
The Early Steps of Molecule-to-Material Conversion in Chemical Vapor Deposition (CVD): A Case Study

 , ,
, ,
Abstract
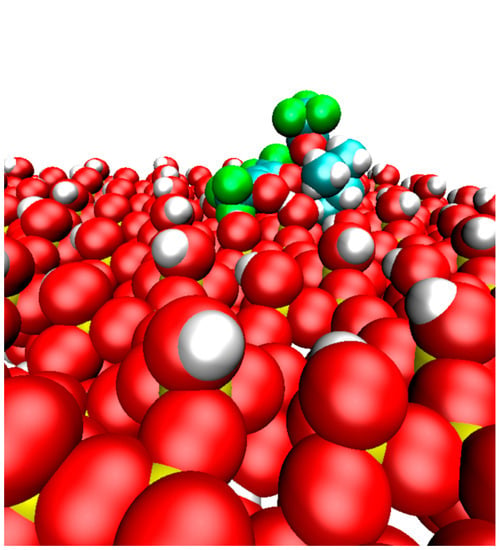
:
1. Introduction
2. Methods
3. Results
3.1. The Square-Pyramidal Zn(hfa)2TMEDA Complex
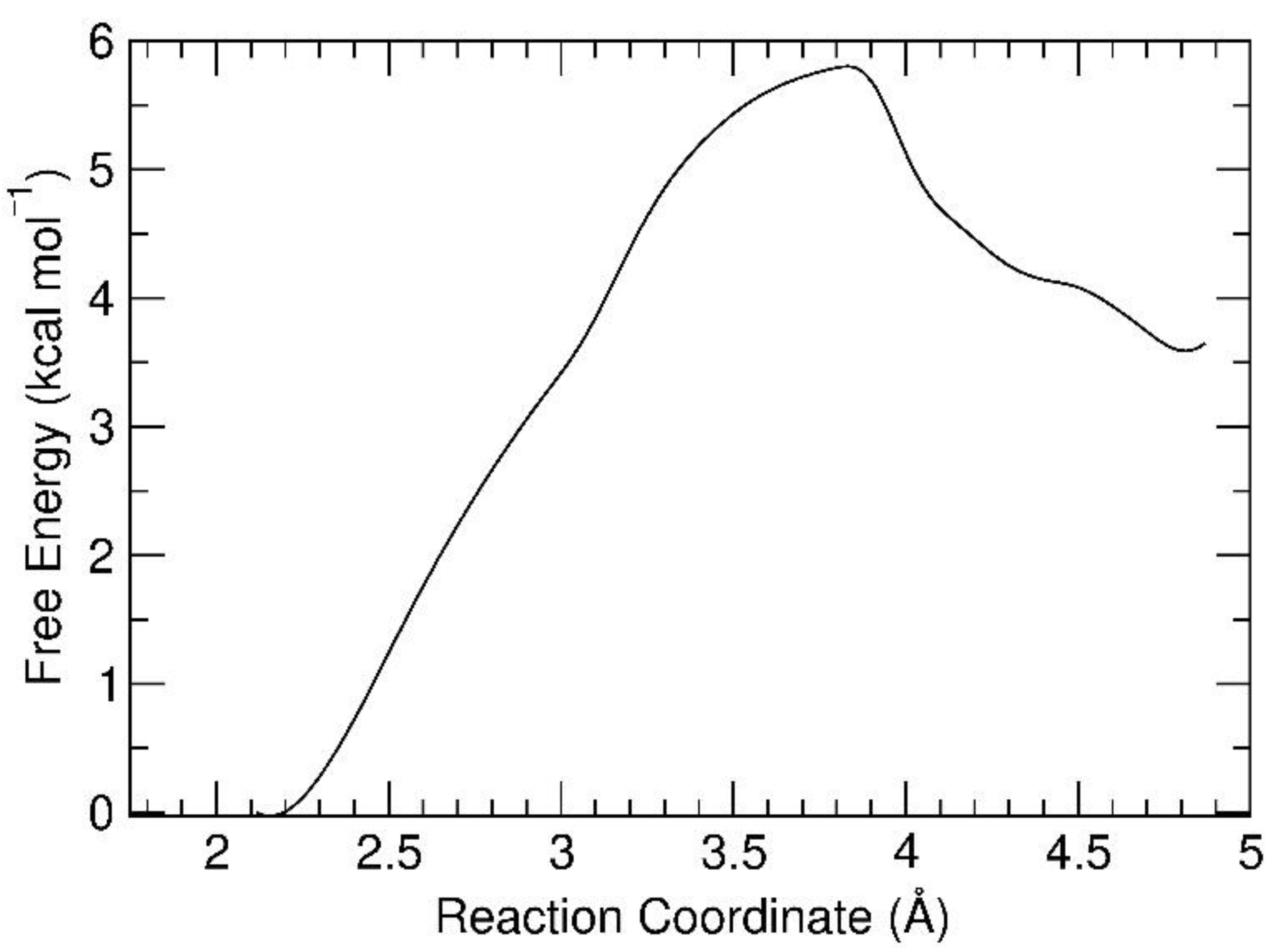
3.2. Free Energy Profile for the Octahedral-to-Pyramidal Conversion on the Hydroxylated Silica Surface
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ariga, K.; Ji, Q.; Nakanishi, W.; Hill, J.P.; Aono, M. Nanoarchitectonics: A new materials horizon for nanotechnology. Mater. Horiz. 2015, 2, 406–413. [Google Scholar] [CrossRef]
- Ariga, K.; Li, J. Nanoarchitectonics for advanced materials: Strategy beyond nanotechnology. Adv. Mater. 2016, 28, 987–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, M.; Mori, T.; Ariga, K. Molecular imprinting: Materials nanoarchitectonics with molecular information. Bull. Chem. Soc. Jpn. 2018, 91, 1075–1111. [Google Scholar] [CrossRef]
- Bekermann, D.; Barreca, D.; Gasparotto, A.; Maccato, C. Multi-component oxide nanosystems by Chemical Vapor Deposition and related routes: Challenges and perspectives. CrystEngComm 2012, 14, 6347–6358. [Google Scholar] [CrossRef]
- Mohd Abdah, M.A.A.; Azman, N.H.N.; Kulandaivalu, S.; Sulaiman, Y. Review of the use of transition-metal-oxide and conducting polymer-based fibres for high-performance supercapacitors. Mater. Des. 2020, 186, 108199. [Google Scholar] [CrossRef]
- Maduraiveeran, G.; Sasidharan, M.; Jin, W. Earth-abundant transition metal and metal oxide nanomaterials: Synthesis and electrochemical applications. Prog. Mater. Sci. 2019, 106, 100574. [Google Scholar] [CrossRef]
- Zappa, D.; Galstyan, V.; Kaur, N.; Munasinghe Arachchige, H.M.M.; Sisman, O.; Comini, E. “Metal oxide -based heterostructures for gas sensors”—A review. Anal. Chim. Acta 2018, 1039, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.K.; Tsang, S.C.E. Facet-dependent photocatalysis of nanosize semiconductive metal oxides and progress of their characterization. Nano Today 2018, 18, 15–34. [Google Scholar] [CrossRef]
- Gasparotto, A.; Barreca, D.; Bekermann, D.; Devi, A.; Fischer, R.A.; Maccato, C.; Tondello, E. Plasma processing of nanomaterials: Emerging technologies for sensing and energy applications. J. Nanosci. Nanotechnol. 2011, 11, 8206–8213. [Google Scholar] [CrossRef]
- McElwee-White, L. Design of precursors for the CVD of inorganic thin films. Dalton Trans. 2006, 5327–5333. [Google Scholar] [CrossRef]
