
Deciphering the Interactions of Bioactive Compounds in Selected Traditional Medicinal Plants against Alzheimer’s Diseases via Pharmacophore Modeling, Auto-QSAR, and Molecular Docking Approaches

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
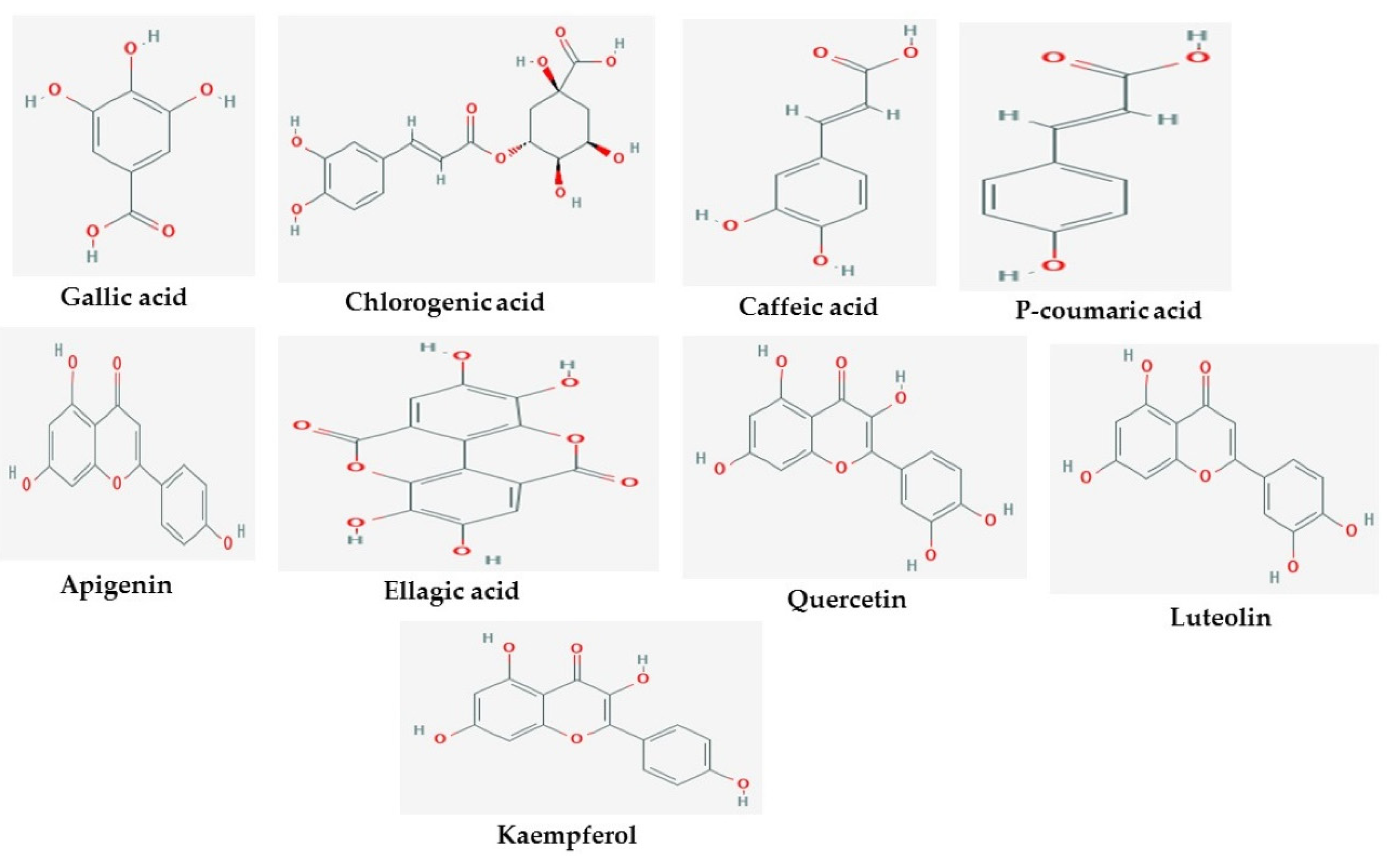
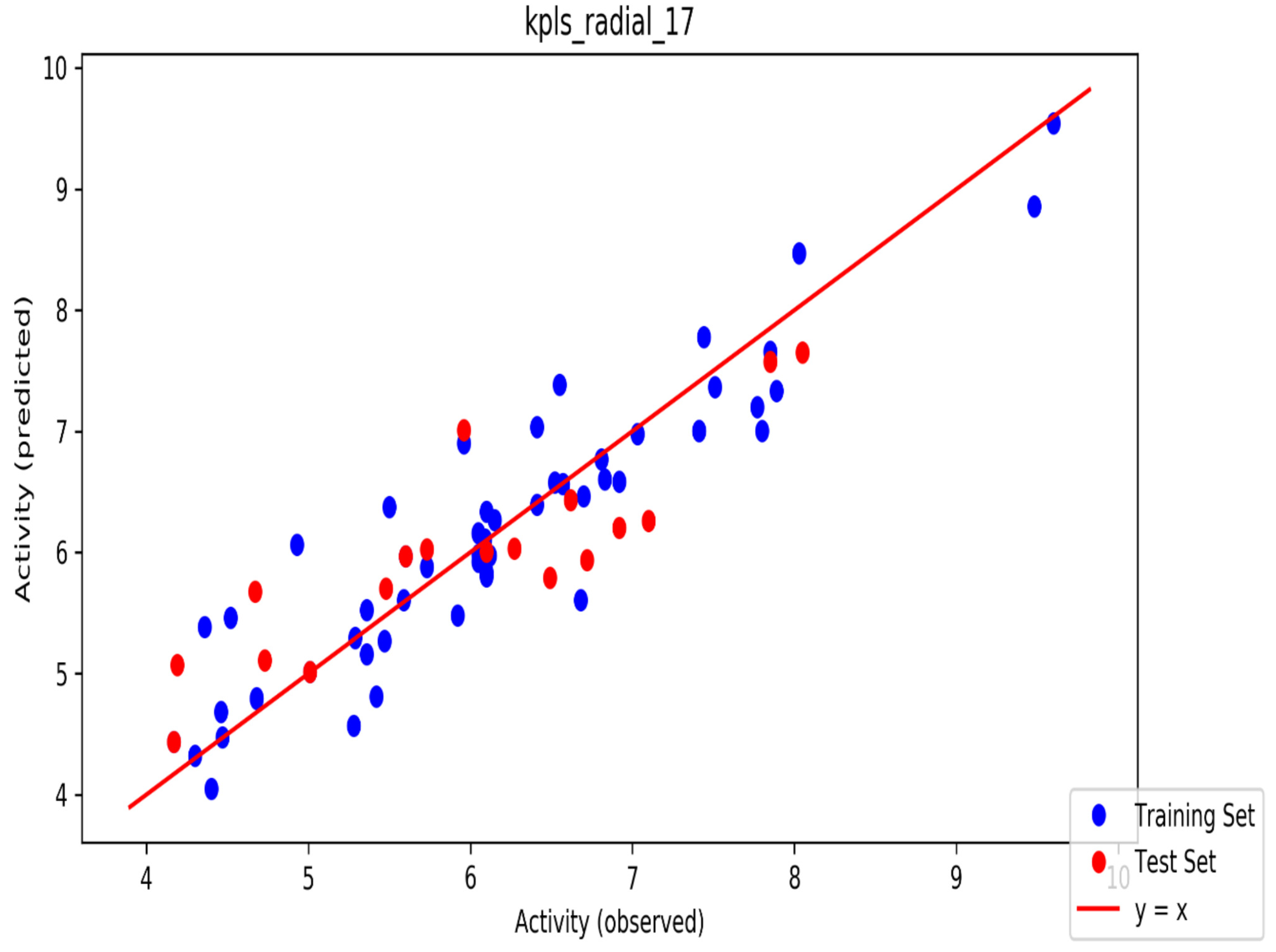
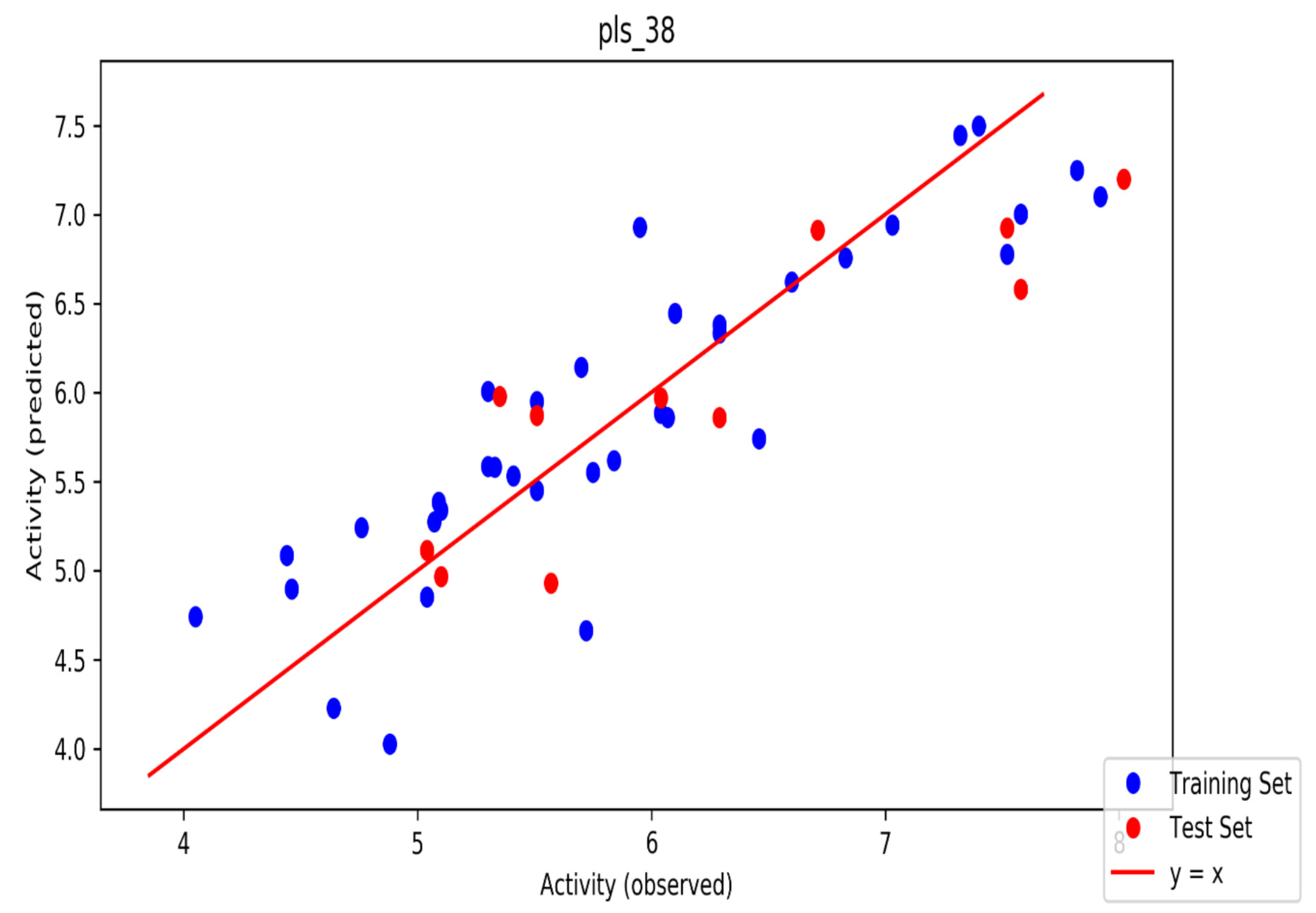
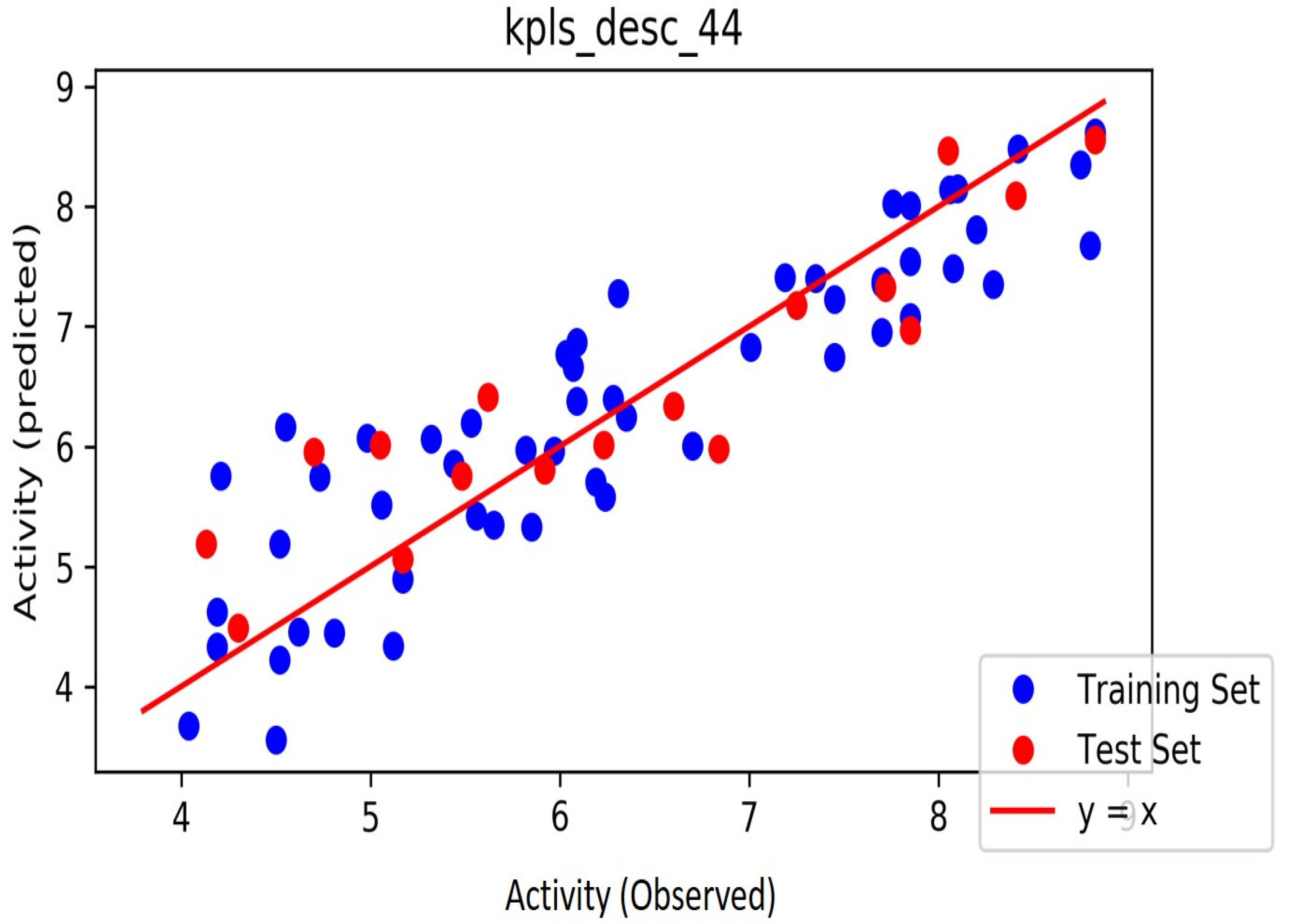
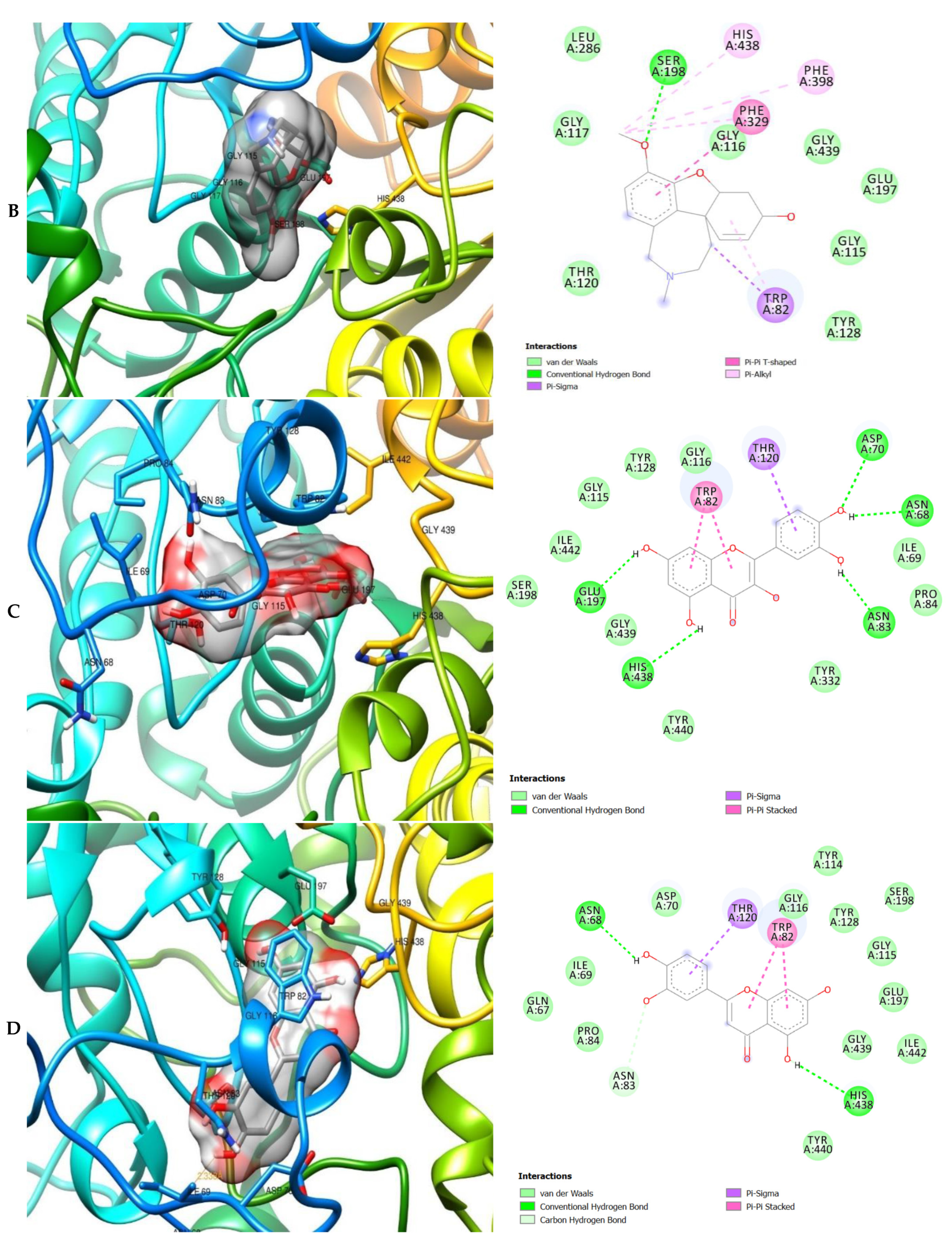
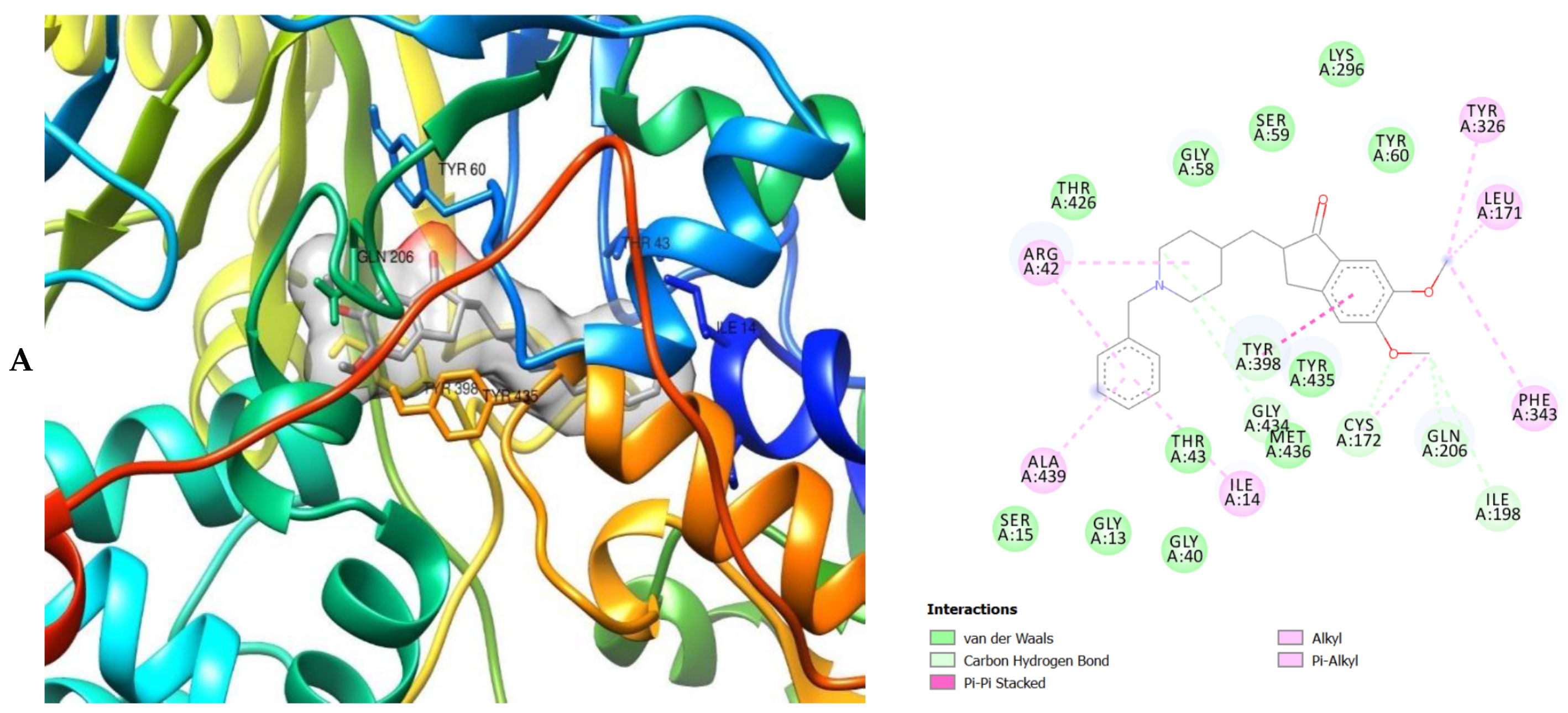
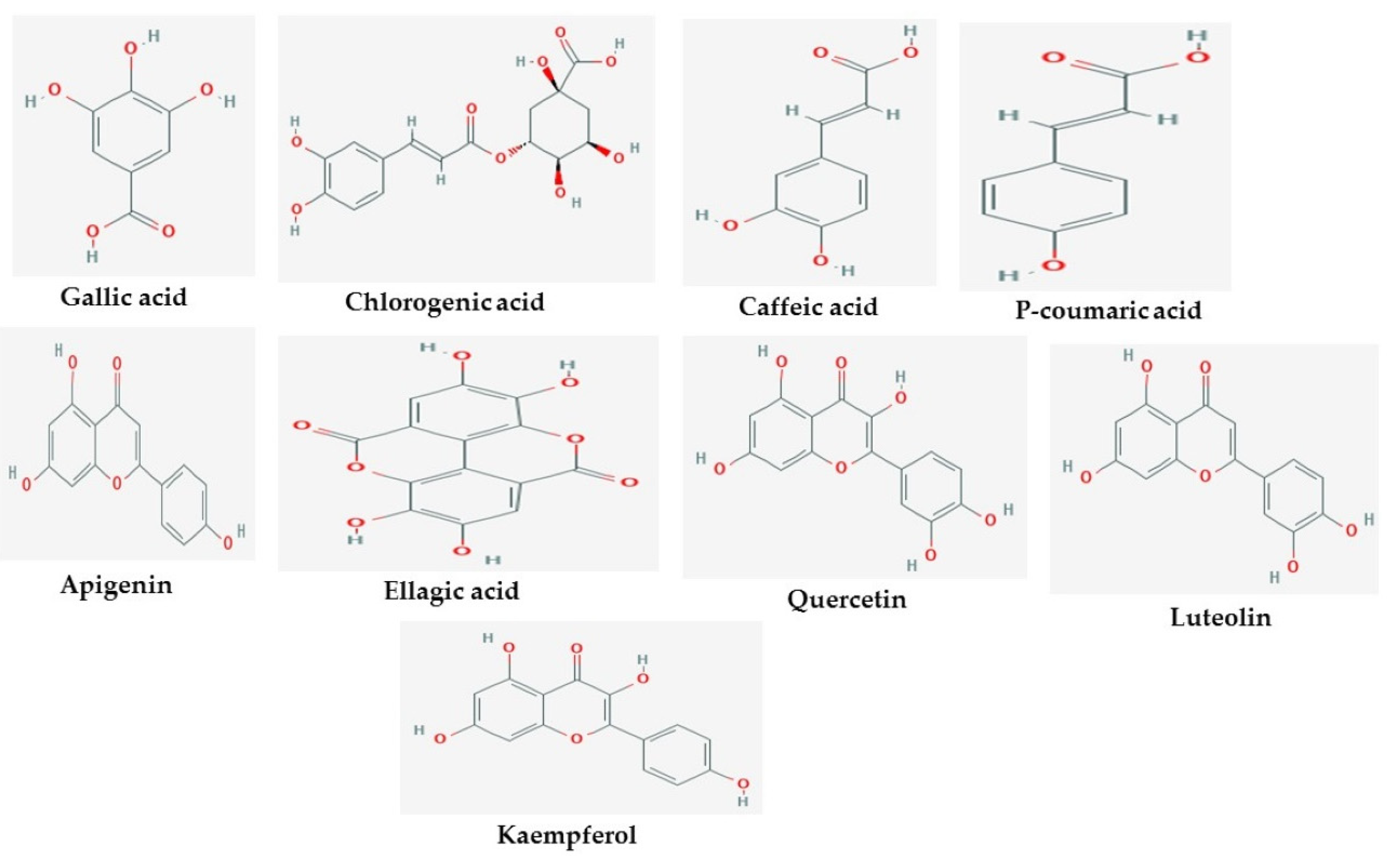
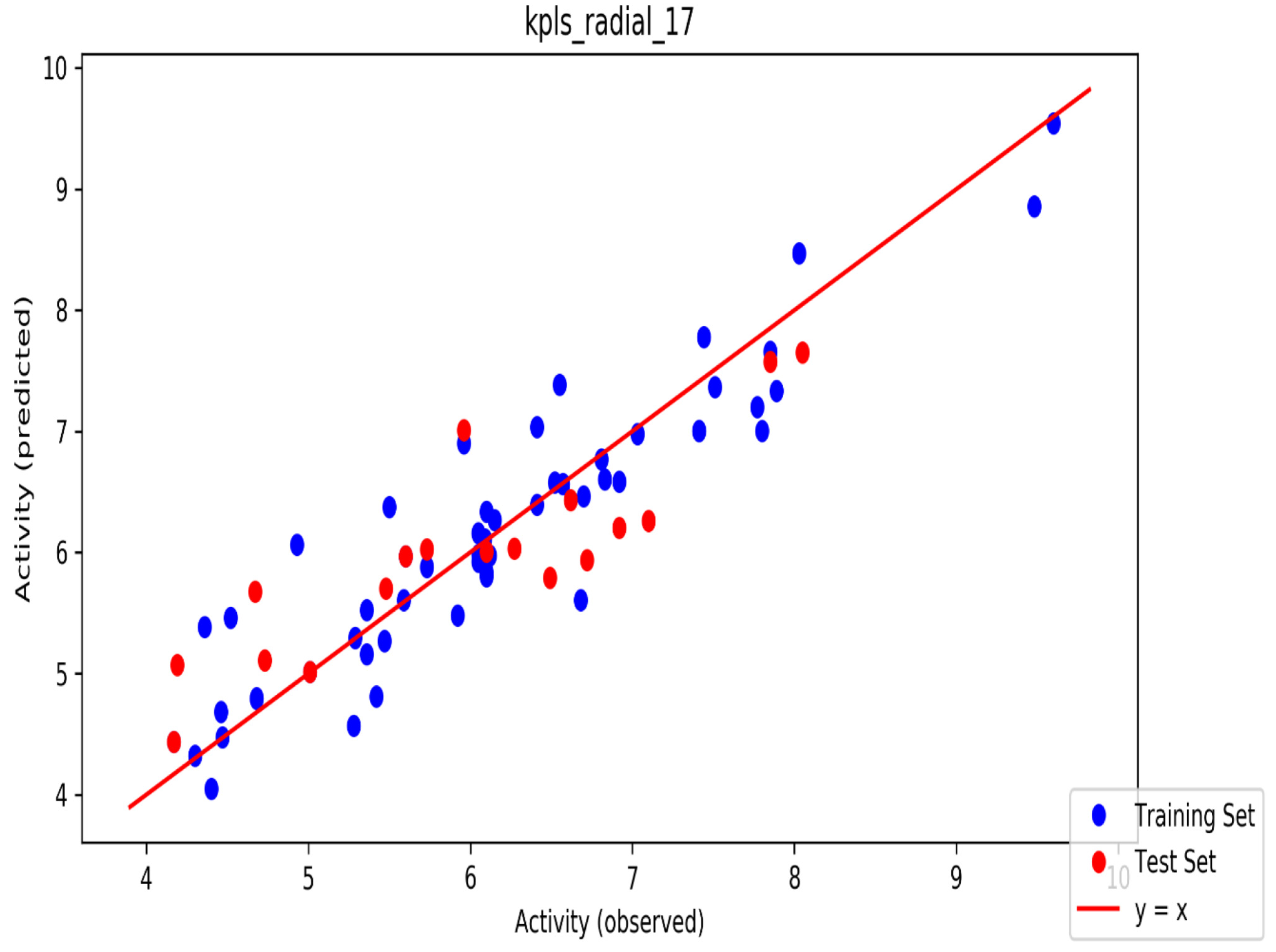
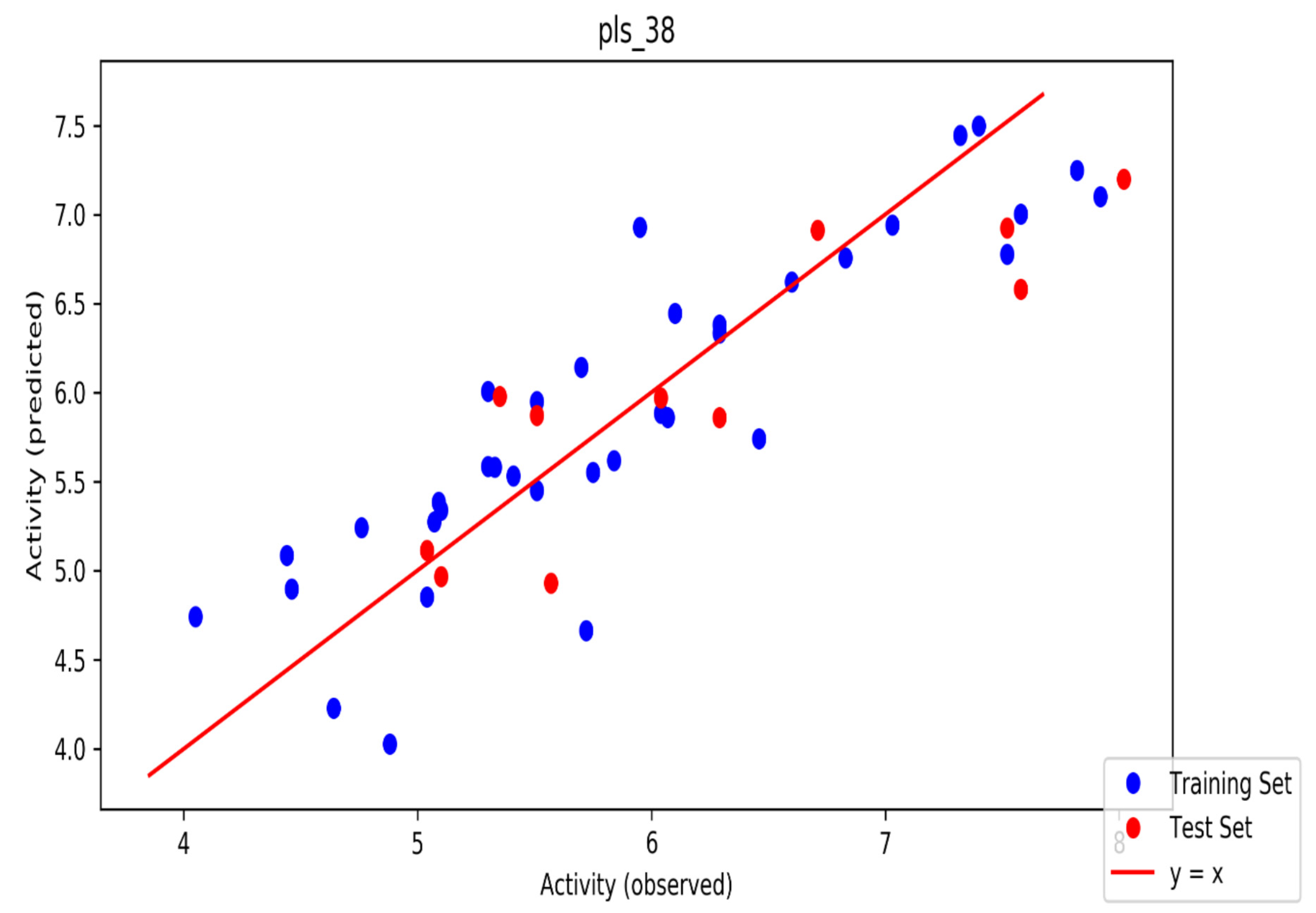
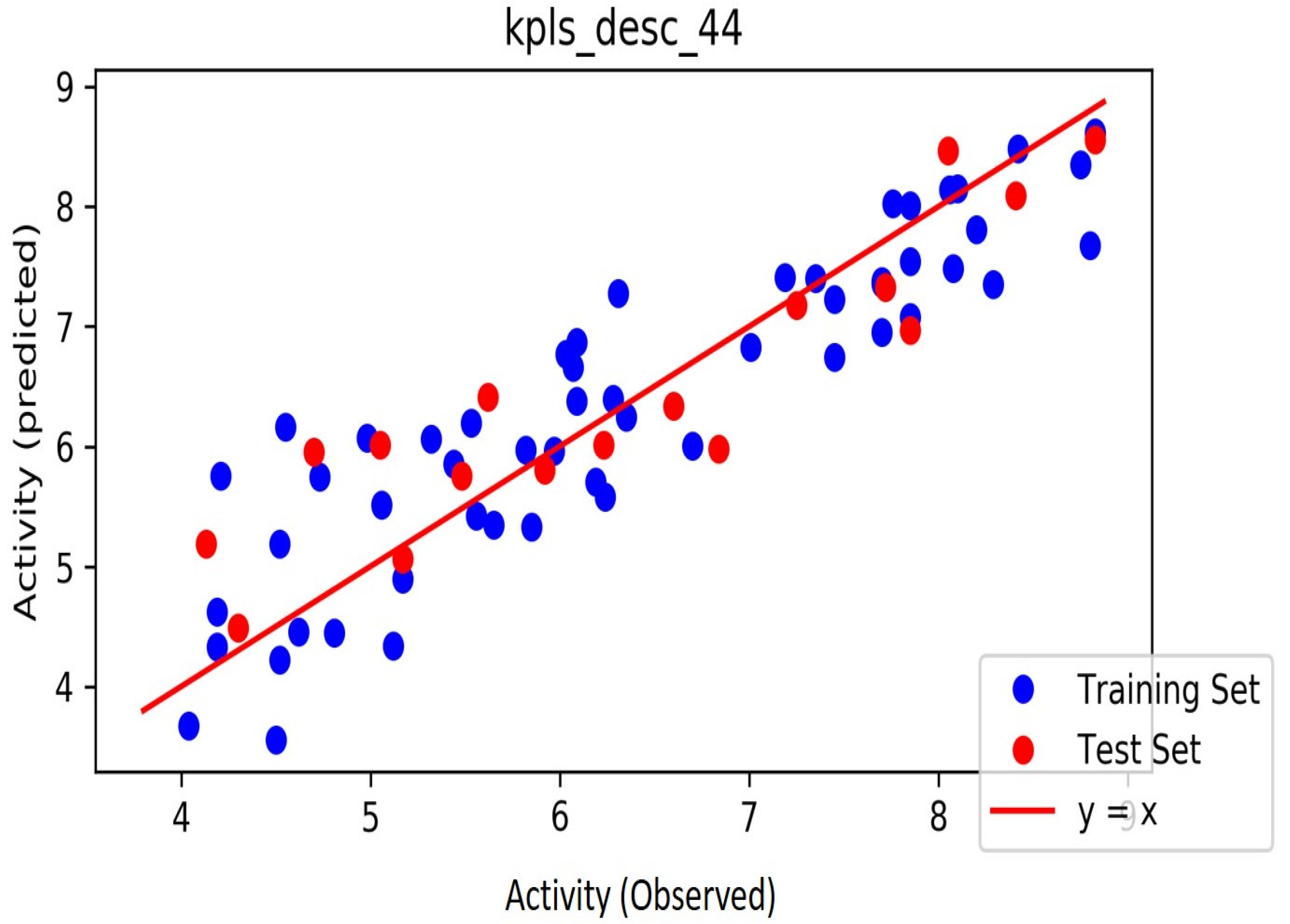
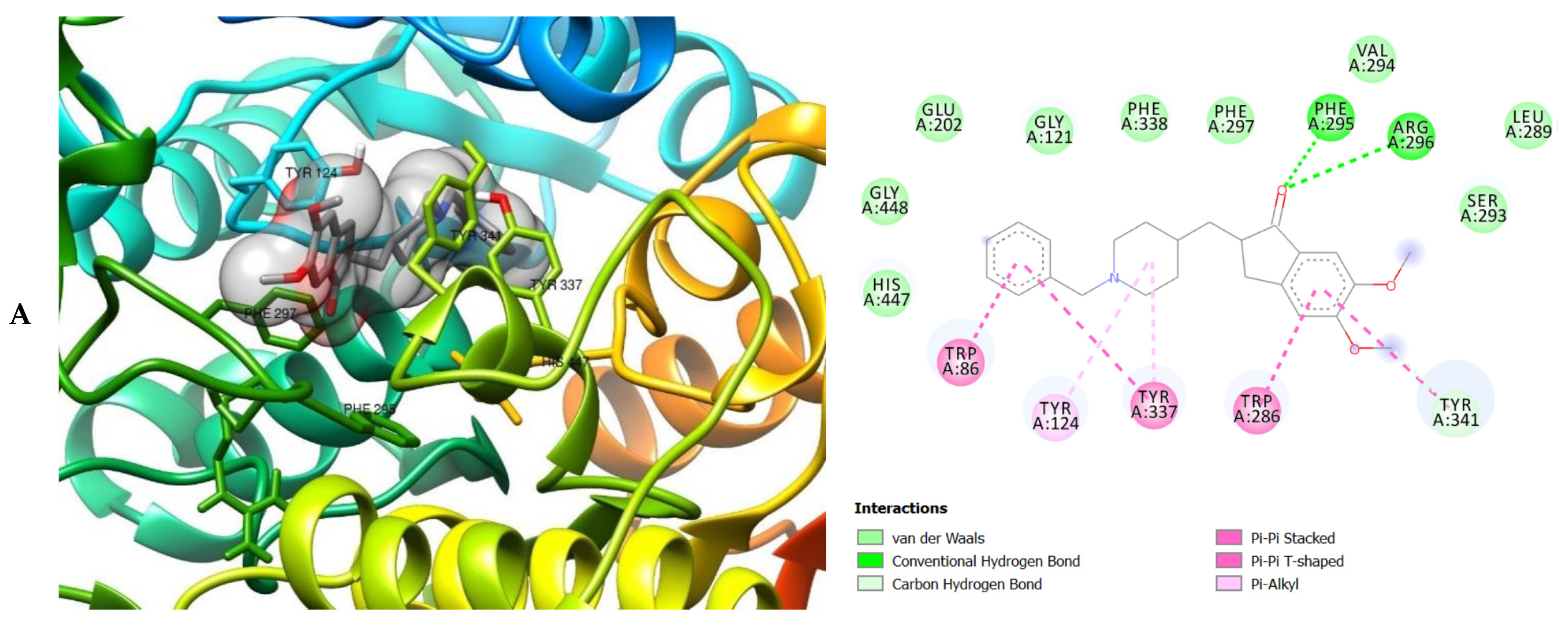
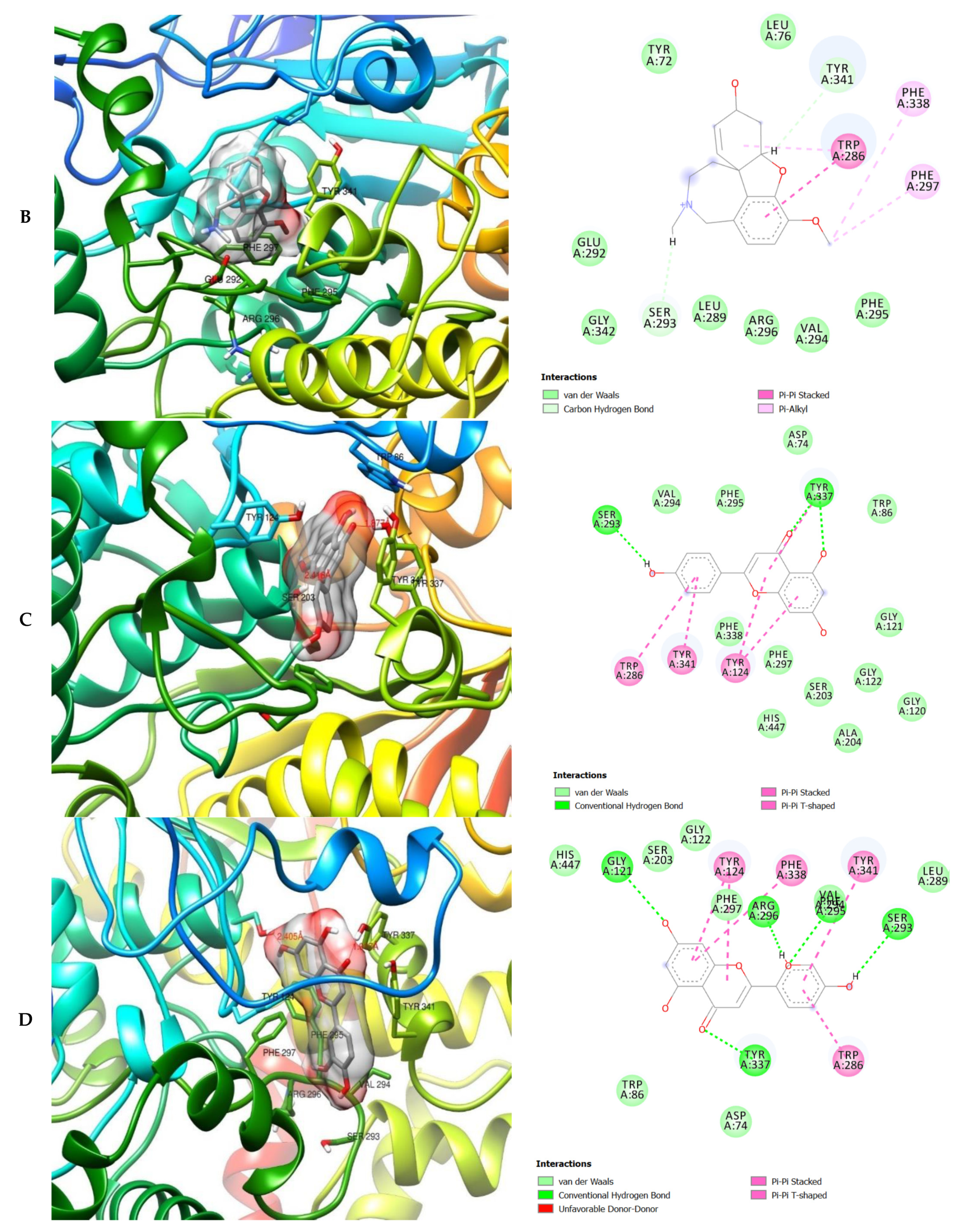
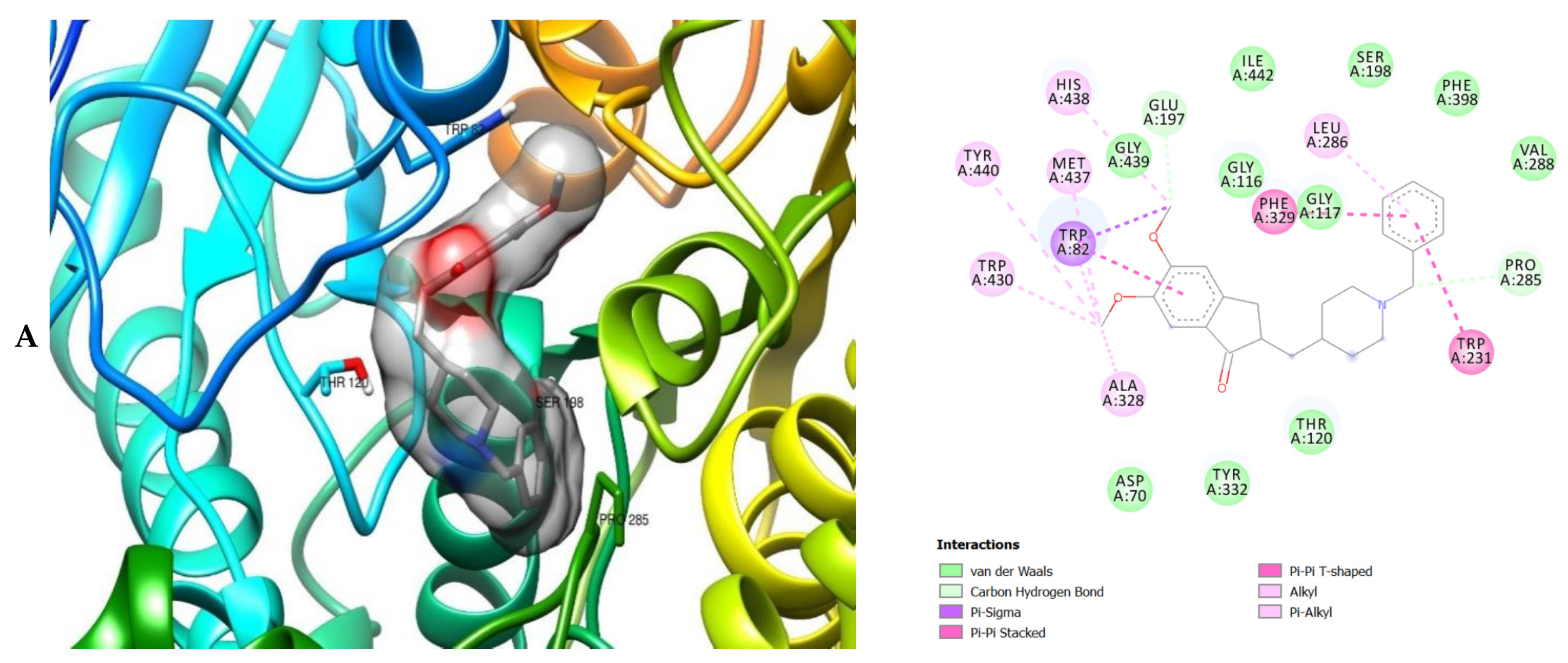
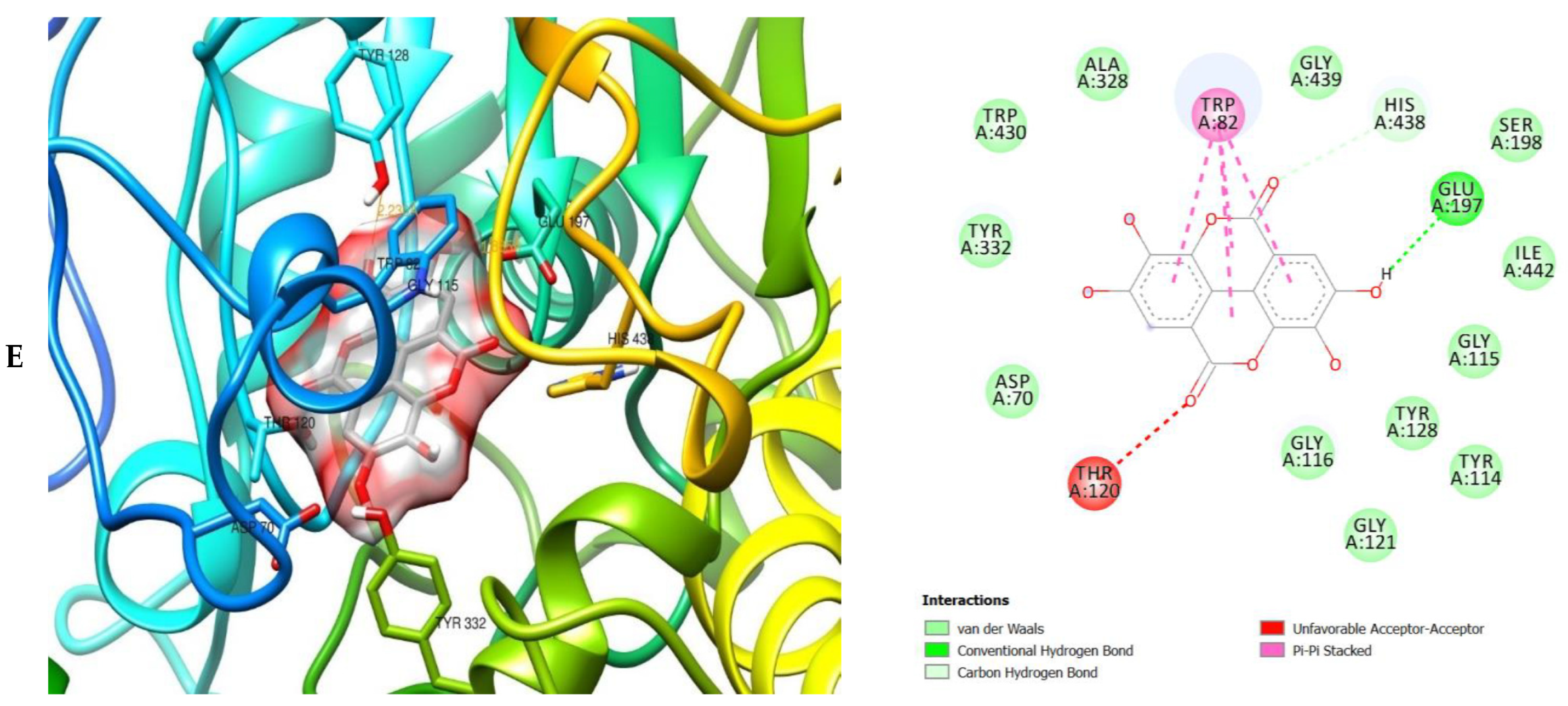
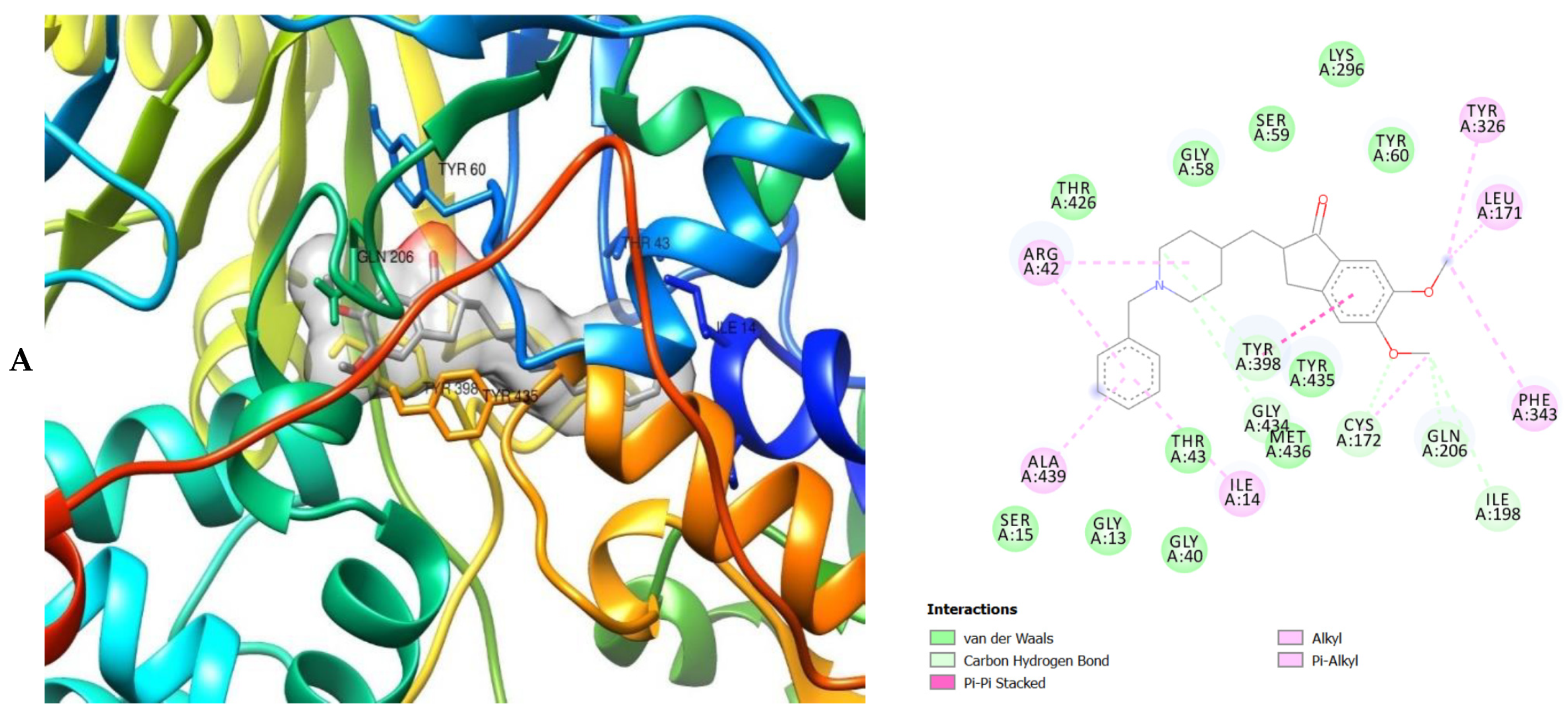
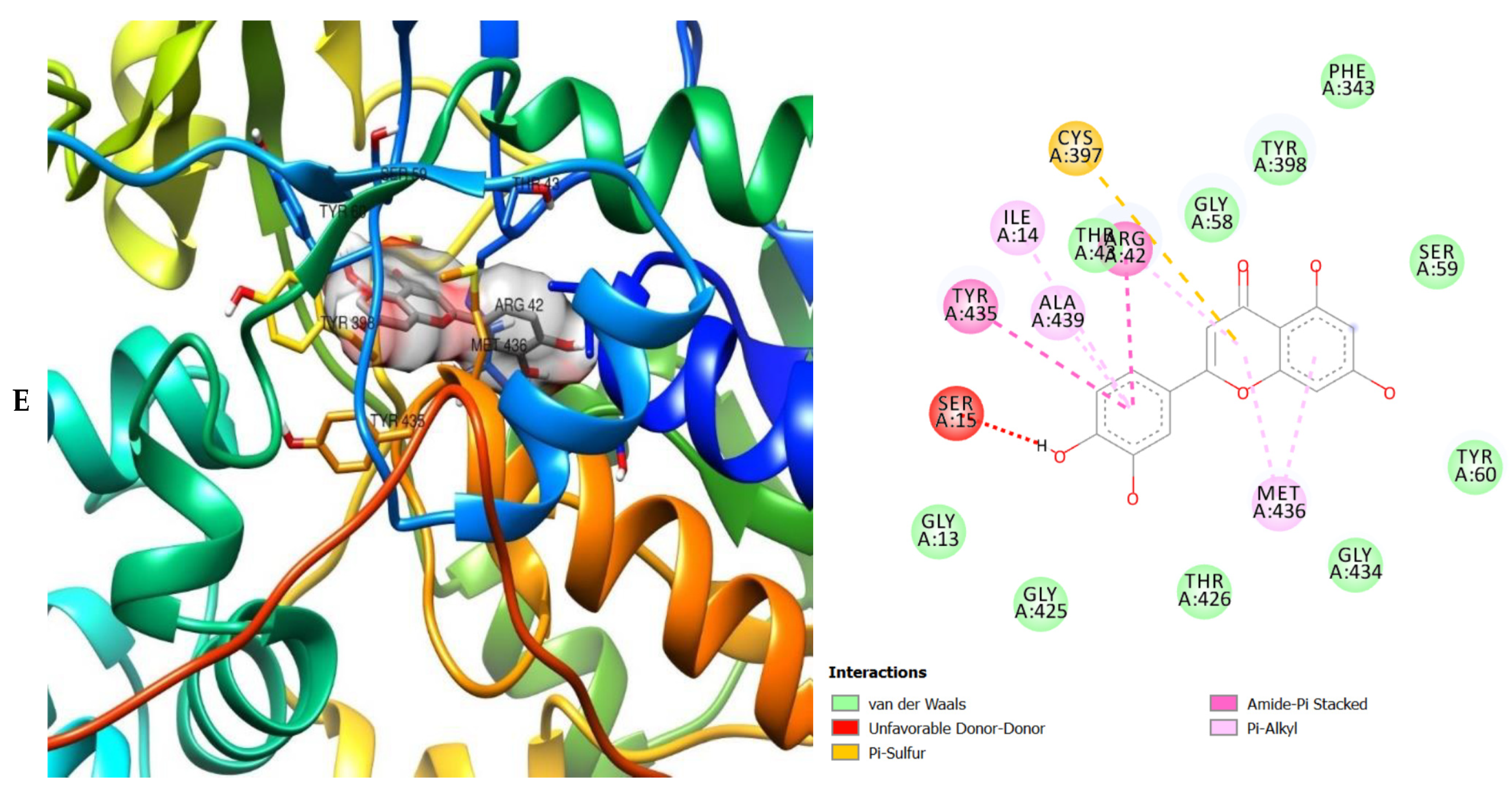
2. Results
3. Discussion
4. Materials and Methods
4.1. Proteins and Ligands Collection
4.2. Generation of Ligand-Based Pharmacophore Model
4.3. Machine Learning Development of Automated QSAR Model
Dataset Generation and Preparation
4.4. Virtual Screening
4.5. ADMET Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Akobundu, C.; Agyakwa, C.W. A Handbook of West African Weeds, Intec Printers, Ibadan; International Institute of Tropical Agriculture: Ibadan, Nigeria, 1998. [Google Scholar]
- Goffin, E.; Ziemons, E.; De Mol, P.; De Madureira, M.D.C.; Martins, A.P.; Da Cunha, A.P.; Philippe, G.; Tits, M.; Angenot, L.; Frédérich, M. In Vitro Antiplasmodial Activity of Tithonia diversifolia and Identification of its Main Active Constituent: Tagitinin C. Planta Medica 2002, 68, 543–545. [Google Scholar] [CrossRef]
- Ojo, O.A.; Ojo, A.B.; Ajiboye, B.O.; Olaiya, O.; Okesola, M.A.; Boligon, A.A.; De Campos, M.M.A.; Oyinloye, B.E.; Kappo, A.P. HPLC-DAD fingerprinting analysis, antioxidant activities of Tithonia diversifolia (Hemsl.) A. Gray leaves and its inhibition of key enzymes linked to Alzheimer’s disease. Toxicol. Rep. 2018, 5, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.A.; Ojo, A.B. Inhibitory activity of Tithonia diversifolia (Hemsl.) A. Gray leaves on some pro-oxidant induced oxidative stress in rat brain. Toxicol. Int. 2016, 23, 254–259. [Google Scholar] [CrossRef]
