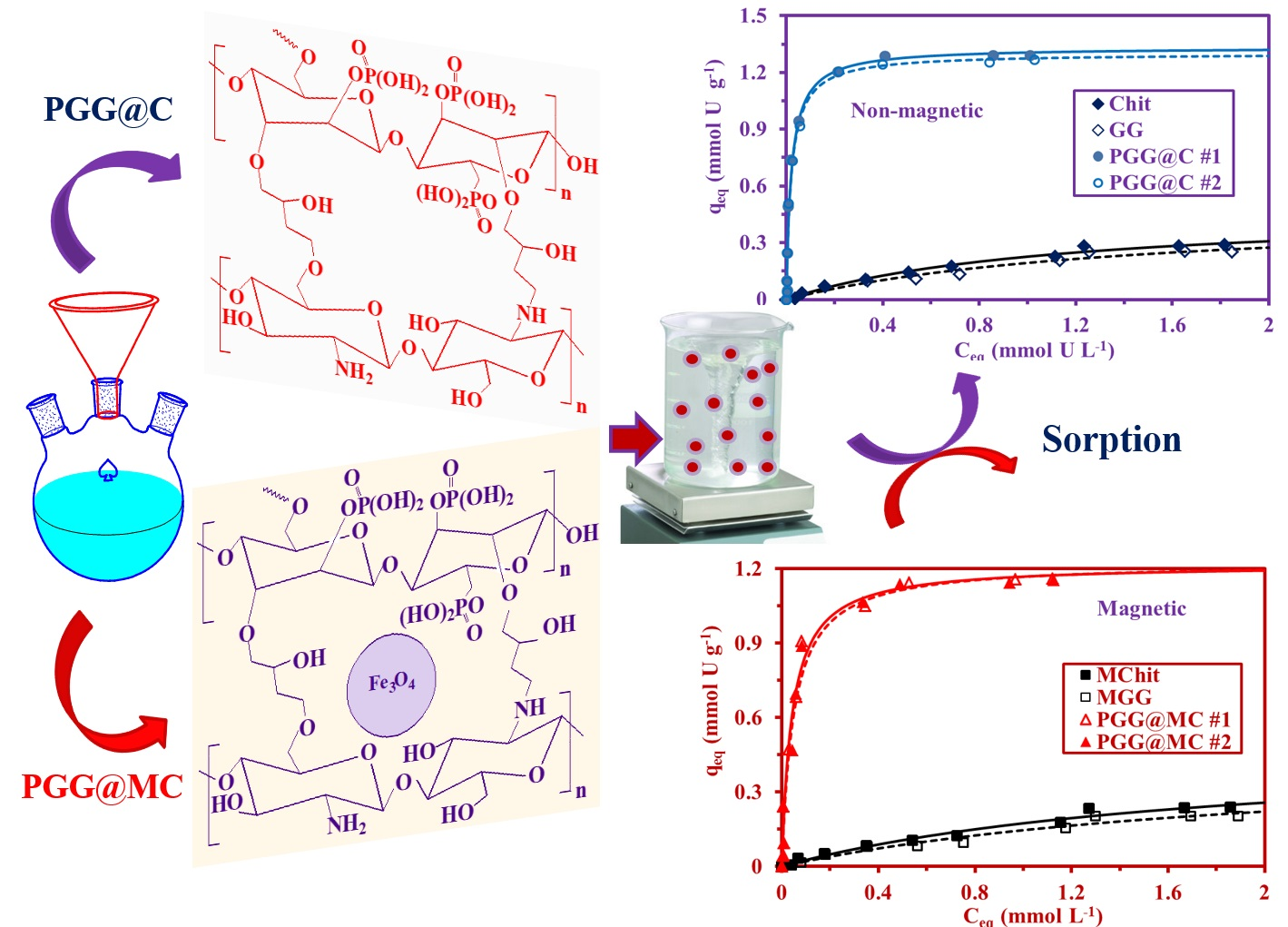
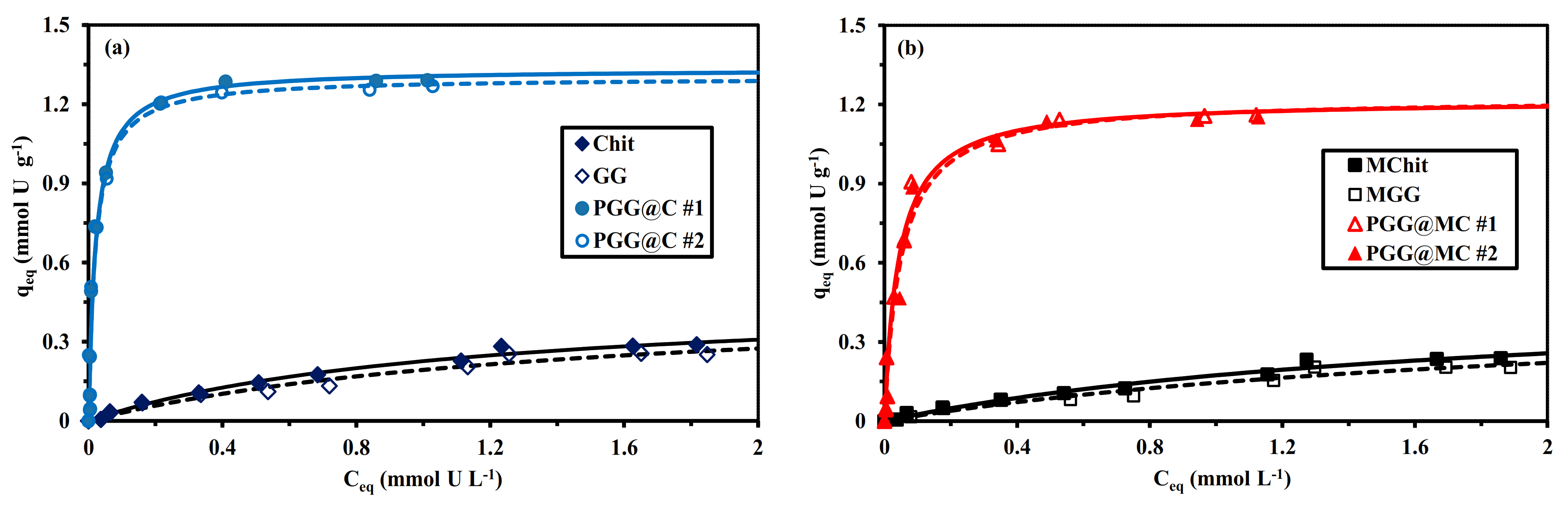
3.2.4. Sorption Isotherms
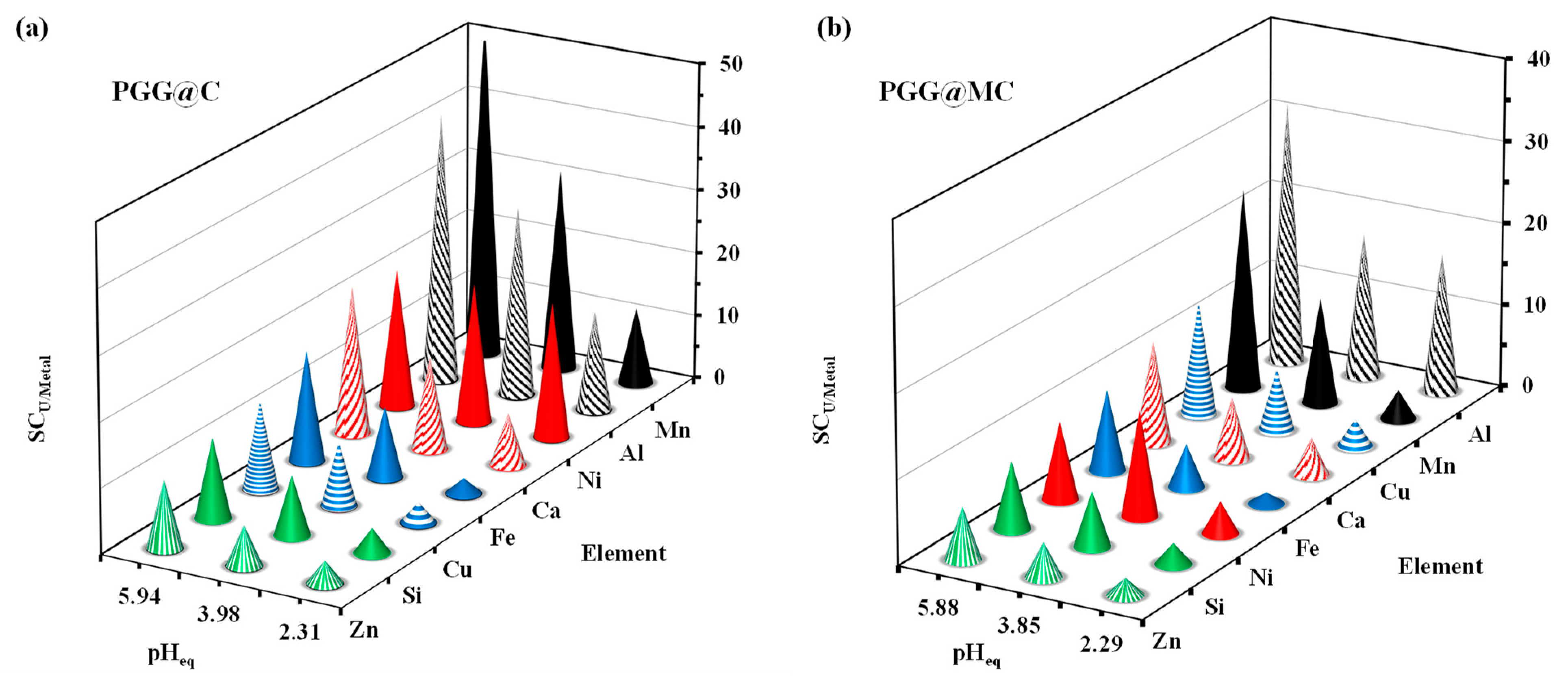
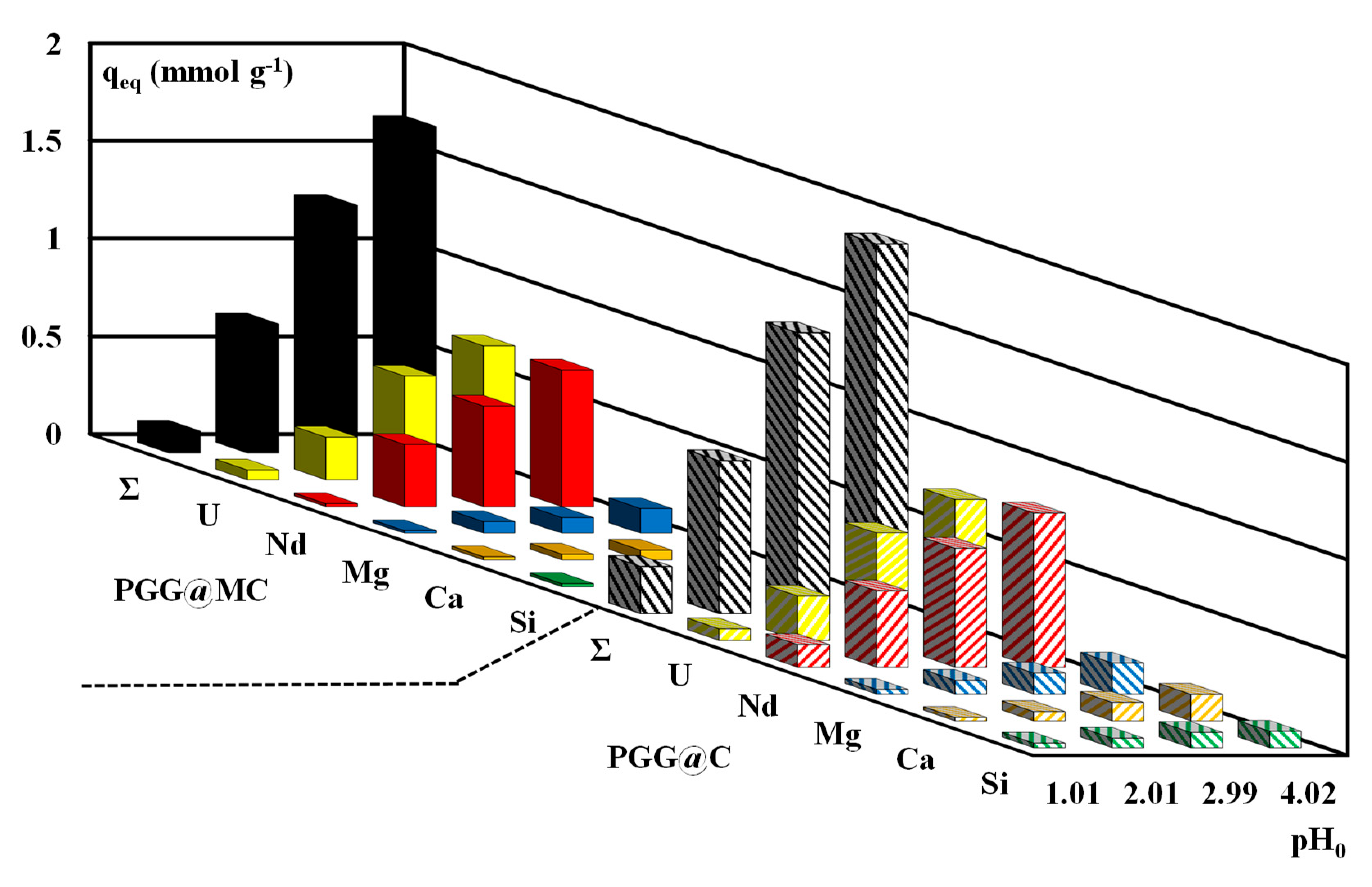
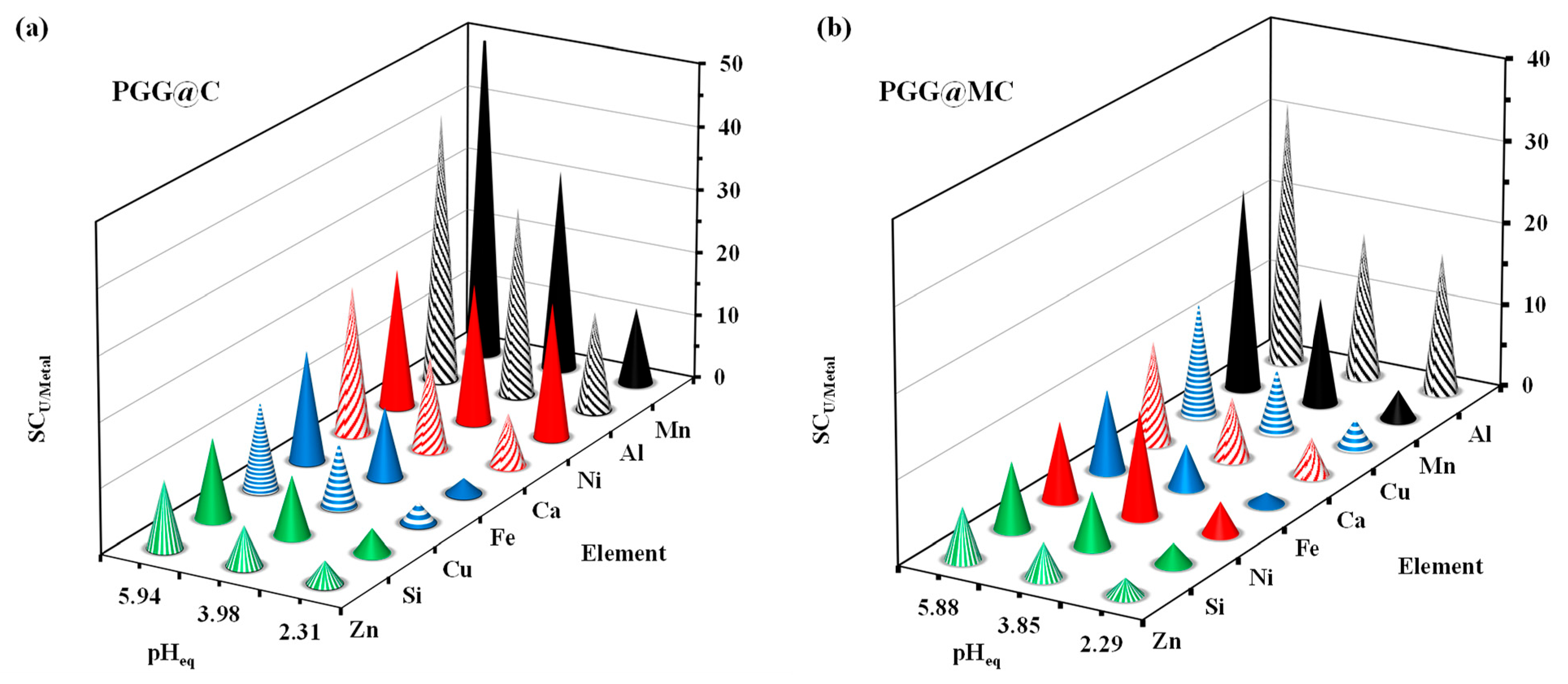
3.2.5. Sorption in Multi-Component Solutions–Selectivity
3.2.6. Uranium Desorption and Sorbent Recycling
3.3. Treatment of Ore Leachate
3.3.1. Ore Origin and Pre-Treatment
3.3.2. Treatment of WPS (Washing Pregnant Solution)
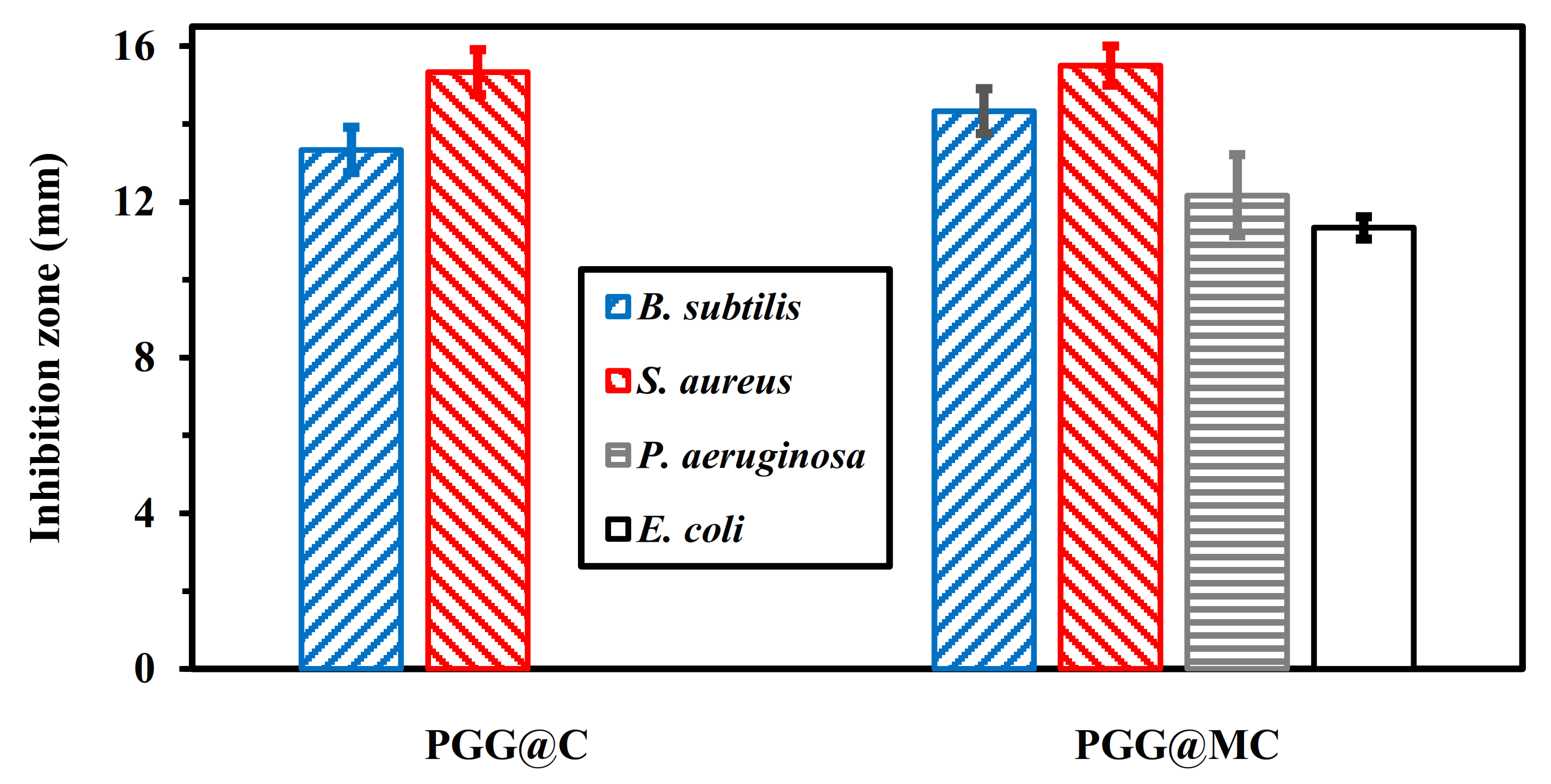
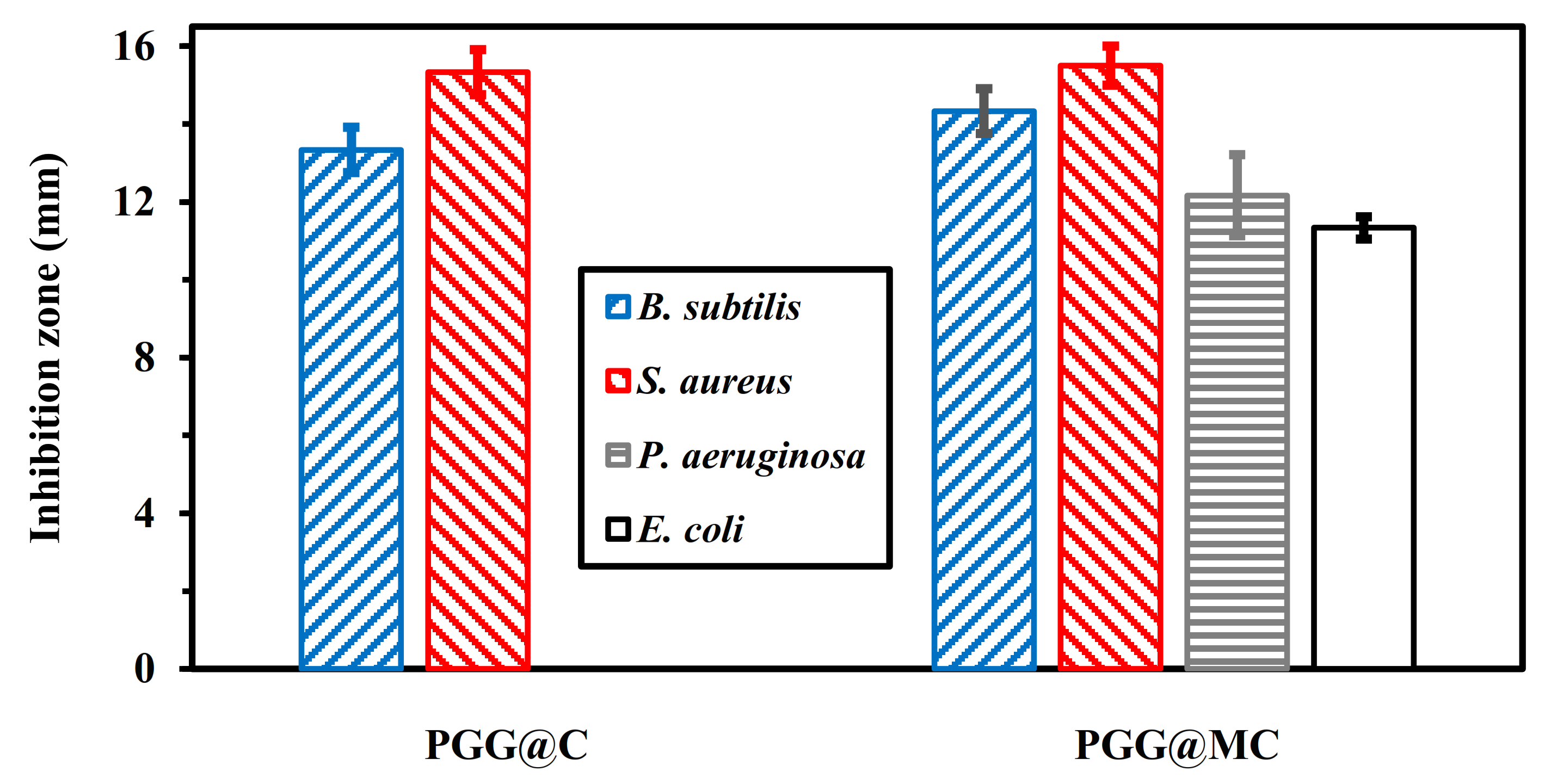
3.4. Antibacterial Application
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wu, S.; Zhao, L.; Wang, L.; Huang, X.; Zhang, Y.; Feng, Z.; Cui, D. Simultaneous recovery of rare earth elements and phosphorus from phosphate rock by phosphoric acid leaching and selective precipitation: Towards green process. J. Rare Earths 2019, 37, 652–658. [Google Scholar] [CrossRef]
- Pavon, S.; Fortuny, A.; Coll, M.T.; Sastre, A.M. Rare earths separation from fluorescent lamp wastes using ionic liquids as extractant agents. Waste Manag. 2018, 82, 241–248. [Google Scholar] [CrossRef]
- Ni’am, A.C.; Wang, Y.-F.; Chen, S.-W.; Chang, G.-M.; You, S.-J. Simultaneous recovery of rare earth elements from waste permanent magnets (WPMs) leach liquor by solvent extraction and hollow fiber supported liquid membrane. Chem. Eng. Process. Process Intensif. 2020, 148, 107831. [Google Scholar] [CrossRef]
- Wu, S.; Wang, L.; Zhang, P.; El-Shall, H.; Moudgil, B.; Huang, X.; Zhao, L.; Zhang, L.; Feng, Z. Simultaneous recovery of rare earths and uranium from wet process phosphoric acid using solvent extraction with D2EHPA. Hydrometallurgy 2018, 175, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Torkabad, M.G.; Keshtkar, A.R.; Safdari, S.J. Comparison of polyethersulfone and polyamide nanofiltration membranes for uranium removal from aqueous solution. Prog. Nucl. Energy 2017, 94, 93–100. [Google Scholar] [CrossRef]
- Pavon, S.; Fortuny, A.; Call, M.T.; Sastre, A.M. Improved rare earth elements recovery from fluorescent lamp wastes applying supported liquid membranes to the leaching solutions. Sep. Purif. Technol. 2019, 224, 332–339. [Google Scholar] [CrossRef]
- Erkaya, I.A.; Arica, M.Y.; Akbulut, A.; Bayramoglu, G. Biosorption of uranium(VI) by free and entrapped Chlamydomonas reinhardtii: Kinetic, equilibrium and thermodynamic studies. J. Radioanal. Nucl. Chem. 2014, 299, 1993–2003. [Google Scholar] [CrossRef]
- Lapo, B.; Bou, J.J.; Hoyo, J.; Carrillo, M.; Pena, K.; Tzanov, T.; Sastre, A.M. A potential lignocellulosic biomass based on banana waste for critical rare earths recovery from aqueous solutions. Environ. Pollut. 2020, 264, 114409. [Google Scholar] [CrossRef]
- Ferreira, R.V.d.P.; de Araujo, L.G.; Canevesi, R.L.S.; da Silva, E.A.; Ferreira, E.G.A.; Palmieri, M.C.; Marumo, J.T. The use of rice and coffee husks for biosorption of U (total), Am-241, and(137)Cs in radioactive liquid organic waste. Environ. Sci. Pollut. Res. 2020, 27, 36651–36663. [Google Scholar] [CrossRef]
- Javadian, H.; Ruiz, M.; Saleh, T.A.; Sastre, A.M. Ca-alginate/carboxymethyl chitosan/Ni0.2Zn0.2F2.6O4 magnetic bionanocomposite: Synthesis, characterization and application for single adsorption of Nd+3, Tb+3, and Dy+3 rare earth elements from aqueous media. J. Mol. Liq. 2020, 306, 112760. [Google Scholar] [CrossRef]
- Javadian, H.; Ruiz, M.; Sastre, A.M. Response surface methodology based on central composite design for simultaneous adsorption of rare earth elements using nanoporous calcium alginate/carboxymethyl chitosan microbiocomposite powder containing Ni0.2Zn0.2Fe2.6O4 magnetic nanoparticles: Batch and column studies. Int. J. Biol. Macromol. 2020, 154, 937–953. [Google Scholar]
- Ma, F.Q.; Dong, B.R.; Gui, Y.Y.; Cao, M.; Han, L.; Jiao, C.S.; Lv, H.T.; Hou, J.J.; Xue, Y. Adsorption of low-concentration uranyl ion by amidoxime polyacrylonitrile fibers. Ind. Eng. Chem. Res. 2018, 57, 17384–17393. [Google Scholar] [CrossRef]