- Tabacchi, G.; Fois, E.; Barreca, D.; Gasparotto, A. Opening the Pandora’s jar of molecule-to-material conversion in chemical vapor deposition: Insights from theory. Int. J. Quantum Chem. 2014, 114, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Devi, A. “Old chemistries” for new applications: Perspectives for development of precursors for MOCVD and ALD applications. Coord. Chem. Rev. 2013, 257, 3332–3384. [Google Scholar] [CrossRef]
- Mishra, S.; Daniele, S. Metal-organic derivatives with fluorinated ligands as precursors for inorganic nanomaterials. Chem. Rev. 2015, 115, 8379–8448. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Daniele, S. Molecular Engineering of Metal Alkoxides for Solution Phase Synthesis of High-Tech Metal Oxide Nanomaterials. Chem. A Eur. J. 2020, 26, 9292–9303. [Google Scholar] [CrossRef]
- Ferreira Da Silva, F.; Thorman, R.M.; Bjornsson, R.; Lu, H.; Mcelwee-White, L.; Ingólfsson, O. Dissociation of the FEBID precursor: Cis-Pt(CO)2Cl2 driven by low-energy electrons. Phys. Chem. Chem. Phys. 2020, 22, 6100–6108. [Google Scholar] [CrossRef]
- Cipriani, M.; Thorman, R.M.; Brewer, C.R.; McElwee-White, L.; Ingólfsson, O. Dissociative ionization of the potential focused electron beam induced deposition precursor π-allyl ruthenium(II) tricarbonyl bromide, a combined theoretical and experimental study. Eur. Phys. J. D 2019, 73, 1–7. [Google Scholar] [CrossRef]
- Bigiani, L.; Andreu, T.; Maccato, C.; Fois, E.; Gasparotto, A.; Sada, C.; Tabacchi, G.; Krishnan, D.; Verbeeck, J.; Morante, J.R.; et al. Engineering Au/MnO2 hierarchical nanoarchitectures for ethanol electrochemical valorization. J. Mater. Chem. A 2020, 8, 16902. [Google Scholar] [CrossRef]
- Bigiani, L.; Zappa, D.; Barreca, D.; Gasparotto, A.; Sada, C.; Tabacchi, G.; Fois, E.; Comini, E.; Maccato, C. Sensing nitrogen mustard gas simulant at the ppb scale via selective dual-site activation at Au/Mn3O4 interfaces. ACS Appl. Mater. Interfaces 2019, 11, 23692–23700. [Google Scholar] [CrossRef]
- Barreca, D.; Carraro, G.; Devi, A.; Fois, E.; Gasparotto, A.; Seraglia, R.; Maccato, C.; Sada, C.; Tabacchi, G.; Tondello, E.; et al. β-Fe2O3 nanomaterials from an iron(ii) diketonate-diamine complex: A study from molecular precursor to growth process. Dalt. Trans. 2012, 41, 149–155. [Google Scholar] [CrossRef]
- Barreca, D.; Carraro, G.; Gasparotto, A.; Maccato, C.; Seraglia, R.; Tabacchi, G. An iron(II) diamine diketonate molecular complex: Synthesis, characterization and application in the CVD of Fe2O3 thin films. Inorg. Chim. Acta 2012, 380, 161–166. [Google Scholar] [CrossRef]