- Ojo, O.A.; Ajiboye, B.O.; Ojo, A.B.; Oyinloye, B.E.; Imiere, O.D.; Adeyonu, O. Ameliorative potential of Blighia sapida K.D. Koenig bark against pancreatic β-cell dysfunction in alloxan-induced diabetic rats. J. Complement. Integr. Med. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Saidu, A.N.; Mann, A.; Ndako, M. Phytochemical studies and effect of the aqueous extract of Blighia sapida stem bark on the liver enzymes of albino rats. Inter. Res. J. Biochem. Bioinform. 2013, 3, 104–108. [Google Scholar]
- Hamzah, R.; Egwim, E.; Kabiru, A.; Muazu, M. Phytochemical and in vitro antioxidant properties of the methanolic extract of fruits of Blighia sapida, Vitellaria paradoxa and Vitex doniana. Oxid. Antioxidants Med. Sci. 2012, 2, 215–221. [Google Scholar] [CrossRef]
- Elufioye, T. Ethnomedicinal study and screening of plants used for memory enhancement and Antiaging in Sagamu, Nigeria. Eur. J. Med. Plants 2012, 2, 262–275. [Google Scholar] [CrossRef]
- Udobi, C.E.; Ubulom, P.M.; Akpabio, E.I.; Eshiet, U. Antimicrobial Activities of Leaf and Stem Bark Extracts of Blighia. sapida. J. Plant Stud. 2013, 2, 47–52. [Google Scholar]
- Ojo, O.A.; Ajiboye, B.O.; Ojo, A.B.; Olayide, I.I.; Akinyemi, A.J.; Fadaka, A.O.; Adedeji, E.A.; Boligon, A.A.; de Campos, M.M. HPLC-DAD fingerprinting analysis, antioxidant activity of phenolic extracts from Blighia sapida bark and its inhibition of cholinergic enzymes linked to Alzheimer’s disease. Jordan J. Biol. Sci. 2017, 10, 257–264. [Google Scholar]
- Ojo, O.A.; Ajiboye, B.O.; Imiere, O.D.; Adeyonu, O.; Olayide, I.I.; Fadaka, O.A. Antioxidative Properties of Blighia sapida K.D. Koenig Stem Bark Extract and Inhibitory Effects on Carbohydrate Hydrolyzing Enzymes Associated with Non-Insulin Dependent Diabetes Mellitus. Pharmacogn. J. 2018, 10, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Dienagha, A.R.; Miebi, T.O. Energy requirements for cracking dika (Ogbono) nuts (Irvingia gabonensis). Eur. J. Sci Res. 2011, 59, 208–215. [Google Scholar]
- Ojo, O.A.; Oyinloye, B.E.; Ajiboye, B.O.; Onikanni, S.A. Neuroprotective Mechanism of Ethanolic Extract of Irvingia gabonensis Stem Bark against Cadmium-induced Neurotoxicity in Rats. Br. J. Med. Med Res. 2014, 4, 5793–5805. [Google Scholar] [CrossRef]
