
Structural Evidence for Pnictogen-Centered Lewis Acidity in Cationic Platinum-Stibine Complexes Featuring Pendent Amino or Ammonium Groups †


Abstract
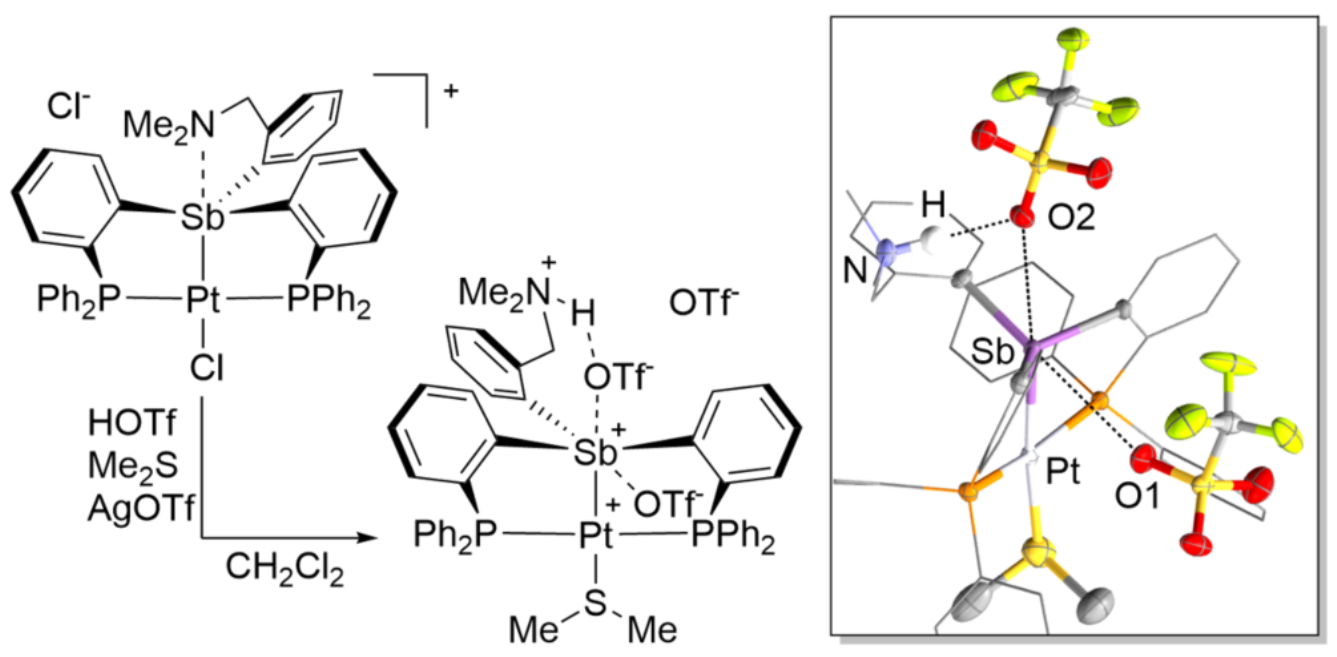
1. Introduction
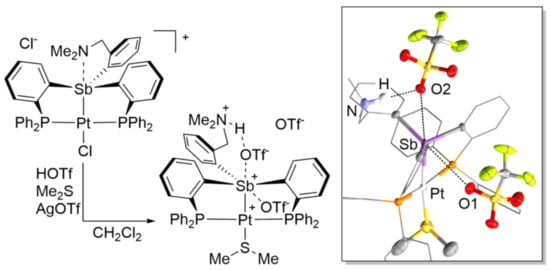
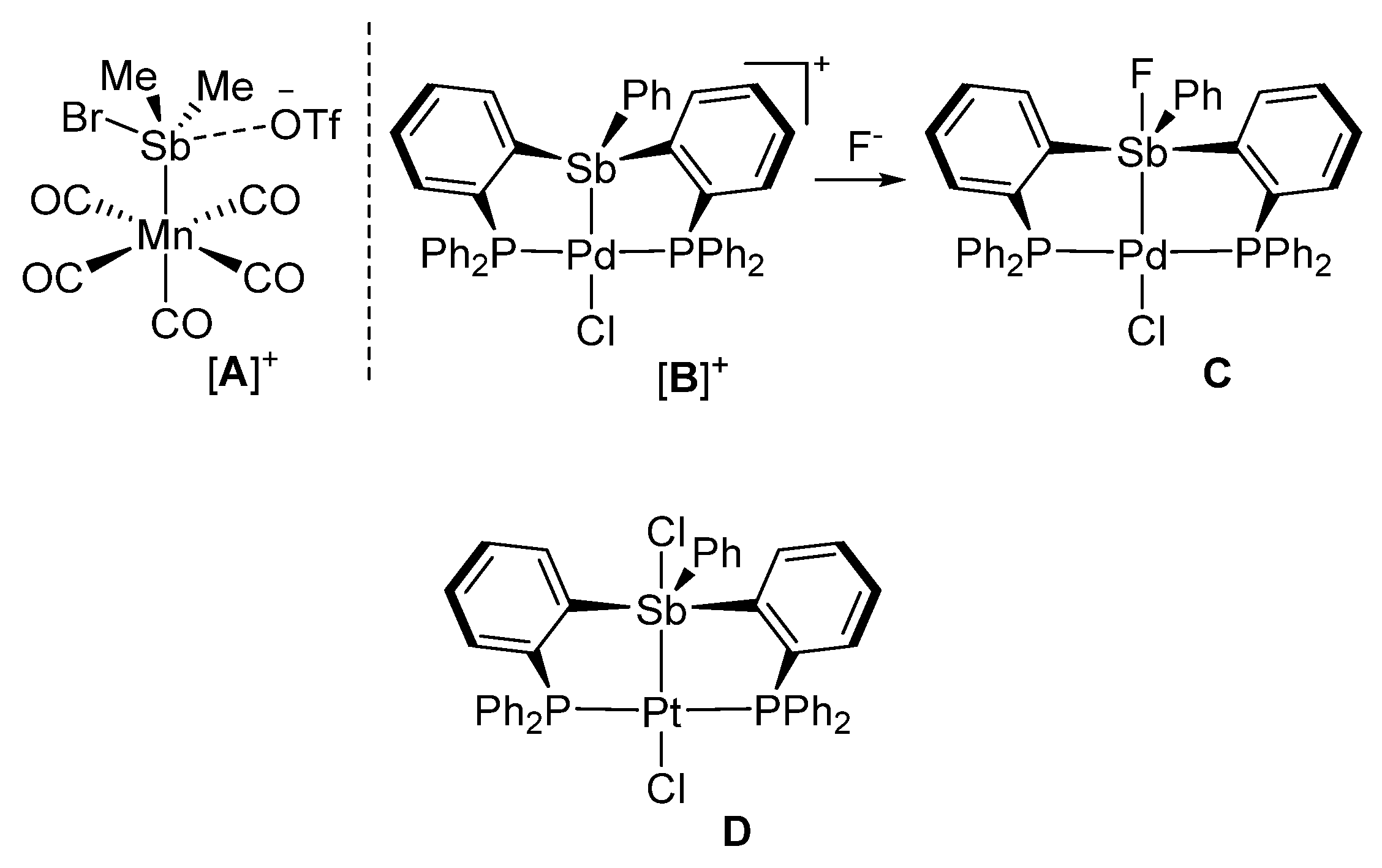
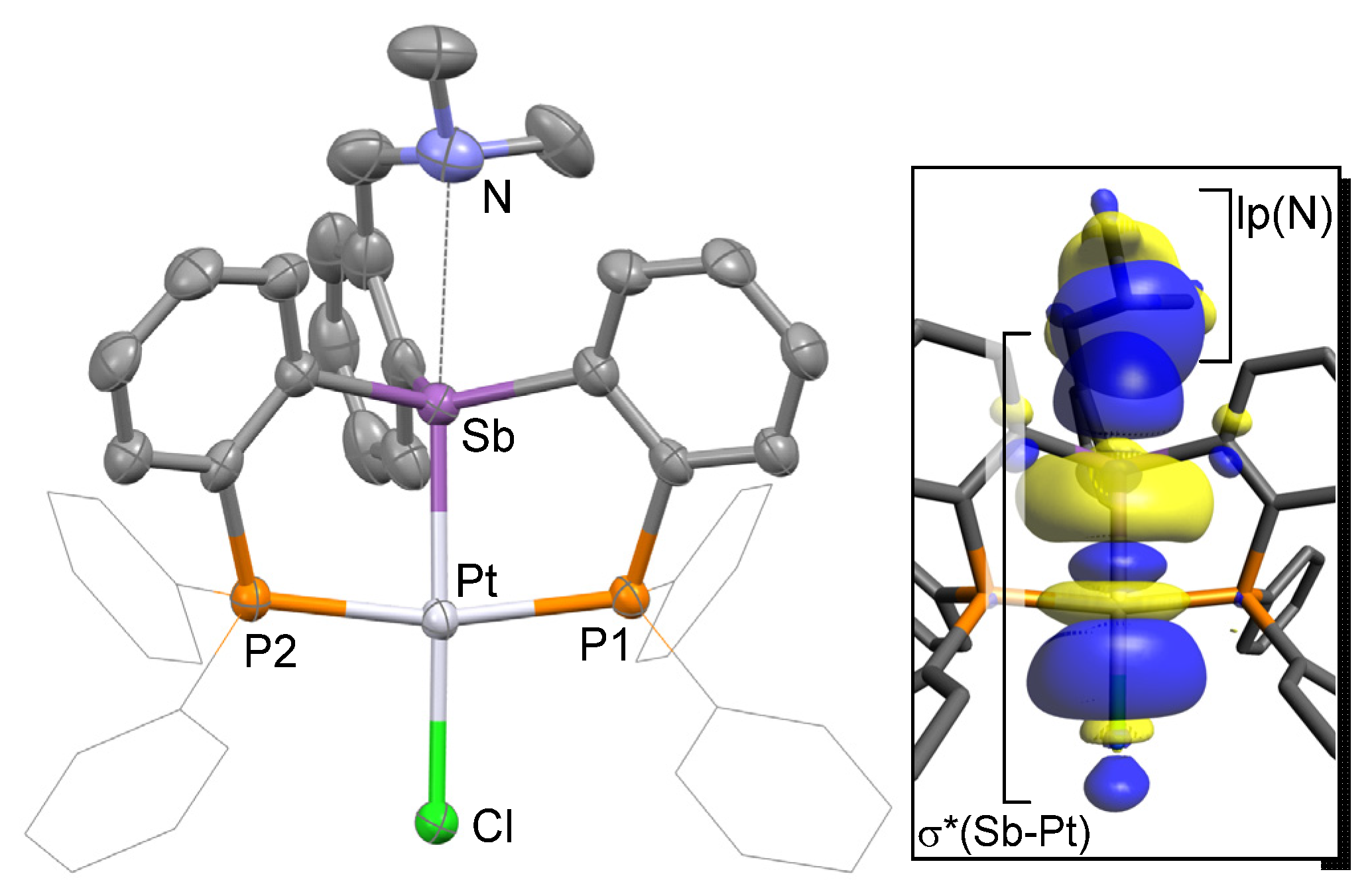
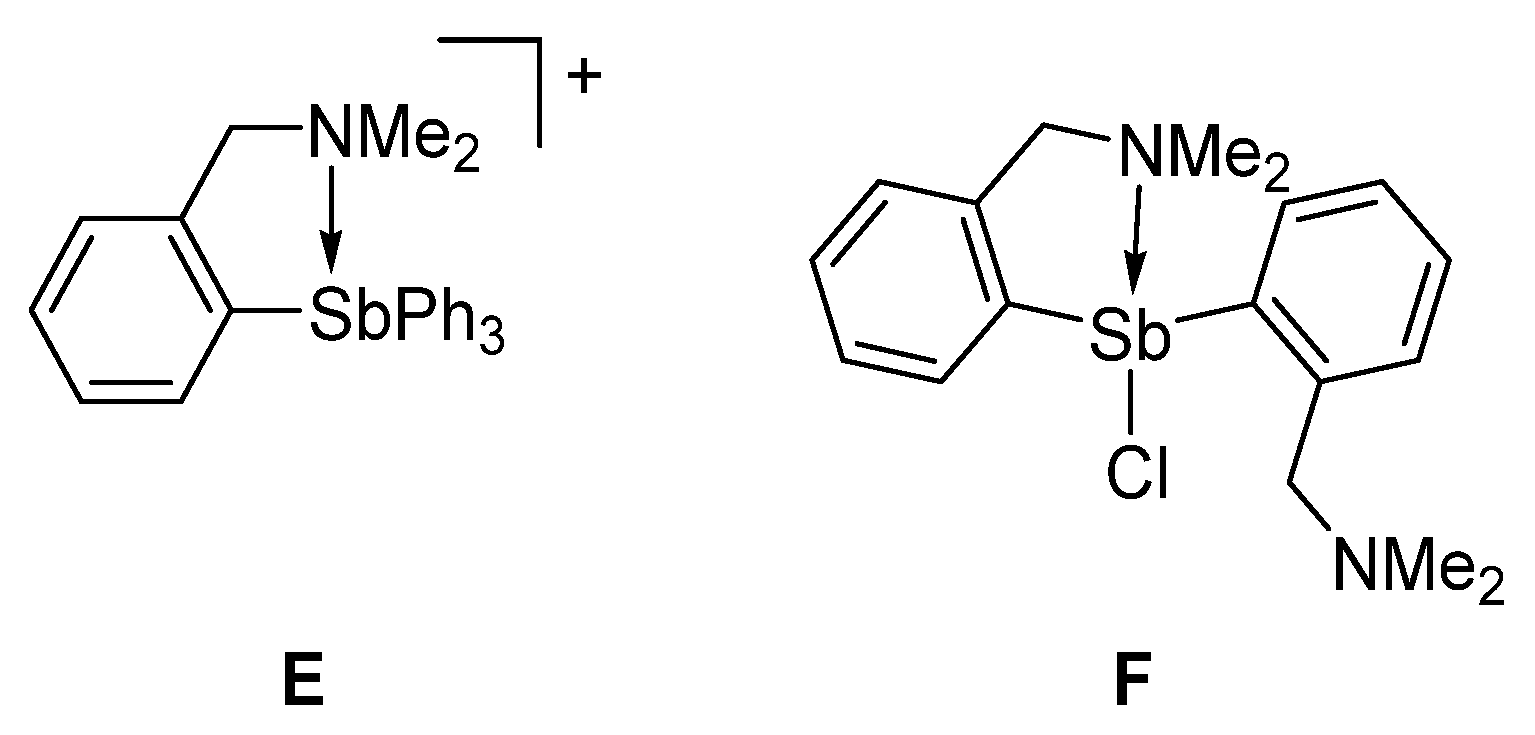
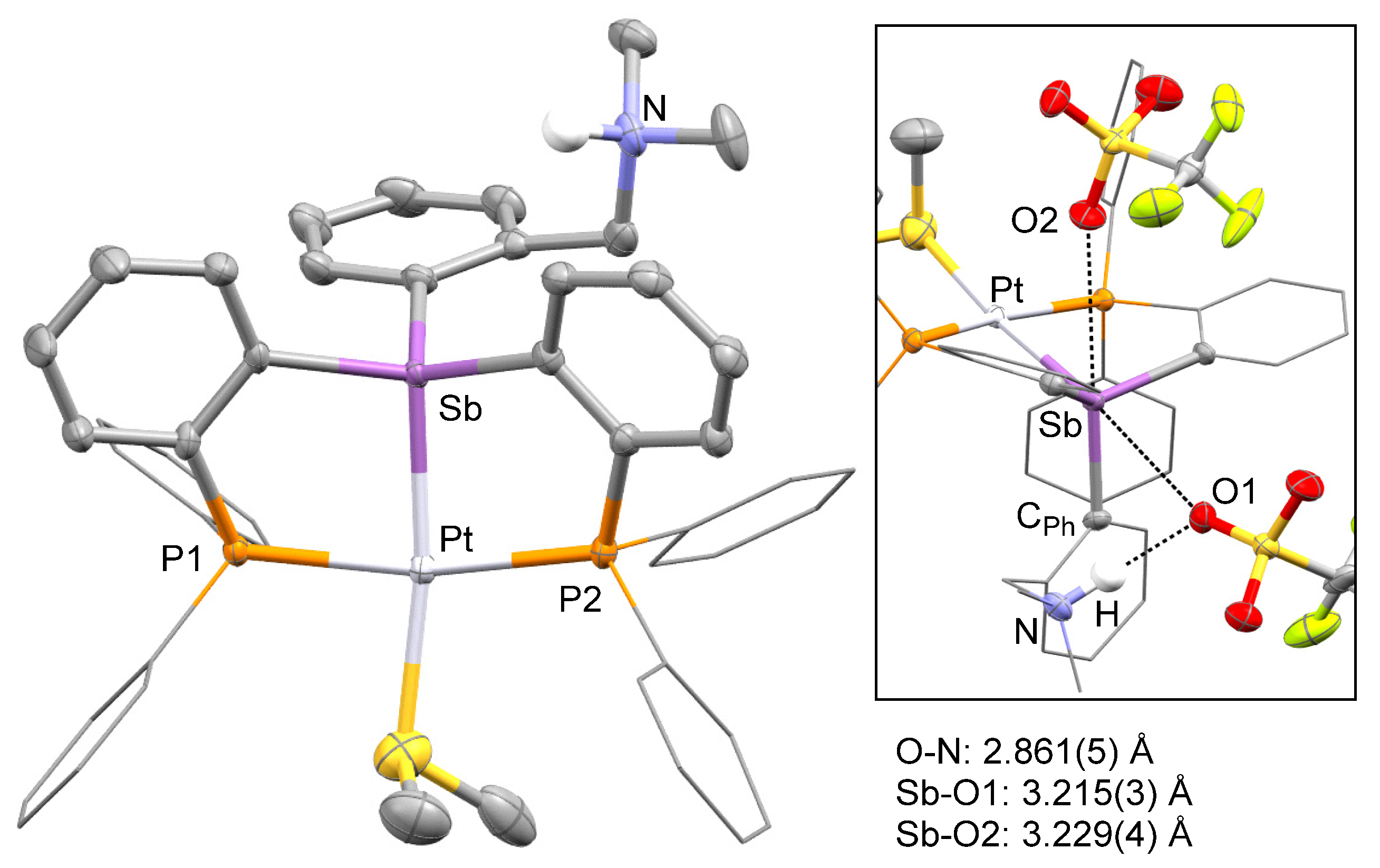
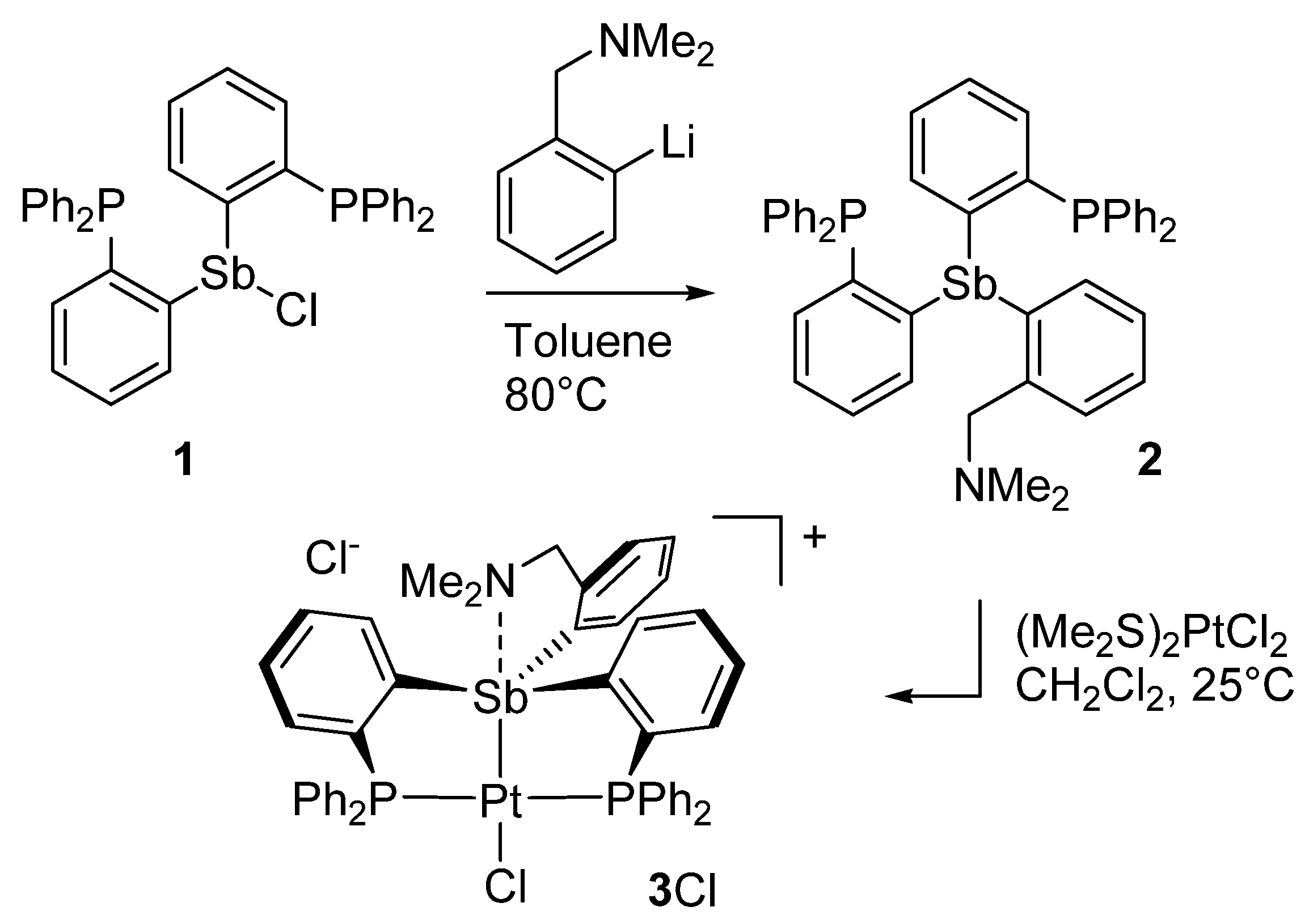
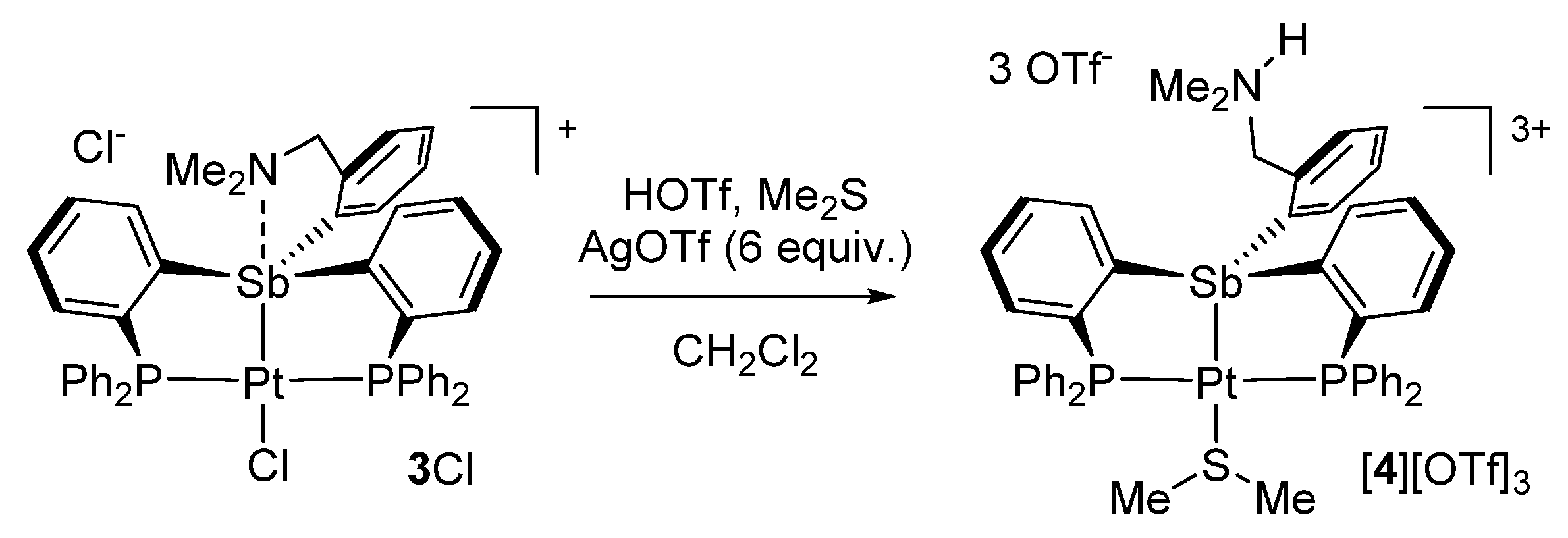
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Considerations
3.2. Synthesis of Compound 2
3.3. Synthesis of [3][Cl]
3.4. Synthesis of [4][OTf]3
3.5. Conductance Measurements
3.6. Crystallographic Measurements
3.7. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Schulz, S. The chemistry of Group 13/15 compounds (III-V compounds) with the higher homologues of Group 15, Sb and Bi. Coord. Chem. Rev. 2001, 215, 1. [Google Scholar] [CrossRef]
- Levason, W.; Reid, G. Developments in the coordination chemistry of stibine ligands. Coord. Chem. Rev. 2006, 250, 2565–2594. [Google Scholar] [CrossRef]
- Burt, J.; Levason, W.; Reid, G. Coordination chemistry of the main group elements with phosphine, arsine and stibine ligands. Coord. Chem. Rev. 2014, 260, 65–115. [Google Scholar] [CrossRef]
- Greenacre, V.K.; Levason, W.; Reid, G. Developments in the chemistry of stibine and bismuthine complexes. Coord. Chem. Rev. 2021, 432, 213698. [Google Scholar] [CrossRef]