- Wiechert, A.I.; Liao, W.-P.; Hong, E.; Halbert, C.E.; Yiacoumi, S.; Saito, T.; Tsouris, C. Influence of hydrophilic groups and metal-ion adsorption on polymer-chain conformation of amidoxime-based uranium adsorbents. J. Colloid Interface Sci. 2018, 524, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Wongjaikham, W.; Wongsawaeng, D.; Hosemann, P. Synthesis of amidoxime polymer gel to extract uranium compound from seawater by UV radiation curing. J. Nucl. Sci. Technol. 2019, 56, 541–552. [Google Scholar] [CrossRef]
- Wu, H.Y.; Chi, F.T.; Zhang, S.; Wen, J.; Xiong, J.; Hu, S. Control of pore chemistry in metal-organic frameworks for selective uranium extraction from seawater. Microporous Mesoporous Mater. 2019, 288, 109567. [Google Scholar] [CrossRef]
- Hamza, M.F.; Gamal, A.; Hussein, G.; Nagar, M.S.; Abdel-Rahman, A.A.H.; Wei, Y.; Guibal, E. Uranium(VI) and zirconium(IV) sorption on magnetic chitosan derivatives-effect of different functional groups on separation properties. J. Chem. Technol. Biotechnol. 2019, 94, 3866–3882. [Google Scholar] [CrossRef]
- He, D.X.; Tan, N.; Luo, X.M.; Yang, X.C.; Ji, K.; Han, J.W.; Chen, C.; Liu, Y.Q. Preparation, uranium (VI) absorption and reuseability of marine fungus mycelium modified by the bis-amidoxime-based groups. Radiochim. Acta 2020, 108, 37–49. [Google Scholar] [CrossRef]
- Dousti, Z.; Dolatyari, L.; Yaftian, M.R.; Rostamnia, S. Adsorption of Eu(III), Th(IV), and U(VI) by mesoporous solid materials bearing sulfonic acid and sulfamic acid functionalities. Sep. Sci. Technol. 2019, 54, 2609–2624. [Google Scholar] [CrossRef]
- Taha, M.H. Solid-liquid extraction of uranium from industrial phosphoric acid using macroporous cation exchange resins: MTC1600H, MTS9500, and MTS9570. Sep. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Ahmad, M.; Yang, K.; Li, L.; Fan, Y.; Shah, T.; Zhang, Q.; Zhang, B. Modified tubular carbon nanofibers for adsorption of uranium(VI) from water. ACS Appl. Nano Mater. 2020, 3, 6394–6405. [Google Scholar] [CrossRef]
- Riegel, M.; Schlitt, V. Sorption dynamics of uranium onto anion exchangers. Water 2017, 9, 268. [Google Scholar] [CrossRef] [Green Version]
- Masoud, A.M. Sorption behavior of uranium from sulfate media using purolite A400 as a strong base anion exchange resin. Int. J. Environ. Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Hamza, M.F.; Mubark, A.E.; Wei, Y.; Vincent, T.; Guibal, E. Quaternization of composite algal/PEI beads for enhanced uranium sorption-application to ore acidic leachate. Gels 2020, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Ang, K.L.; Li, D.; Nikoloski, A.N. The effectiveness of ion exchange resins in separating uranium and thorium from rare earth elements in acidic aqueous sulfate media. Part 2. Chelating resins. Miner. Eng. 2018, 123, 8–15. [Google Scholar] [CrossRef]
- Guo, X.; Feng, Y.; Ma, L.; Gao, D.; Jing, J.; Yu, J.; Sun, H.; Gong, H.; Zhang, Y. Phosphoryl functionalized mesoporous silica for uranium adsorption. Appl. Surf. Sci 2017, 402, 53–60. [Google Scholar]
- Sarafraz, H.; Minuchehr, A.; Alahyarizadeh, G.; Rahimi, Z. Synthesis of enhanced phosphonic functional groups mesoporous silica for uranium selective adsorption from aqueous solutions. Sci. Rep. 2017, 7, 11675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.Y.; Han, X.L.; Wang, T.; Li, S.H.; Hua, D.B. Conjugated microporous polymers bearing phosphonate ligands as an efficient sorbent for potential uranium extraction from high- level liquid wastes. J. Mater. Chem. A 2018, 6, 13894–13900. [Google Scholar] [CrossRef]
- Duval, C.E.; Hardy, W.A.; Pellizzeri, S.; DeVol, T.A.; Husson, S.M. Phosphonic acid and alkyl phosphate-derivatized resins for the simultaneous concentration and detection of uranium in environmental waters. React. Funct. Polym. 2019, 137, 133–139. [Google Scholar] [CrossRef]