- Bandoli, G.; Barreca, D.; Gasparotto, A.; Seraglia, R.; Tondello, E.; Devi, A.; Fischer, R.A.; Winter, M.; Fois, E.; Tabacchi, G. An integrated experimental and theoretical investigation on Cu (hfa)2‧TMEDA: Structure, bonding and reactivity. Phys. Chem. Chem. Phys. 2009, 11, 5998–6007. [Google Scholar] [CrossRef]
- Fois, E.; Tabacchi, G.; Barreca, D.; Gasparotto, A.; Tondello, E. “Hot” surface activation of molecular complexes: Insight from modeling studies. Angew. Chem. Int. Ed. 2010, 49, 1944–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabacchi, G.; Fois, E.; Barreca, D.; Gasparotto, A. CVD precursors for transition metal oxide nanostructures: Molecular properties, surface behavior and temperature effects. Phys. Status Solidi A 2014, 211, 251–259. [Google Scholar] [CrossRef]
- Tabacchi, G.; Fois, E.; Barreca, D.; Carraro, G.; Gasparotto, A.; Maccato, C. Modeling the first activation stages of the Fe(hfa)2‧TMEDA CVD precursor on a heated growth surface. In Advanced Processing and Manufacturing Technologies for Nanostructured and Multifunctional Materials II; Ceramic Engineering and Science, Proceedings; Ohji, T., Singh, M., Halbig, M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 83–90. ISBN 9781119211662. [Google Scholar]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, G.; Xu, L.; Cao, Y.; Li, A. Theoretical design and computational screening of precursors for atomic layer deposition. Coord. Chem. Rev. 2016, 322, 94–103. [Google Scholar] [CrossRef]
- Pedersen, H.; Elliott, S.D. Studying chemical vapor deposition processes with theoretical chemistry. Theor. Chem. Acc. 2014, 133, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, N.; Ventura-Macias, E.; Guerrero-Sánchez, J.; Zaera, F. Density functional theory study of the adsorption and dissociation of copper(I) acetamidinates on Ni(110): The effect of the substrate. J. Phys. Chem. C 2020, 124, 15366–15376. [Google Scholar] [CrossRef]
- Sukkaew, P.; Danielsson, Ö.; Kordina, O.; Janzén, E.; Ojamäe, L. Ab initio study of growth mechanism of 4H-SiC: Adsorption and Surface Reaction of C2H2, C2H4, CH4, and CH3. J. Phys. Chem. C 2017, 121, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.R.; Peters, A.W.; Li, Z.; Ortuño, M.A.; Martinson, A.B.F.; Cramer, C.J.; Hupp, J.T.; Farha, O.K. Atomic layer deposition of Cu(I) oxide films using Cu(II) bis(dimethylamino-2-propoxide) and water. Dalton Trans. 2017, 46, 5790–5795. [Google Scholar] [CrossRef]