- Wade, C.R.; Gabbaï, F.P. Two-Electron Redox Chemistry and Reversible Umpolung of a Gold–Antimony Bond. Angew. Chem. Int. Ed. 2011, 50, 7369–7372. [Google Scholar] [CrossRef]
- Jones, J.S.; Wade, C.R.; Gabbaï, F.P. Redox and anion exchange chemistry of a stibine nickel complex: Writing the L, X, Z ligand alphabet with a single element. Angew. Chem. Int. Ed. 2014, 53, 8876–8879. [Google Scholar] [CrossRef]
- Jones, J.S.; Wade, C.R.; Gabbaï, F.P. Guilty on Two Counts: Stepwise Coordination of Two Fluoride Anions to the Antimony Atom of a Noninnocent Stibine Ligand. Organometallics 2015, 34, 2647–2654. [Google Scholar] [CrossRef]
- Jones, J.S.; Gabbaï, F.P. Coordination- and Redox-Noninnocent Behavior of Ambiphilic Ligands Containing Antimony. Acc. Chem. Res. 2016, 49, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Ke, I.-S.; Gabbaï, F.P. T-Shaped Gold→Stiborane Complexes as Carbophilic Catalysts: Influence of the Peripheral Substituents. Organometallics 2017, 36, 4224–4230. [Google Scholar] [CrossRef]
- Wächtler, E.; Oro, L.A.; Iglesias, M.; Gerke, B.; Pöttgen, R.; Gericke, R.; Wagler, J. Synthesis and Oxidation of a Paddlewheel-Shaped Rhodium/Antimony Complex Featuring Pyridine-2-Thiolate Ligands. Chem. Eur. J. 2017, 23, 3447–3454. [Google Scholar] [CrossRef]
- Benjamin, S.L.; Levason, W.; Reid, G.; Warr, R.P. Halostibines SbMeX2 and SbMe2X: Lewis Acids or Lewis Bases? Organometallics 2012, 31, 1025–1034. [Google Scholar] [CrossRef]