- Yang, P.P.; Liu, Q.; Liu, J.Y.; Chen, R.R.; Li, R.M.; Bai, X.F.; Wang, J. Highly efficient immobilization of uranium(VI) from aqueous solution by phosphonate-functionalized dendritic fibrous nanosilica (DFNS). J. Hazard. Mater. 2019, 363, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.Z.; Zhang, S.A.; Tan, J.L.; Dai, Y.H.; Wang, Y.; He, Y.; Liu, Y.; Zhao, X.H.; Zhang, M.M.; Zhang, Q.H. Highly efficacious entrapment of Th (IV) and U (VI) from rare earth elements in concentrated nitric acid solution using a phosphonic acid functionalized porous organic polymer adsorbent. Sep. Purif. Technol. 2020, 237, 116379. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Chiarizia, R.; Diamond, H.; Gatrone, R.C.; Alexandratos, S.D.; Trochimczuk, A.Q.; Crick, D.W. Uptake of metal-ions by a new chelating ion-exchange resin. 1. Acid dependencies of actinide ions. Solvent Extr. Ion Exch. 1993, 11, 943–966. [Google Scholar] [CrossRef]
- Sabharwal, K.N.; Nandy, K.K.; Srinivasan, T.G.; Rao, P.R.V. Recovery of uranium from acid media by macroporous bifunctional phosphinic acid resin. Solvent Extr. Ion Exch. 1996, 14, 1101–1114. [Google Scholar] [CrossRef]
- Sabharwal, K.N.; Rao, P.R.V.; Srinivasan, M. Extraction of uranium by macroporous bifunctional phosphinic acid resin. Solvent Extr. Ion Exch. 1995, 13, 561–574. [Google Scholar] [CrossRef]
- Bhanushali, R.D.; Pius, I.C.; Chetty, K.V.; Vaidya, V.N.; Venugopal, V.; Rao, P.R.V. Sorption of Pu(IV) on macroporous bifunctional phosphinic acid resin from uranium analytical waste solution. J. Radioanal. Nucl. Chem. 2005, 265, 389–394. [Google Scholar] [CrossRef]
- Venkatesan, K.A.; Patre, D.K.; Sabharwal, K.N.; Srinivasan, T.G.; Rao, P.R.V. Kinetics of uranium extraction by macroporous bifunctional phosphinic acid resin. Solvent Extr. Ion Exch. 2000, 18, 551–565. [Google Scholar] [CrossRef]
- Piechowicz, M.; Abney, C.W.; Zhou, X.; Thacker, N.C.; Li, Z.; Lin, W. Design, synthesis, and characterization of a bifunctional chelator with ultrahigh capacity for uranium uptake from seawater simulant. Ind. Eng. Chem. Res. 2016, 55, 4170–4178. [Google Scholar] [CrossRef]
- Wei, Y.Q.; Qian, J.; Huang, L.; Hua, D.B. Bifunctional polymeric microspheres for efficient uranium sorption from aqueous solution: Synergistic interaction of positive charge and amidoxime group. RSC Adv. 2015, 5, 64286–64292. [Google Scholar] [CrossRef]
- Alexandratos, S.D.; Zhu, X. Polymer-supported aminomethylphosphinate as a ligand with a high affinity for U(VI) from phosphoric acid solutions: Combining variables to optimize ligand-ion communication. Solvent Extr. Ion Exch. 2016, 34, 290–295. [Google Scholar] [CrossRef]
- Rashad, M.M.; El-Sayed, I.E.; Galhoum, A.A.; Abdeen, M.M.; Mira, H.I.; Elshehy, E.A.; Zhang, S.; Lu, X.; Xin, J.; Guibal, E. Synthesis of α-aminophosphonate based sorbents–Influence of inserted groups (carboxylic vs. amine) on uranyl sorption. Chem. Eng. J. 2020, 127830. [Google Scholar] [CrossRef]
- Cheira, M.F. Synthesis of aminophosphonate-functionalised ZnO/polystyrene-butadiene nanocomposite and its characteristics for uranium adsorption from phosphoric acid. Int. J. Environ. Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Imam, E.A.; El-Tantawy El-Sayed, I.; Mahfouz, M.G.; Tolba, A.A.; Akashi, T.; Galhoum, A.A.; Guibal, E. Synthesis of α-aminophosphonate functionalized chitosan sorbents: Effect of methyl vs phenyl group on uranium sorption. Chem. Eng. J. 2018, 352, 1022–1034. [Google Scholar] [CrossRef]
- Vaaramaa, K.; Pulli, S.; Lehto, J. Effects of pH and uranium concentration on the removal of uranium from drinking water by ion exchange. Radiochim. Acta 2000, 88, 845–849. [Google Scholar] [CrossRef]
- Pawar, S.N.; Edgar, K.J. Alginate derivatization: A review of chemistry, properties and applications. Biomaterials 2012, 33, 3279–3305. [Google Scholar] [CrossRef] [PubMed]
- Guibal, E. Interactions of metal ions with chitosan-based sorbents: A review. Sep. Purif. Technol. 2004, 38, 43–74. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Hu, J.; Wang, J.L. Competitive adsorption of Pb(II), Cu(II) and Zn(II) onto xanthate-modified magnetic chitosan. J. Hazard. Mater. 2012, 221, 155–161. [Google Scholar] [CrossRef]