- Shen, J.; Muthukumar, K.; Jeschke, H.O.; Valentí, R. Physisorption of an organometallic platinum complex on silica: An ab initio study. New J. Phys. 2012, 14, 073040. [Google Scholar] [CrossRef]
- Krasnov, P.O.; Mikhaleva, N.S.; Kuzubov, A.A.; Nikolaeva, N.S.; Zharkova, G.I.; Sheludyakova, L.A.; Morozova, N.B.; Basova, T.V. Prediction of the relative probability and the kinetic parameters of bonds breakage in the molecules of palladium MOCVD precursors. J. Mol. Struct. 2017, 1139, 269–274. [Google Scholar] [CrossRef]
- Lang, H.; Adner, D.; Georgi, C. Advances in deposition of transition metal oxides from metal enolates. In PATAI’S Chemistry of Functional Groups; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; pp. 1–38. [Google Scholar]
- Carter, E.A.; Ciccotti, G.; Hynes, J.T.; Kapral, R. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 1989, 156, 472–477. [Google Scholar] [CrossRef]
- Barreca, D.; Fois, E.; Gasparotto, A.; Seraglia, R.; Tondello, E.; Tabacchi, G. How does CuII convert into CuI? An unexpected ring-mediated single-electron reduction. Chem. A Eur. J. 2011, 17, 10864–10870. [Google Scholar] [CrossRef] [PubMed]
- Theerthagiri, J.; Salla, S.; Senthil, R.A.; Nithyadharseni, P.; Madankumar, A.; Arunachalam, P.; Maiyalagan, T.; Kim, H.S. A review on ZnO nanostructured materials: Energy, environmental and biological applications. Nanotechnology 2019, 30, 392001. [Google Scholar] [CrossRef]
- Le, A.T.; Ahmadipour, M.; Pung, S.Y. A review on ZnO-based piezoelectric nanogenerators: Synthesis, characterization techniques, performance enhancement and applications. J. Alloys Compd. 2020, 844, 156172. [Google Scholar] [CrossRef]
- Constantinoiu, I.; Viespe, C. ZnO metal oxide semiconductor in surface acoustic wave sensors: A Review. Sensors 2020, 20, 5118. [Google Scholar] [CrossRef]
- Özgür, Ü.; Alivov, Y.I.; Liu, C.; Teke, A.; Reshchikov, M.A.; Doǧan, S.; Avrutin, V.; Cho, S.J.; Morko, H. A comprehensive review of ZnO materials and devices. J. Appl. Phys. 2005, 98, 1–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, R.; Xiang, L.; Komarneni, S. Synthesis, properties and applications of ZnO nanomaterials with oxygen vacancies: A review. Ceram. Int. 2018, 44, 7357–7377. [Google Scholar] [CrossRef]