- Benjamin, S.L.; Levason, W.; Light, M.E.; Reid, G.; Rogers, S.M. Bromostibine Complexes of Iron(II): Hypervalency and Reactivity. Organometallics 2014, 33, 2693–2695. [Google Scholar] [CrossRef]
- Ke, I.-S.; Jones, J.S.; Gabbaï, F.P. Anion-Controlled Switching of an X Ligand into a Z Ligand: Coordination Non-innocence of a Stiboranyl Ligand. Angew. Chem. Int. Ed. 2014, 53, 2633–2637. [Google Scholar] [CrossRef]
- Sen, S.; Ke, I.-S.; Gabbaï, F.P. Anion-Controlled Positional Switching of a Phenyl Group about the Dinuclear Core of a AuSb Complex. Inorg. Chem. 2016, 55, 9162–9172. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Wade, C.R.; Yang, M.; Gabbaï, F.P. On the coordination non-innocence of antimony in nickel(II) complexes of the tetradentate (o-(Ph2P)C6H4)3Sb ligand. Dalton Trans. 2017, 46, 5598–5604. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Gabbaï, F.P. Activation of an Au-Cl Bond by a Pendent SbIII Lewis Acid: Impact on Structure and Catalytic Activity. Chem. Eur. J. 2017, 23, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Plajer, A.J.; Colebatch, A.L.; Rizzuto, F.J.; Pröhm, P.; Bond, A.D.; García-Rodríguez, R.; Wright, D.S. How Changing the Bridgehead Can Affect the Properties of Tripodal Ligands. Angew. Chem. Int. Ed. 2018, 57, 6648–6652. [Google Scholar] [CrossRef]