- Yuvaraja, G.; Su, M.; Chen, D.-Y.; Pang, Y.; Kong, L.-J.; Subbaiah, M.V.; Wen, J.-C.; Reddy, G.M. Impregnation of magnetic–Momordica charantia leaf powder into chitosan for the removal of U(VI) from aqueous and polluted wastewater. Int. J. Biol. Macromol. 2020, 149, 127–139. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Li, L.; Kong, F.; Lin, S.; Wang, Z.; Li, W. Preparation of three-dimensional fiber-network chitosan films for the efficient treatment of uranium-contaminated effluents. Water Sci. Technol. 2020, 81, 52–61. [Google Scholar] [CrossRef]
- Hamza, M.F.; Roux, J.-C.; Guibal, E. Uranium and europium sorption on amidoxime-functionalized magnetic chitosan micro-particles. Chem. Eng. J. 2018, 344, 124–137. [Google Scholar] [CrossRef]
- Jakobik-Kolon, A.; Bok-Badura, J.; Milewski, A.; Karon, K. Long term and large-scale continuous studies on zinc(II) sorption and desorption on hybrid pectin-guar gum biosorbent. Polymers 2019, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Singha, N.R.; Mahapatra, M.; Karmakar, M.; Dutta, A.; Mondal, H.; Chattopadhyay, P.K. Synthesis of guar gum-g-(acrylic acid-co-acrylamide-co-3-acrylamido propanoic acid) IPN via in situ attachment of acrylamido propanoic acid for analyzing superadsorption mechanism of Pb(II)/Cd(II)/Cu(II)/MB/MV. Polym. Chem. 2017, 8, 6750–6777. [Google Scholar] [CrossRef]
- Patra, A.S.; Ghorai, S.; Sarkar, D.; Das, R.; Sarkar, S.; Pal, S. Anionically functionalized guar gum embedded with silica nanoparticles: An efficient nanocomposite adsorbent for rapid adsorptive removal of toxic cationic dyes and metal ions. Bioresour. Technol. 2017, 225, 367–376. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, L.; Wang, B.; Yang, B.; Khan, I.; Khan, A.; Ni, Y. Multifunctional self-assembling hydrogel from guar gum. Chem. Eng. J. 2017, 330, 1044–1051. [Google Scholar] [CrossRef]
- Hiremath, J.N.; Vishalakshi, B. Effective removal of divalent metal ions: Synthesis and characterization of pH-sensitive guar gum based hydrogels. Desalin. Water Treat. 2016, 57, 4018–4027. [Google Scholar] [CrossRef]
- Thakur, S.; Kumari, S.; Dogra, P.; Chauhan, G.S. A new guar gum-based adsorbent for the removal of Hg(II) from its aqueous solutions. Carbohydr. Polym. 2014, 106, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Fresco-Cala, B.; Batista, A.D.; Cardenas, S. Molecularly imprinted polymer micro- and nano-particles: A review. Molecules 2020, 25, 4740. [Google Scholar] [CrossRef]
- Habila, M.A.; Alothman, Z.A.; El-Toni, A.M.; Labis, J.P.; Khan, A.; Al-Marghany, A.; Elafifi, H.E. One-step carbon coating and polyacrylamide functionalization of Fe3O4 nanoparticles for enhancing magnetic adsorptive-remediation of heavy metals. Molecules 2017, 22, 2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manousi, N.; Rosenberg, E.; Deliyanni, E.; Zachariadis, G.A.; Samanidou, V. Magnetic solid-phase extraction of organic compounds based on graphene oxide nanocomposites. Molecules 2020, 25, 1148. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Han, X.; Hua, D. Polyoxime-functionalized magnetic nanoparticles for uranium adsorption with high selectivity over vanadium. J. Mater. Chem. A 2017, 5, 12278–12284. [Google Scholar] [CrossRef]
- Hamza, M.F.; Wei, Y.; Mira, H.I.; Abdel-Rahman, A.A.H.; Guibal, E. Synthesis and adsorption characteristics of grafted hydrazinyl amine magnetite-chitosan for Ni(II) and Pb(II) recovery. Chem. Eng. J. 2019, 362, 310–324. [Google Scholar] [CrossRef] [Green Version]
- Liakos, I.; Grumezescu, A.M.; Holban, A.M. Magnetite nanostructures as novel strategies for anti-infectious therapy. Molecules 2014, 19, 12710–12726. [Google Scholar] [CrossRef]
- Haldorai, Y.; Kharismadewi, D.; Tuma, D.; Shim, J.-J. Properties of chitosan/magnetite nanoparticles composites for efficient dye adsorption and antibacterial agent. Korean J. Chem. Eng. 2015, 32, 1688–1693. [Google Scholar] [CrossRef]
- Salem, S.S.; Fouda, A. Green synthesis of metallic nanoparticles and their prospective biotechnological applications: An overview. Biol. Trace Elem. Res. 2021, 199, 344–370. [Google Scholar] [CrossRef]