- Ojo, O.A.; Ajiboye, B.O.; Oyinloye, B.E.; Ojo, A.B. Prophylactic Effects of Ethanolic Extract of Irvingia gabonensis Stem Bark against Cadmium-Induced Toxicity in Albino Rats. Adv. Pharm. 2014, 2014, 1–8. [Google Scholar]
- Ojo, O.A.; Ajiboye, B.O.; Oyinloye, B.E.; Ojo, O.A.; Olarewaju, O.I. Protective effect of Irvingia gabonensis stem bark extract on cadmium induced nephrotoxicity in rats. Interdiscip. Toxicol. 2014, 7, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojo, O.A.; Ojo, A.B.; Ajiboye, B.O.; Oyinloye, B.E.; Akinyemi, A.J.; Okesola, M.A.; Boligon, A.A.; de Campos, M.M. Chromatographic fingerprint analysis, antioxidant properties, and inhibition of cholinergic enzymes (acetylcholinesterase and butyrylcholinesterase) of phenolic extracts from Irvingia gabonensis (Aubry-Lecomte ex O’Rorke) Baill bark. J. Basic Clin. Physiol. Pharmacol. 2018, 29, 217–224. [Google Scholar] [CrossRef]
- Mucke, L. Neuroscience: Alzheimer’s disease. Nature 2009, 461, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Fargo, K.; Bleiler, L. Alzheimer’s association report: 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014, 10, 47–92. [Google Scholar]
- Ojo, O.A.; Afon, A.A.; Ojo, A.B.; Ajiboye, B.O.; Okesola, M.A.; Aruleba, R.T.; Adekiya, T.A.; Oyinloye, B.E. Spondias mombim L. (Anacardiaceae): Chemical fingerprints, inhibitory activities and molecular docking on key enzymes relevant to erectile dysfunction and Alzheimer’s diseases. J. Food Biochem. 2019, 43, e12772. [Google Scholar] [CrossRef]
- Konrath, E.L.; Passos, C.S.; Klein-Junior, L.C.; Henriques, A.T. Alkaloids as a source of potential anticholinesterase inhibitors for the treatment of Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1701–1725. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.A.; Ojo, A.B.; Ajiboye, B.O.; Olaiya, O.; Akawa, A.; Olaoye, O.; Anifowose, O.O.; Idowu, O.; Olasehinde, O.; Obafemi, T.; et al. Inhibitory effect of Bryophyllum pinnatum (Lam.) Oken leaf extract and their fractions on α-amylase, α-glucosidase and cholinesterase enzyme. Pharmacogn. J. 2018, 10, 497–506. [Google Scholar]
- Jacob, S.N.; Nienborg, H. Monoaminergic Neuromodulation of Sensory Processing. Front. Neural Circuits 2018, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.J.; Lamperski, C.S.; Waterhouse, B.D. Identification and distribution of projections from monoaminergic and cholinergic nuclei to functionally differentiated subregions of prefrontal cortex. Brain Res. 2013, 1522, 38–58. [Google Scholar] [CrossRef] [Green Version]