- Kołodziejczak-Radzimska, A.; Jesionowski, T. Zinc Oxide—from synthesis to application: A review. Materials 2014, 7, 2833–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Yan, H.; Wang, A.; Yang, Y.; Stern, C.L.; Metz, A.W.; Jin, S.; Wang, L.; Marks, T.J.; Ireland, J.R.; et al. MOCVD-derived highly transparent, conductive zinc- and tin-doped indium oxide thin films: Precursor synthesis, metastable phase film growth and characterization, and application as anodes in polymer light-emitting diodes. J. Am. Chem. Soc. 2005, 127, 5613–5624. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Tabacchi, G.; Gianotti, E.; Fois, E.; Martra, G.; Marchese, L.; Coluccia, S.; Gamba, A. Understanding the vibrational and electronic features of Ti(IV) sites in mesoporous silicas by integrated ab initio and spectroscopic investigations. J. Phys. Chem. C 2007, 111, 4946–4955. [Google Scholar] [CrossRef]
- Fois, E.; Gamba, A.; Tabacchi, G.; Coluccia, S.; Martra, G. Ab initio study of defect sites at the inner surfaces of mesoporous silicas. J. Phys. Chem. B 2003, 107, 10767–10772. [Google Scholar] [CrossRef]
- IBM Corp. MPI für Festkörperforschung Stuttgart 1997–2001 CPMD: Car Parrinello Molecular Dynamics 2019, Version 4.1; IBM Corp: Zürich, Switzerland, 2019. [Google Scholar]
- Tabacchi, G. Supramolecular organization in confined nanospaces. ChemPhysChem 2018, 19, 1249–1297. [Google Scholar] [CrossRef]
- Kleinman, L.; Bylander, D.M. Efficacious form for model pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425–1428. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Martínez-Suarez, L.; Siemer, N.; Frenzel, J.; Marx, D. Reaction Network of Methanol Synthesis over Cu/ZnO Nanocatalysts. ACS Catal. 2015, 5, 4201–4218. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, G.D.; Lotti, P.; Tabacchi, G. The effect of pressure on open-framework silicates: Elastic behaviour and crystal–fluid interaction. Phys. Chem. Miner. 2018, 45, 115–138. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac-Hartree-Fock data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Gaussian Basis Sets for Molecular Calculations. In Methods of Electronic Structure Theory; Springer: New York, NY, USA, 1977; pp. 1–27. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Halz, J.H.; Heiser, C.; Wagner, C.; Merzweiler, K. Syntheses and crystal structures of three [ M(acac)2(TMEDA)] complexes (M = Mn, Fe and Zn). Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 66–71. [Google Scholar] [CrossRef]
- Tabacchi, G.; Fabbiani, M.; Mino, L.; Martra, G.; Fois, E. The case of formic acid on anatase TiO2 (101): Where is the acid proton? Angew. Chem. Int. Ed. 2019, 58, 12431–12434. [Google Scholar] [CrossRef] [PubMed]