- Jolleys, A.; Lake, B.R.M.; Krämer, T.; Benjamin, S.L. A Five-Membered PdSbn Coordination Series. Organometallics 2018, 37, 3854–3862. [Google Scholar] [CrossRef]
- You, D.; Yang, H.; Sen, S.; Gabbaï, F.P. Modulating the σ-Accepting Properties of an Antimony Z-type Ligand via Anion Abstraction: Remote-Controlled Reactivity of the Coordinated Platinum Atom. J. Am. Chem. Soc. 2018, 140, 9644–9651. [Google Scholar] [CrossRef]
- Furan, S.; Hupf, E.; Boidol, J.; Brünig, J.; Lork, E.; Mebs, S.; Beckmann, J. Transition metal complexes of antimony centered ligands based upon acenaphthyl scaffolds. Coordination non-innocent or not? Dalton Trans. 2019, 48, 4504–4513. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. Ruthenium Complexes of Stibino Derivatives of Carboxylic Amides: Synthesis and Characterization of Bidentate Sb,E, Tridentate Sb,E2, and Tetradentate Sb,E3 (E = N and O) Ligands and Their Reactivity Toward [RuCl2(PPh3)3]. Inorg. Chem. 2020, 59, 6359–6375. [Google Scholar] [CrossRef]
- Wachtler, E.; Gericke, R.; Block, T.; Pottgen, R.; Wagler, J. Trivalent Antimony as L-, X-, and Z-Type Ligand: The Full Set of Possible Coordination Modes in Pt-Sb Bonds. Inorg. Chem. 2020, 59, 15541–15552. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.R.; Ke, I.-S.; Gabbaï, F.P. Sensing of Aqueous Fluoride Anions by Cationic Stibine–Palladium Complexes. Angew. Chem. Int. Ed. 2012, 51, 478–481. [Google Scholar] [CrossRef]