- Toda, N.; Kaneko, T.; Kogen, H. Development of an efficient therapeutic agent for Alzheimer’s disease: Design and synthesis of dual inhibitors of acetylcholinesterase and serotonin transporter. Chem. Pharm. Bull. 2010, 58, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohanka, M. Inhibitors of acetylcholinesterase and butyrylcholinesterase meet immunity. Int. J. Mol. Sci. 2014, 15, 9809–9825. [Google Scholar] [CrossRef] [Green Version]
- Yılmaz, S.; Akbaba, Y.; Ozgeris, B. Synthesis and inhibitory properties of some carbamates on carbonic anhydrase and acetylcholin esterase. J. Enzyme Inhib. Med. Chem. 2016, 31, 1484–1491. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “rm2” metrics for judging quality of quantitative structure–Activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef]
- Puzyn, T.; Leszczynski, J.; Cronin, M.T. (Eds.) Recent Advances in QSAR Studies: Methods and Applications; Springer Science & Business Media: Berlin, Germany, 2010. [Google Scholar]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic, P.T.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, T.; Islam, M.A.; Pal, R.; Saha, A. Exploring structural requirement and binding interactions of β-amyloid cleavage enzyme inhibitors using molecular modeling techniques. Med. Chem. Res. 2013, 22, 4766–4774. [Google Scholar] [CrossRef]
- Jain, P.; Jadhav, H.R. Quantitative structure activity relationship analysis of aminoimidazoles as BACE-I inhibitors. Med. Chem. Res. 2013, 22, 1740–1746. [Google Scholar] [CrossRef]
- Ambure, P.; Roy, K. Understanding the structural requirements of cyclic sulfone hydroxyethylamines as hBACE1 inhibitors against Aβ plaques in Alzheimer’s disease: A predictive QSAR approach. RSC Adv. 2016, 6, 28171–28186. [Google Scholar] [CrossRef]
- Shen, L.L.; Liu, G.X.; Tang, Y. Molecular docking and 3DQSAR studies of 2-substituted 1-indanone derivatives as acetylcholinesterase inhibitors. Acta Pharmacol. Sin. 2007, 28, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Weng, X.; Ning, F.X.; Ou, J.B.; Hou, J.Q.; Luo, H.B.; Li, D.; Huang, Z.S.; Huang, S.L.; Gu, L.Q. 3D-QSAR studies of azaoxoisoaporphine, oxoaporphine, and oxoisoaporphine derivatives as antiAChE and anti-AD agents by the CoMFA method. J. Mol. Graph. Model. 