- Yeswanth, S.; Sekhar, K.C.; Chaudhary, A.; Sarma, P. Anti-microbial and Anti-biofilm activity of a novel Dibenzyl (benzo d thiazol-2-yl (hydroxy) methyl) phosphonate by inducing protease expression in Staphylococcus aureus. Med. Chem. Res. 2018, 27, 785–795. [Google Scholar] [CrossRef]
- Fouda, A.; Hassan, S.E.-D.; Abdo, A.M.; El-Gamal, M.S. Antimicrobial, antioxidant and larvicidal activities of spherical silver nanoparticles synthesized by endophytic Streptomyces spp. Biol. Trace Elem. Res. 2020, 195, 707–724. [Google Scholar] [CrossRef]
- Massart, R. Preparation of aqueous magnetic liquids in alkaline and acidic media. IEEE Trans. Magn. 1981, 17, 1247–1249. [Google Scholar] [CrossRef]
- Marczenko, Z.; Balcerzak, M. Chapter 54-Uranium. In Analytical Spectroscopy Library; Marczenko, Z., Balcerzak, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2000; Volume 10, pp. 446–455. [Google Scholar]
- Marczenko, Z.; Balcerzak, M. Chapter 39-Rare-earth Elements. In Analytical Spectroscopy Library; Marczenko, Z., Balcerzak, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2000; Volume 10, pp. 341–349. [Google Scholar]
- Davies, W.; Gray, W. A rapid and specific titrimetric method for the precise determination of uranium using iron(II) sulphate as reductant. Talanta 1964, 11, 1203–1211. [Google Scholar] [CrossRef]
- Mathew, K.J.; Buerger, S.; Vogt, S.; Mason, P.; Morales-Arteaga, M.E.; Narayanan, U.I. Uranium assay determination using Davies and Gray titration: An overview and implementation of GUM for uncertainty evaluation. J. Radioanal. Nucl. Chem. 2009, 282, 939–944. [Google Scholar] [CrossRef]
- Nehra, P.; Chauhan, R.P.; Garg, N.; Verma, K. Antibacterial and antifungal activity of chitosan coated iron oxide nanoparticles. Br. J. Biomed. Sci. 2018, 75, 13–18. [Google Scholar] [CrossRef]
- Salem, N.; Ahmad, A.; Awwad, A. New route for synthesis magnetite nanoparticles from ferrous ions and Pistachio leaf extract. J. Nanosci. Nanotechnol. 2013, 3, 48–51. [Google Scholar]
- Stoia, M.; Istratie, R.; Păcurariu, C. Investigation of magnetite nanoparticles stability in air by thermal analysis and FTIR spectroscopy. J. Therm. Anal. Calorim. 2016, 125, 1185–1198. [Google Scholar] [CrossRef]
- Wang, S.; Yu, D. Adsorption of Cd(II), Pb(II), and Ag(I) in aqueous solution on hollow chitosan microspheres. J. Appl. Polym. Sci. 2010, 118, 733–739. [Google Scholar] [CrossRef]
- Duarte, M.L.; Ferreira, M.C.; Marvao, M.R.; Rocha, J. An optimised method to determine the degree of acetylation of chitin and chitosan by FTIR spectroscopy. Int. J. Biol. Macromol. 2002, 31, 1–8. [Google Scholar] [CrossRef]
- Mudgil, D.; Barak, S.; Khatkar, B.S. X-ray diffraction, IR spectroscopy and thermal characterization of partially hydrolyzed guar gum. Int. J. Biol. Macromol. 2012, 50, 1035–1039. [Google Scholar] [CrossRef]
- Coleman, R.J.; Lawrie, G.; Lambert, L.K.; Whittaker, M.; Jack, K.S.; Grøndahl, L. Phosphorylation of alginate: Synthesis, characterization, and evaluation of in vitro mineralization capacity. Biomacromolecules 2011, 12, 889–897. [Google Scholar] [CrossRef]
- Petcharoen, K.; Sirivat, A. Synthesis and characterization of magnetite nanoparticles via the chemical co-precipitation method. Mater. Sci. Eng. B 2012, 177, 421–427. [Google Scholar] [CrossRef]
- Sorlier, P.; Denuzière, A.; Viton, C.; Domard, A. Relation between the degree of acetylation and the electrostatic properties of chitin and chitosan. Biomacromolecules 2001, 2, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Das, T.; Sengupta, S.; Sardar, S.; Mondal, S.; Bandyopadhyay, A. An elastic semi IPN polymer hybrid for enhanced adsorption of heavy metals. Carbohydr. Polym. 2020, 236, 116055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bu, A.; Ji, Q.Y.; Min, L.F.; Zhao, S.; Wang, Y.X.; Chen, J. pKa-directed incorporation of phosphonates into MOF-808 via ligand exchange: Stability and adsorption properties for uranium. ACS Appl. Mater. Interfaces 2019, 11, 33931–33940. [Google Scholar] [CrossRef]