- Opris, L.M.; Silvestru, A.; Silvestru, C.; Breunig, H.J.; Lork, E. Solid-state structure and solution behaviour of hypervalent organoantimony halides containing 2-(Me2NCH2)C6H4–moieties. Dalton Trans. 2003, 32, 4367–4374. [Google Scholar] [CrossRef]
- Sharma, P.; Castillo, D.; Rosas, N.; Cabrera, A.; Gomez, E.; Toscano, A.; Lara, F.; Hernández, S.; Espinosa, G. Synthesis and structures of organoantimony compounds containing intramolecular Sb–N interactions. J. Organomet. Chem. 2004, 689, 2593–2600. [Google Scholar] [CrossRef]
- Carmalt, C.J.; Cowley, A.H.; Culp, R.D.; Jones, R.A.; Kamepalli, S.; Norman, N.C. Synthesis and Structures of Intramolecularly Base-Coordinated Group 15 Aryl Halides. Inorg. Chem. 1997, 36, 2770–2776. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Chen, X.; Kojima, S.; Ohdoi, K.; Kitano, M.; Doi, Y.; Akiba, K.-Y. Experimental Investigation on Edge Inversion at Trivalent Bismuth and Antimony: Great Acceleration by Intra- and Intermolecular Nucleophilic Coordination. J. Am. Chem. Soc. 1995, 117, 3922–3932. [Google Scholar] [CrossRef]
- Yang, H.; Gabbaï, F.P. Solution and Solid-State Photoreductive Elimination of Chlorine by Irradiation of a [PtSb]VII Complex. J. Am. Chem. Soc. 2014, 136, 10866–10869. [Google Scholar] [CrossRef] [PubMed]
- Wendt, O.F.; Scodinu, A.; Elding, L.I. Trans influence of triphenylstibine. Crystal and molecular structures of cis-[PtCl2(SbPh3)2] and trans-[PtI2(SbPh3)2]. Inorg. Chim. Acta 1998, 277, 237–241. [Google Scholar] [CrossRef]