2013, 41, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Akula, N.; Lecanu, L.; Greeson, J.; Papadopoulos, V. 3D QSAR studies of AChE inhibitors based on molecular docking scores and CoMFA. Bioorganic Med. Chem. Lett. 2006, 16, 6277–6280. [Google Scholar] [CrossRef]
- Gharaghani, S.; Khayamian, T.; Ebrahimi, M. Molecular dynamics simulation study and molecular docking descriptors in structure-based QSAR on acetylcholinesterase (AChE) inhibitors. SAR QSAR Environ. Res. 2013, 24, 773–794. [Google Scholar] [CrossRef]
- Islam, M.R.; Zaman, A.; Jahan, I.; Chakravorty, R.; Chakrabo, S. In silico QSAR analysis of quercetin reveals its potential as therapeutic drug for Alzheimer’s disease. J. Young Pharma. 2013, 5, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatkar, A.; Nanda, A.; Kumar, P.; Narasimhan, B. Synthesis, antimicrobial evaluation and QSAR studies of p-coumaric acid derivatives. Arab. J. Chem. 2017, 10, S3804–S3815. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodabadi, N.; Ajloo, D. QSAR, docking and Molecular dynamic studies on the polyphenolic as inhibitors of β-amyloid aggregation. Med. Chem. Res. 2016, 25, 2104–2118. [Google Scholar] [CrossRef]
- Malik, N.; Khatkar, A. 3D-QSAR and in-silico Studies of Natural Products and Related Derivatives as Monoamine Oxidase Inhibitors. Curr. Neuropharmacol. 2018, 16, 881–900. [Google Scholar]
- Das, S.; Laskar, M.A.; Sarker, S.D.; Choudhury, M.D.; Choudhury, P.R.; Mitra, A.; Jamil, S.; Lathiff, S.M.A.; Abdullah, S.A.; Basar, N.; et al. Prediction of Anti-Alzheimer’s Activity of Flavonoids Targeting Acetylcholinesterase in silico. Phytochem. Anal. 2017, 28, 324–331. [Google Scholar] [CrossRef]
- Mukesh, S.; Dharmendra, A. QSAR Studies of Flavonoids Derivatives for Antioxidant and Antimicrobial Activity. J. Drug Deliv. Ther. 2019, 9, 765–773. [Google Scholar]