- Singha, N.R.; Dutta, A.; Mahapatra, M.; Karmakar, M.; Mondal, H.; Chattopadhyay, P.K.; Maiti, D.K. Guar gum-grafted terpolymer hydrogels for ligand-selective individual and synergistic adsorption: Effect of comonomer composition. ACS Omega 2018, 3, 472–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yean, S.; Cong, L.; Yavuz, C.T.; Mayo, J.T.; Yu, W.W.; Kan, A.T.; Colvin, V.L.; Tomson, M.B. Effect of magnetite particle size on adsorption and desorption of arsenite and arsenate. J. Mater. Res. 2005, 20, 3255–3264. [Google Scholar] [CrossRef]
- Thomas, G.; Demoisson, F.; Boudon, J.; Millot, N. Efficient functionalization of magnetite nanoparticles with phosphonate using a one-step continuous hydrothermal process. Dalton Trans. 2016, 45, 10821–10829. [Google Scholar] [CrossRef]
- Abu-Dalo, M.; Al-Rawashdeh, N.; Al-Mheidat, I.; Nassory, N. Construction of uranyl selective electrode based on complex of uranyl ion with new ligand carboxybenzotriazole in PVC matrix membrane. In IOP Conference Series Materials Science and Engineering; IOP Publishing: Bristol, UK, 2015; Volume 92, p. 012023. [Google Scholar]
- Krishna, G.M.; Suneesh, A.S.; Kumaresan, R.; Venkatesan, K.A.; Antony, M.P. Electrochemical behavior of U(VI) in imidazolium ionic liquid medium containing tri-n-butyl phosphate and chloride ion and spectroscopic characterization of uranyl species. ChemistrySelect 2017, 2, 8706–8715. [Google Scholar] [CrossRef]
- Guibal, E.; Roulph, C.; Lecloirec, P. Uranium biosorption by a filamentous fungus Mucor miehei: pH effect on mechanisms and performances of uptake. Water Res. 1992, 26, 1139–1145. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Oxford University Press: Oxford, UK, 1975; p. 414. [Google Scholar]
- Ho, Y.S.; McKay, G. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Simonin, J.-P. On the comparison of pseudo-first order and pseudo-second order rate laws in the modeling of adsorption kinetics. Chem. Eng. J. 2016, 300, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Hubbe, M.A.; Azizian, S.; Douven, S. Implications of apparent pseudo-second-order adsorption kinetics onto cellulosic materials: A review. BioResources 2019, 14, 7582–7626. [Google Scholar] [CrossRef]
- Awakura, Y.; Sato, K.; Majima, H.; Hirono, S. The measurement of the diffusion coefficient of U(VI) in aqueous uranyl sulfate solutions. Metall. Trans. B 1987, 18, 19–23. [Google Scholar] [CrossRef]
- Wen, Z.; Huang, K.; Niu, Y.; Yao, Y.; Wang, S.; Cao, Z.; Zhong, H. Kinetic study of ultrasonic-assisted uranium adsorption by anion exchange resin. Colloids Surf. A 2020, 585, 124021. [Google Scholar] [CrossRef]
- Elwakeel, K.Z.; Hamza, M.F.; Guibal, E. Effect of agitation mode (mechanical, ultrasound and microwave) on uranium sorption using amine- and dithizone-functionalized magnetic chitosan hybrid materials. Chem. Eng. J. 2021, 411, 128553. [Google Scholar] [CrossRef]
- Amesh, P.; Venkatesan, K.A.; Suneesh, A.S.; Samanta, N. Diethylenetriamine tethered mesoporous silica for the sequestration of uranium from aqueous solution and seawater. J. Environ. Chem. Eng. 2020, 8, 103995. [Google Scholar] [CrossRef]
- Huo, Z.; Zhao, S.; Yi, J.; Zhang, H.; Li, J. Biomass-based cellulose functionalized by phosphonic acid with high selectivity and capacity for capturing U(VI) in aqueous solution. Appl. Sci. 2020, 10, 5455. [Google Scholar] [CrossRef]
- Venkatesan, K.A.; Shyamala, K.V.; Antony, M.P.; Srinivasan, T.G.; Rao, P.R.V. Batch and dynamic extraction of uranium(VI) from nitric acid medium by commercial phosphinic acid resin, Tulsion CH-96. J. Radioanal. Nucl. Chem. 2008, 275, 563–570. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, D.; Zhang, Q.; Wang, Y.; Liu, Y.; Zhao, J.; Chen, B. Highly efficient removal of uranium from highly acidic media achieved using a phosphine oxide and amino functionalized superparamagnetic composite polymer adsorbent. J. Mater. Chem. A 2020, 8, 10925–10934. [Google Scholar] [CrossRef]
- Persson, I. Hydrated metal ions in aqueous solution: How regular are their structures? Pure Appl. Chem. 2010, 82, 1901–1917. [Google Scholar] [CrossRef]
- Marcus, Y. Ion Properties; Marcel Dekker, Inc.: New York, NY, USA, 1997; p. 259. [Google Scholar]