- Fois, E.; Gamba, A.; Medici, C.; Tabacchi, G.; Quartieri, S.; Mazzucato, E.; Arletti, R.; Vezzalini, G.; Dmitriev, V. High pressure deformation mechanism of Li-ABW: Synchrotron XRPD study and ab initio molecular dynamics simulations. Microporous Mesoporous Mater. 2008, 115, 267–280. [Google Scholar] [CrossRef]
- Frank, I. A single-molecule reaction cascade: First-principles molecular dynamics simulation. Int. J. Quantum Chem. 2017, 117, e25395. [Google Scholar] [CrossRef]
- Fois, E.; Gamba, A.; Tabacchi, G. Structure and dynamics of a Brønsted acid site in a zeolite: An ab initio study of hydrogen sodalite. J. Phys. Chem. B 1998, 102, 3974–3979. [Google Scholar] [CrossRef]
- Litman, Y.; Donadio, D.; Ceriotti, M.; Rossi, M. Decisive role of nuclear quantum effects on surface mediated water dissociation at finite temperature. J. Chem. Phys. 2018, 148, 102320. [Google Scholar] [CrossRef] [Green Version]
- Arletti, R.; Fois, E.; Gigli, L.; Vezzalini, G.; Quartieri, S.; Tabacchi, G. Irreversible conversion of a water–ethanol solution into an organized two-dimensional network of alternating supramolecular units in a hydrophobic zeolite under pressure. Angew. Chem. Int. Ed. 2017, 56, 2105–2109. [Google Scholar] [CrossRef]
- Konuk, M.; Sellschopp, K.; Vonbun-Feldbauer, G.B.; Meißner, R.H. Modeling charge redistribution at magnetite interfaces in empirical force fields. J. Phys. Chem. C 2021. [Google Scholar] [CrossRef]
- Harju, M.; Mäntylä, T.; Vähä-Heikkilä, K.; Lehto, V.P. Water adsorption on plasma sprayed transition metal oxides. Appl. Surf. Sci. 2005, 249, 115–126. [Google Scholar] [CrossRef]
- Boehm, H.P.; Knoezinger, H. Nature and estimation of functional groups on solid surfaces. In Catalysis—Science and Technology; Anderson, J.R., Boudart, M., Eds.; Springer: Berlin/Heidelberg, Germany, 1983; Volume 4, pp. 39–209. [Google Scholar] [CrossRef]
- Védrine, J.C. Recent developments and prospectives of acid-base and redox catalytic processes by metal oxides. Appl. Catal. A Gen. 2019, 575, 170–179. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance | <r> | σ | rmin | rmax |
|---|---|---|---|---|
| Zn-O1 | 2.23 | 0.25 | 1.82 | 3.24 |
| Zn-O1′ | 2.04 | 0.12 | 1.75 | 2.43 |
| Zn-O2 | 2.15 | 0.21 | 1.82 | 2.89 |
| Zn-O2′ | 3.62 | 0.91 | 1.86 | 5.21 |
| Zn-N | 2.26 | 0.18 | 1.85 | 2.93 |
| Zn-N′ | 2.33 | 0.19 | 1.94 | 2.89 |