- Kondeva-Burdina, M.; Doytchinova, I.; Krasteva, I.; Ionkova, I.; Manov, V. Hepato-, neuroprotective effects and QSAR studies on flavoalkaloids and flavonoids from Astragalus monspessulanus. Biotechnol. Biotechnol. Equip. 2019, 33, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Basu, S. Multi-functional activities of citrus flavonoid narirutin in Alzheimer’s disease therapeutics: An integrated screening approach and in vitro validation. Int. J. Biol. Macromol. 2017, 103, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, R.; Hadizadeh, F.; Abdizadeh, T. Molecular modeling studies of Anti-Alzheimer Agents by QSAR, Molecular Docking and Molecular Dynamics Simulations Techniques. Med. Chem. 2020, 16, 903–927. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Loving, K.; Salam, N.K.; Sherman, W. Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef]
- Ojo, O.A. Puerarin as a potential drug candidate for the treatment of Diabetes mellitus: Molecular docking and Pharmacophore Modelling studies. Biointerface Res. Appl. Chem. 2021, 11, 8751–8759. [Google Scholar]
- Palakurti, R.; Sriram, D.; Yogeeswari, P.; Vadrevu, R. Multiple e-Pharmacophore Modeling Combined with High-Throughput Virtual Screening and Docking to Identify Potential Inhibitors of β-Secretase(BACE1). Mol. Inform. 2013, 32, 385–398. [Google Scholar] [CrossRef]
- Tripathia, A.C.; Sonara, P.K.; Rathoreb, R.; Sarafa, S.K. Structural Insights into the Molecular Design of HER2 Inhibitors. Open Pharm. Sci. J. 2016, 3, 164–181. [Google Scholar] [CrossRef] [Green Version]
- Arthur, D.E.; Ejeh, S.; Uzairu, A. Quantitative structure-activity relationship (QSAR) and design of novel ligands that demonstrate high potency and target selectivity as protein tyrosine phosphatase 1B (PTP 1B) inhibitors as an effective strategy used to model anti-diabetic agents. J. Recept. Signal Transduct. 2020, 40, 501–520. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Sheridan, R.P.; Singh, S.B.; Fluder, E.M.; Kearsley, S.K. Protocols for bridging the peptide to nonpeptide gap in topological similarity searches. J. Chem. Inf. Comp. Sci. 2001, 41, 1395–1406. [Google Scholar] [CrossRef]
- Kumar, V.; Ojha, P.K.; Saha, A.; Roy, K. Exploring 2D-QSAR for prediction of beta-secretase 1 (BACE1) inhibitory activity against Alzheimer’s disease. SAR QSAR Environ. Res. 2020, 31, 87–133. [Google Scholar] [CrossRef] [PubMed]