- Li, K.; Li, M.; Xue, D. Solution-phase electronegativity scale: Insight into the chemical behaviors of metal ions in solution. J. Phys. Chem. A 2012, 116, 4192–4198. [Google Scholar] [CrossRef]
- Merdivan, M.; Buchmeiser, M.R.; Bonn, G. Phosphonate-based resins for the selective enrichment of uranium(VI). Anal. Chim. Acta 1999, 402, 91–97. [Google Scholar] [CrossRef]
- Xu, H.; Xu, D.C.; Wang, Y. Natural indices for the chemical hardness/softness of metal cations and ligands. ACS Omeg 2017, 2, 7185–7193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Alexandratos, S.D. Affinity of polymer-supported reagents for lanthanides as a function of donor atom polarizability. Ind. Eng. Chem. Res. 2009, 48, 6173–6187. [Google Scholar] [CrossRef]
- Sirry, S.M.; Aldakhil, F.; Alharbi, O.M.L.; Ali, I. Chemically treated date stones for uranium (VI) uptake and extraction in aqueous solutions. J. Mol. Liq. 2019, 273, 192–202. [Google Scholar] [CrossRef]
- Oyola, Y.; Vukovic, S.; Dai, S. Elution by Le Chatelier’s principle for maximum recyclability of adsorbents: Applied to polyacrylamidoxime adsorbents for extraction of uranium from seawater. Dalton Trans. 2016, 45, 8532–8540. [Google Scholar] [CrossRef]
- Kabay, N.; Demircioglu, M.; Yayli, S.; Gunay, E.; Yuksel, M.; Saglam, M.; Streat, M. Recovery of uranium from phosphoric acid solutions using chelating ion-exchange resins. Ind. Eng. Chem. Res. 1998, 37, 1983–1990. [Google Scholar] [CrossRef]
- Stopa, L.C.B.; Yamaura, M. Uranium removal by chitosan impregnated with magnetite nanoparticles: Adsorption and desorption. Int. J. Nucl. Energy Sci. Technol. 2010, 5, 283–289. [Google Scholar] [CrossRef]
- Pan, H.-B.; Wai, C.M.; Kuo, L.-J.; Gill, G.; Tian, G.; Rao, L.; Das, S.; Mayes, R.T.; Janke, C.J. Bicarbonate elution of uranium from amidoxime-based polymer adsorbents for sequestering uranium from seawater. Chemistryselect 2017, 2, 3769–3774. [Google Scholar] [CrossRef]
- Hamza, M.F. Uranium recovery from concentrated chloride solution produced from direct acid leaching of calcareous shale, Allouga ore materials, southwestern Sinai, Egypt. J. Radioanal. Nucl. Chem. 2018, 315, 613–626. [Google Scholar] [CrossRef]
- Merritt, R.C. The Extractive Metallurgy of Uranium; Colorado School of Mines Research Institute: Golden, Korea, 1971. [Google Scholar]
- Pearson, R.G. Acids and bases. Science 1966, 151, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.C.; da Silva, D.P.; Signini, R.; Naves, P.L.F. Inhibition of bacterial biofilms by carboxymethyl chitosan combined with silver, zinc and copper salts. Int. J. Biol. Macromol. 2017, 105, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadegan, A.; Ghahramani, Y.; Gholami, A.; Hemmateenejad, B.; Dorostkar, S.; Nabavizadeh, M.; Sharghi, H. The effect of charge at the surface of silver nanoparticles on antimicrobial activity against Gram-positive and Gram-negative bacteria: A peliminary study. J. Nanomater. 2015. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Bulut, O.; Some, S.; Mandal, A.K.; Yilmaz, M.D. Green synthesis of silver nanoparticles: Biomolecule-nanoparticle organizations targeting antimicrobial activity. RSC Adv. 2019, 9, 2673–2702. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.E.-D.; Fouda, A.; Radwan, A.A.; Salem, S.S.; Barghoth, M.G.; Awad, M.A.; Abdo, A.M.; El-Gamal, M.S. Endophytic actinomycetes Streptomyces spp mediated biosynthesis of copper oxide nanoparticles as a promising tool for biotechnological applications. J. Biol. Inorg. Chem. 2019, 24, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Shrifian-Esfahni, A.; Salehi, M.T.; Nasr-Esfahni, M.; Ekramian, E. Chitosan-modified superparamgnetic iron oxide nanoparticles: Design, fabrication, characterization and antibacterial activity. Chemik 2015, 69, 19–32. [Google Scholar]
- Fu, Y.; Wang, F.; Sheng, H.; Xu, M.; Liang, Y.; Bian, Y.; Hashsham, S.A.; Jiang, X.; Tiedje, J.M. Enhanced antibacterial activity of magnetic biochar conjugated quaternary phosphonium salt. Carbon 2020, 163, 360–369. [Google Scholar] [CrossRef]