| Parameter | Oct- PBE-D2/PW | Pyr- PBE-D2/PW | Oct- M06/D95+(d,p) | Pyr- M06/D95+(d,p) |
|---|---|---|---|---|
| Zn-O1 | 2.115 | 2.057 | 2.074 | 2.031 |
| Zn-O1′ | 2.116 | 2.064 | 2.097 | 2.039 |
| Zn-O2 | 2.115 | 1.964 | 2.074 | 1.942 |
| Zn-O2′ | 2.116 | 4.334 | 2.097 | 4.302 |
| Zn-N | 2.196 | 2.188 | 2.162 | 2.152 |
| Zn-N’ | 2.196 | 2.167 | 2.162 | 2.123 |
| O2′-C2′ | 1.269 | 1.241 | 1.251 | 1.225 |
| O2-C2 | 1.270 | 1.282 | 1.254 | 1.267 |
| O1′-C1′ | 1.269 | 1.271 | 1.251 | 1.254 |
| O1- C1 | 1.270 | 1.273 | 1.254 | 1.256 |
| C2*-C2′ | 1.408 | 1.437 | 1.404 | 1.437 |
| C2*-C2 | 1.405 | 1.387 | 1.400 | 1.378 |
| C1*-C1′ | 1.408 | 1.405 | 1.404 | 1.399 |
| C1*-C1 | 1.405 | 1.404 | 1.400 | 1.400 |
| O2-Zn-O1 | 88.0 | 111.9 | 87.3 | 111.8 |
| O2-Zn-O1′ | 86.7 | 108.1 | 86.6 | 107.0 |
| O2-Zn-N | 93.8 | 99.3 | 94.2 | 99.8 |
| O2-Zn-N’ | 91.6 | 91.5 | 92.0 | 92.2 |
| (kcal/mol) | Oct@SiO2 | TS@SiO2 | Pyr@SiO2 |
|---|---|---|---|
| ΔG | 0 | +5.80 | +3.57 |
| ΔE | 0 | +7.80 | +2.23 |
| BE | 28.27 | - | 36.07 |
| Δ(BE) | 0 | - | 7.81 |
| Distance | Oct@SiO2 | TS@SiO2 | Pyr@SiO2 | Isolated-Oct | Isolated-Pyr |
|---|---|---|---|---|---|
| Zn-O1 | 2.087 | 2.090 | 2.044 | 2.115 | 2.057 |
| Zn-O1′ | 2.139 | 2.018 | 2.091 | 2.116 | 2.064 |
| Zn-O2 | 2.115 | 2.042 | 2.050 | 2.115 | 1.964 |
| Zn-O2′ | 2.263 | 3.818 | 4.873 | 2.116 | 4.334 |
| Zn-N | 2.177 | 2.141 | 2.160 | 2.196 | 2.188 |
| Zn-N’ | 2.149 | 2.145 | 2.147 | 2.196 | 2.167 |
| O2′-H* | 1.885 | 1.630 | 1.586 | - | - |
| O2′-C2′ | 1.279 | 1.248 | 1.251 | 1.269 | 1.241 |
| O2-C2 | 1.264 | 1.265 | 1.277 | 1.270 | 1.282 |
| O1′-C1′ | 1.267 | 1.277 | 1.269 | 1.269 | 1.271 |
| O1- C1 | 1.271 | 1.269 | 1.272 | 1.270 | 1.273 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreca, D.; Fois, E.; Gasparotto, A.; Maccato, C.; Oriani, M.; Tabacchi, G. The Early Steps of Molecule-to-Material Conversion in Chemical Vapor Deposition (CVD): A Case Study. Molecules 2021, 26, 1988. https://doi.org/10.3390/molecules26071988
Barreca D, Fois E, Gasparotto A, Maccato C, Oriani M, Tabacchi G. The Early Steps of Molecule-to-Material Conversion in Chemical Vapor Deposition (CVD): A Case Study. Molecules. 2021; 26(7):1988. https://doi.org/10.3390/molecules26071988
Chicago/Turabian StyleBarreca, Davide, Ettore Fois, Alberto Gasparotto, Chiara Maccato, Mario Oriani, and Gloria Tabacchi. 2021. "The Early Steps of Molecule-to-Material Conversion in Chemical Vapor Deposition (CVD): A Case Study" Molecules 26, no. 7: 1988. https://doi.org/10.3390/molecules26071988