- Yasri, A.; Hartsough, D.J. Toward an optimal procedure for variable selection and QSAR modeling building. Chem. Inf. Comput. Sci. 2001, 41, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Mol. Des. 2002, 16, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Veerasamy, R.; Rajak, H.; Jain, A.; Sivadasan, S.; Varghese, C.; Agrawal, R. Validation of QSAR models-strategies and importance. Int. J. Drug Des. Discov. 2011, 2, 511–519. [Google Scholar]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminformatics 2009, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ojo, O.A.; Aruleba, R.T.; Adekiya, T.A.; Sibuyi, N.R.S.; Ojo, A.B.; Ajiboye, B.O.; Oyinloye, B.E.; Adeola, H.A.; Fadaka, A.O. Deciphering the interaction of puerarin with cancer macromolecules: An in silico investigation. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Priyadarshini, V.; Pradhan, D.; Munikumar, M.; Swargam, S.; Umamaheswari, A.; Rajasekhar, D. Genome-based approaches to develop epitope-driven subunit vaccines against pathogens of infective endocarditis. J. Biomol. Struct. Dyn. 2014, 32, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Glue, P.; Clement, R.P. Cytochrome P450 Enzymes and Drug Metabolism—Basic Concepts and Methods of Assessment. Cell. Mol. Neurobiol. 1999, 19, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Dixit, B. A review on the effects of CMPF binding with Human Serum Albumin. Bioinform. Rev. 2017, 3, 9–18. [Google Scholar]
- Radchenko, E.V.; Dyabina, A.S.; Palyulin, V.A.; Zefirov, N.S. Prediction of human intestinal absorption of drug compounds. Russ. Chem. Bull. 2016, 65, 576–580. [Google Scholar] [CrossRef]
- Wessel, M.D.; Jurs, P.C.; Tolan, J.W.; Muskal, S.M. Prediction of human intestinal absorption of drug compounds from molecular structure. J. Chem. Inf. Comput. Sci. 1998, 38, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Basant, N.; Gupta, S.; Singh, K.P. Predicting human intestinal absorption of diverse chemicals using ensemble learning based QSAR modeling approaches. Comput. Biol. Chem. 2016, 61, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Swierczewska, M.; Lee, K.C.; Lee, S. What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs 2015, 20, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Chow, H.S.; Garland, L.L.; Hsu, C.H.; Vining, D.R.; Chew, W.M.; Miller, J.A.; Perloff, M.; Crowell, J.A.; Alberts, D.S. Resveratrol modulates drug-and carcinogen metabolizing enzymes in a healthy volunteer study. Cancer Prev. Res. 2010, 3, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.F.; Bari, M.A.; Islam, M.K.; Islam, M.S.; Kayser, M.S.; Nahar, N.; Al-Faruk, M.; Rashid, M.A. The natural anti-tubercular agents: In silico study of physicochemical, pharmacokinetic and toxicological properties. J. App. Pharm. Sci. 2017, 7, 34–38. [Google Scholar]