- Yang, M.; Pati, N.; Belanger-Chabot, G.; Hirai, M.; Gabbaï, F.P. Influence of the catalyst structure in the cycloaddition of isocyanates to oxiranes promoted by tetraarylstibonium cations. Dalton Trans. 2018, 47, 11843–11850. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Robertson, A.P.M.; Chitnis, S.S.; Jenkins, H.A.; McDonald, R.; Ferguson, M.J.; Burford, N. Establishing the Coordination Chemistry of Antimony(V) Cations: Systematic Assessment of Ph4Sb(OTf) and Ph3Sb(OTf)2 as Lewis Acceptors. Chem. Eur. J. 2015, 21, 7902–7913. [Google Scholar] [CrossRef] [PubMed]
- LeSuer, R.J.; Buttolph, C.; Geiger, W.E. Comparison of the Conductivity Properties of the Tetrabutylammonium Salt of Tetrakis(pentafluorophenyl)borate Anion with Those of Traditional Supporting Electrolyte Anions in Nonaqueous Solvents. Anal. Chem. 2004, 76, 6395–6401. [Google Scholar] [CrossRef]
- You, D.; Gabbaï, F.P. Unmasking the Catalytic Activity of a Platinum Complex with a Lewis Acidic, Non-innocent Antimony Ligand. J. Am. Chem. Soc. 2017, 139, 6843–6846. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Smith, J.E.; Sen, S.; Gabbaï, F.P. A Stiboranyl Platinum Triflate Complex as an Electrophilic Catalyst. Organometallics 2020, 39, 4169–4173. [Google Scholar] [CrossRef]
- Wile, B.M.; Burford, R.J.; McDonald, R.; Ferguson, M.J.; Stradiotto, M. Neutral and Cationic Platinum(II) Complexes Supported by a P,N-Functionalized Indene Ligand: Structural and Reactivity Comparisons with a Related Gold(III) Zwitterion. Organometallics 2006, 25, 1028–1035. [Google Scholar] [CrossRef]