- Behera, S.; Patra, J.K.; Pramanik, K.; Panda, N.; Thatoi, H. Characterization and evaluation of antibacterial activities of chemically synthesized iron oxide nanoparticles. World J. Nano Sci. Eng. 2012, 2, 196–200. [Google Scholar] [CrossRef] [Green Version]
- El-Belely, E.F.; Farag, M.M.S.; Said, H.A.; Amin, A.S.; Azab, E.; Gobouri, A.A.; Fouda, A. Green synthesis of zinc oxide nanoparticles (ZnO-NPs) using Arthrospira platensis (Class: Cyanophyceae) and evaluation of their biomedical activities. Nanomaterials 2021, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Wang, J.; Zhao, B.; Zhao, M.; Nie, M.; Wang, X.; Yao, J.; Zhang, J. Regioselective synthesis and antioxidant activities of phosphorylated guar gum. Int. J. Biol. Macromol. 2013, 62, 741–747. [Google Scholar] [CrossRef]
- Wang, J.L.; Yang, T.; Tian, J.; Zeng, T.; Wang, X.F.; Yao, J.; Zhang, J.; Lei, Z.Q. Synthesis and characterization of phosphorylated galactomannan: The effect of DS on solution conformation and antioxidant activities. Carbohydr. Polym. 2014, 113, 325–335. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sorbent | Agitation Mode | qeq,exp. (mmol U g−1) | qeq,1 (mmol U g−1) | k1 × 102 (min−1) | R2 | AIC |
|---|---|---|---|---|---|---|
| Chit | MA | 0.094 | 0.104 | 4.94 | 0.991 | −141 |
| GG | MA | 0.070 | 0.075 | 5.19 | 0.971 | −134 |
| PGG@C #1 | MA | 0.488 | 0.512 | 7.83 | 0.995 | −107 |
| PGG@C #2 | MA | 0.481 | 0.510 | 7.54 | 0.990 | −100 |
| MChit | MA | 0.065 | 0.067 | 10.4 | 0.985 | −141 |
| MGG | MA | 0.062 | 0.064 | 9.34 | 0.989 | −149 |
| PGG@MC #1 | MA | 0.433 | 0.446 | 11.3 | 0.990 | −103 |
| PGG@MC #2 | MA | 0.438 | 0.454 | 10.5 | 0.986 | −99 |
| PGG@C | UT | 0.501 | 0.501 | 25.6 | 0.998 | −123 |
| PGG@MC | UT | 0.397 | 0.398 | 28.7 | 0.989 | −106 |
| Sorbent | qm,exp. (mmol U g−1) | qm,L. (mmol U g−1) | bL (L mmol−1) | qm,L × bL (L g−1) | R2 | AIC |
|---|---|---|---|---|---|---|
| Chit | 0.288 | 0.482 | 0.888 | 0.428 | 0.988 | −92 |
| GG | 0.256 | 0.472 | 0.694 | 0.328 | 0.970 | −85 |
| PGG@C #1 | 1.29 | 1.33 | 46.6 | 62.1 | 0.969 | −47 |
| PGG@C #2 | 1.27 | 1.30 | 47.6 | 62.0 | 0.966 | −47 |
| MChit | 0.239 | 0.494 | 0.541 | 0.267 | 0.979 | −90 |
| MGG | 0.203 | 0.457 | 0.467 | 0.214 | 0.964 | −88 |
| PGG@MC #1 | 1.16 | 1.22 | 23.6 | 28.7 | 0.984 | −57 |
| PGG@MC #2 | 1.15 | 1.22 | 20.3 | 24.9 | 0.979 | −55 |
| Sorption | Desorption | ||||
|---|---|---|---|---|---|
| Sorbent | Cycle | Sorption Efficiency (%) | Std. Dev. | Desorption Efficiency (%) | Std. Dev. |
| PGG@C | 1 | 97.0 | 0.1 | 100.0 | 0.0 |
| 2 | 95.9 | 0.1 | 99.9 | 0.0 | |
| 3 | 94.1 | 0.2 | 100.0 | 0.1 | |
| 4 | 93.1 | 0.2 | 99.5 | 0.5 | |
| 5 | 92.2 [−5%] | 0.1 | 98.9 | 0.3 | |
| PGG@MC | 1 | 89.9 | 0.6 | 99.6 | 0.0 |
| 2 | 89.1 | 0.4 | 100.0 | 0.0 | |
| 3 | 87.5 | 1.0 | 100.3 | 0.4 | |
| 4 | 85.5 | 0.6 | 100.1 | 0.0 | |
| 5 | 84.6 [−6%] | 1.1 | 99.7 | 0.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamza, M.F.; Fouda, A.; Elwakeel, K.Z.; Wei, Y.; Guibal, E.; Hamad, N.A. Phosphorylation of Guar Gum/Magnetite/Chitosan Nanocomposites for Uranium (VI) Sorption and Antibacterial Applications. Molecules 2021, 26, 1920. https://doi.org/10.3390/molecules26071920
Hamza MF, Fouda A, Elwakeel KZ, Wei Y, Guibal E, Hamad NA. Phosphorylation of Guar Gum/Magnetite/Chitosan Nanocomposites for Uranium (VI) Sorption and Antibacterial Applications. Molecules. 2021; 26(7):1920. https://doi.org/10.3390/molecules26071920
Chicago/Turabian StyleHamza, Mohammed F., Amr Fouda, Khalid Z. Elwakeel, Yuezhou Wei, Eric Guibal, and Nora A. Hamad. 2021. "Phosphorylation of Guar Gum/Magnetite/Chitosan Nanocomposites for Uranium (VI) Sorption and Antibacterial Applications" Molecules 26, no. 7: 1920. https://doi.org/10.3390/molecules26071920
APA StyleHamza, M. F., Fouda, A., Elwakeel, K. Z., Wei, Y., Guibal, E., & Hamad, N. A. (2021). Phosphorylation of Guar Gum/Magnetite/Chitosan Nanocomposites for Uranium (VI) Sorption and Antibacterial Applications. Molecules, 26(7), 1920. https://doi.org/10.3390/molecules26071920