- Fürstner, A.; Stelzer, F.; Szillat, H. Platinum-Catalyzed Cycloisomerization Reactions of Enynes. J. Am. Chem. Soc. 2001, 123, 11863–11869. [Google Scholar] [CrossRef]
- Talley, M.R.; Stokes, R.W.; Walker, W.K.; Michaelis, D.J. Electrophilic activation of alkynes for enyne cycloisomerization reactions with in situ generated early/late heterobimetallic Pt–Ti catalysts. Dalton Trans. 2016, 45, 9770–9773. [Google Scholar] [CrossRef]
- Hill, G.S.; Irwin, M.J.; Levy, C.J.; Rendina, L.M.; Puddephatt, R.J.; Andersen, R.A.; Mclean, L. Platinum (II) complexes of dimethyl sulfide. Inorg. Synth. 1998, 32, 149–153. [Google Scholar]
- Bruker. APEX3 (v2019.1); Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 2016. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [3][Cl]-(CH2Cl2)2-(H2O) | [4]OTf3-MeCN | |
|---|---|---|
| Empirical formula | C47H46Cl6NOP2PtSb | C52H50F9N2O9P2PtS4Sb |
| M | 1232.33 g/mol | 1524.96 g/mol |
| Temperature | 110 K | 110 K |
| Wavelength | 1.54178 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| Unit cell dimensions | a = 11.0507(5) Å | a = 13.2984(15) Å |
| b = 18.1574(8) Å | b = 14.3100(17) Å | |
| c = 24.0847(11) Å β = 99.231(2)° | c = 30.743(3) Å β = 101.601(3)° | |
| Volume | 4770.1(4) Å3 | 5730.9(11) Å3 |
| Z | 4 | 4 |
| Density (calculated) | 1.716 g/cm3 | 1.767 g/cm3 |
| Absorption coefficient | 13.906 mm−1 | 3.196 mm−1 |
| Θ range | 3.062 to 66.593°. | 1.963 to 28.281°. |
| Reflections collected | 56,128 | 126,808 |
| Independent reflections | 8379 [Rint = 0.0648] | 14,206 [Rint = 0.0639] |
| Goodness-of-fit on F2 | 1.065 | 1.081 |
| R indices (all data) | R1 = 0.0494, wR2 = 0.1206 | R1 = 0.0530, wR2 = 0.0853 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, R.R.; Gabbaï, F.P. Structural Evidence for Pnictogen-Centered Lewis Acidity in Cationic Platinum-Stibine Complexes Featuring Pendent Amino or Ammonium Groups. Molecules 2021, 26, 1985. https://doi.org/10.3390/molecules26071985
Rodrigues RR, Gabbaï FP. Structural Evidence for Pnictogen-Centered Lewis Acidity in Cationic Platinum-Stibine Complexes Featuring Pendent Amino or Ammonium Groups. Molecules. 2021; 26(7):1985. https://doi.org/10.3390/molecules26071985
Chicago/Turabian StyleRodrigues, Roberta R., and François P. Gabbaï. 2021. "Structural Evidence for Pnictogen-Centered Lewis Acidity in Cationic Platinum-Stibine Complexes Featuring Pendent Amino or Ammonium Groups" Molecules 26, no. 7: 1985. https://doi.org/10.3390/molecules26071985
APA StyleRodrigues, R. R., & Gabbaï, F. P. (2021). Structural Evidence for Pnictogen-Centered Lewis Acidity in Cationic Platinum-Stibine Complexes Featuring Pendent Amino or Ammonium Groups. Molecules, 26(7), 1985. https://doi.org/10.3390/molecules26071985