- Sahin, S.; Benet, L.Z. The operational multiple dosing half-life: A key to defining drug accumulation in patients and to designing extended release dosage forms. Pharm. Res. 2008, 25, 2869–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Dixon, S.L. In silico models for the prediction of dose-dependent human hepatotoxicity. J. Comput. Aid Mol. Des. 2003, 17, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Saddala, M.S.; Obaiah, J.; Rani, A.U. Identification of potent VEGFR-2 Inhibitors of Angiogenesis through homology modeling, structure based virtual screening, docking and molecular dynamics simulations. Int. J. Sci. Eng. Res. 2015, 6, 1382–1390. [Google Scholar]
- Radwan, A.A.; Alanazi, F.K. In silico studies on novel inhibitors of MERS-CoV: Structure-based pharmacophore modeling, database screening and molecular docking. Trop. J. Pharm. Res. 2018, 17, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release. 2020-1: LigPrep 2020 Schrödinger; LLC: New York, NY, USA, 2020. [Google Scholar]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. Data warrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2005, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release. 2020-1: PHASE 2020 Schrödinger; LLC: New York, NY, USA, 2020. [Google Scholar]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints; insights into database screening performance using eight fingerprint method. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef] [PubMed]
- de Oliveria, M.T.; Katekawa, E. On the virtues of automated QSAR—the new kid on the block. Future Med. Chem. 2018, 10, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Sepay, N.; Al Hoque, A.; Mondal, R.; Halder, U.; Muddassir, M. In silico fight against novel coronavirus by finding chromone derivatives as inhibitor of coronavirus main proteases enzyme. Struct. Chem. 2020, 31, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
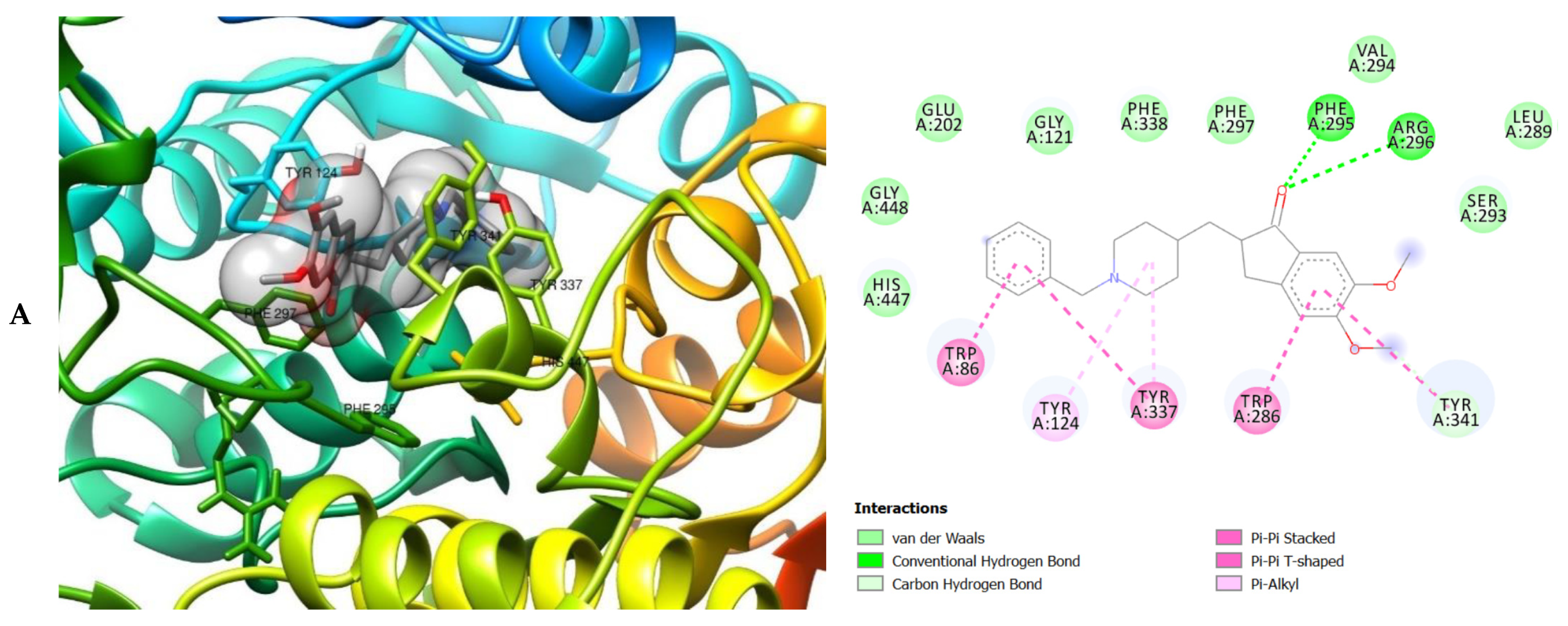
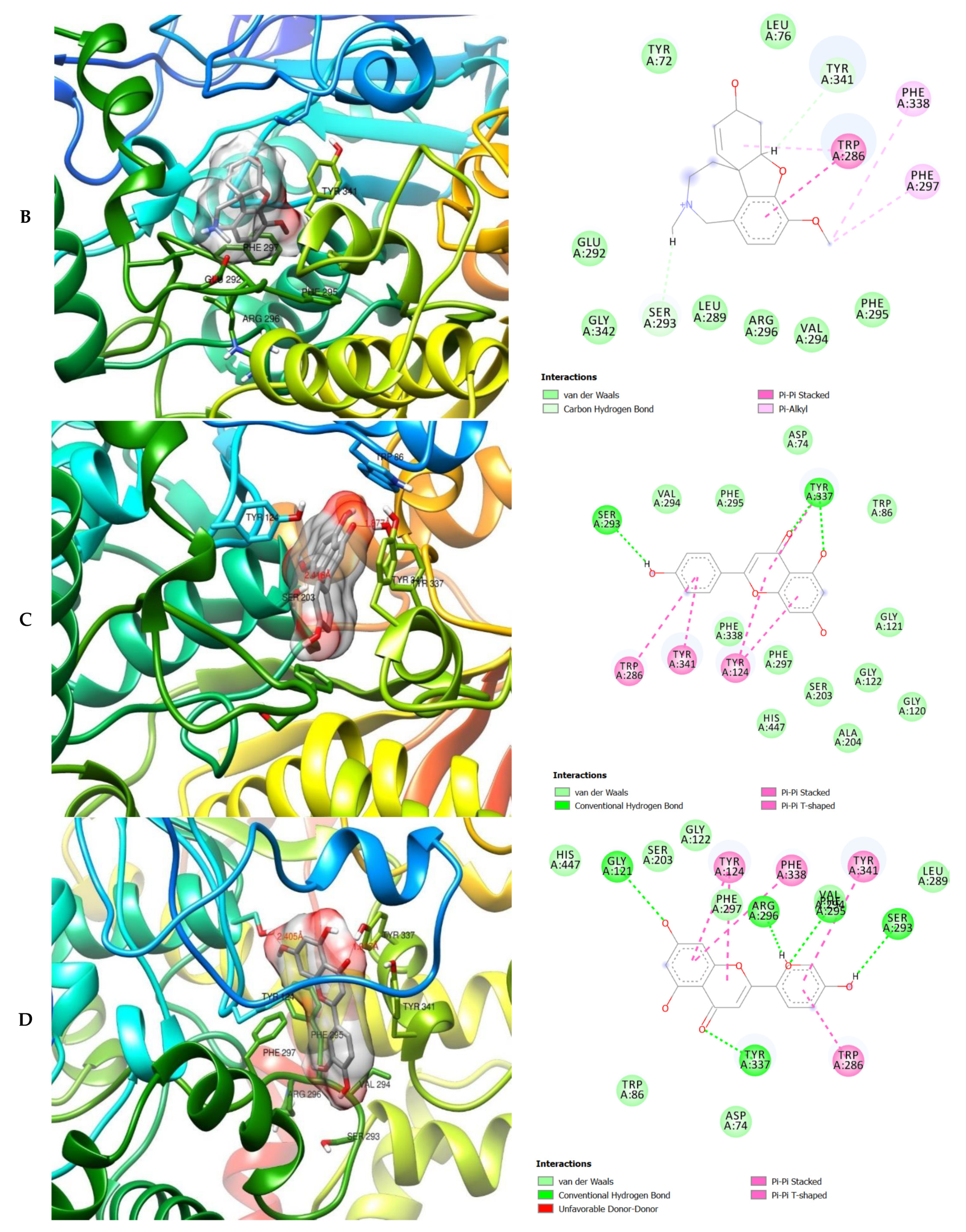
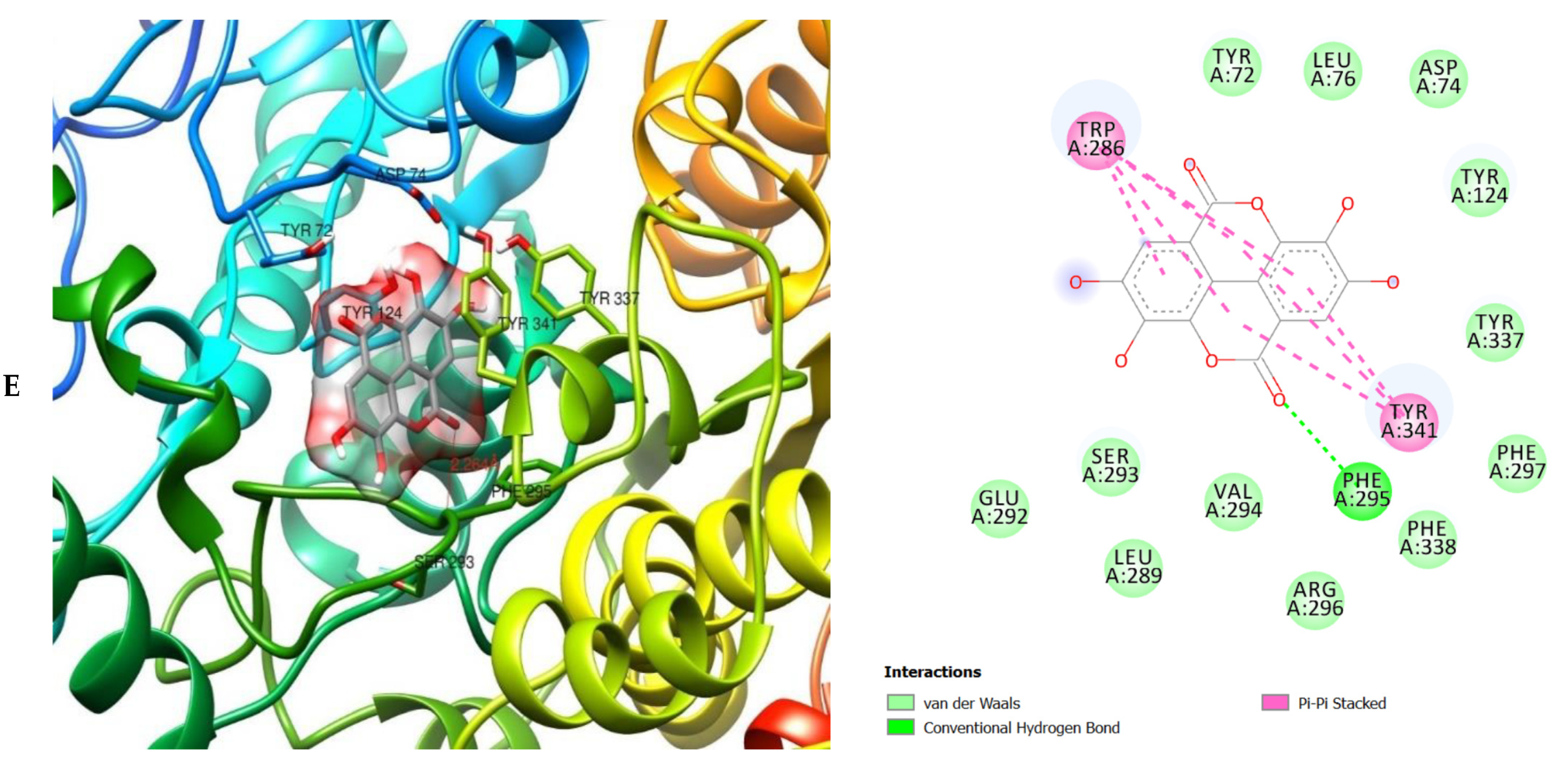
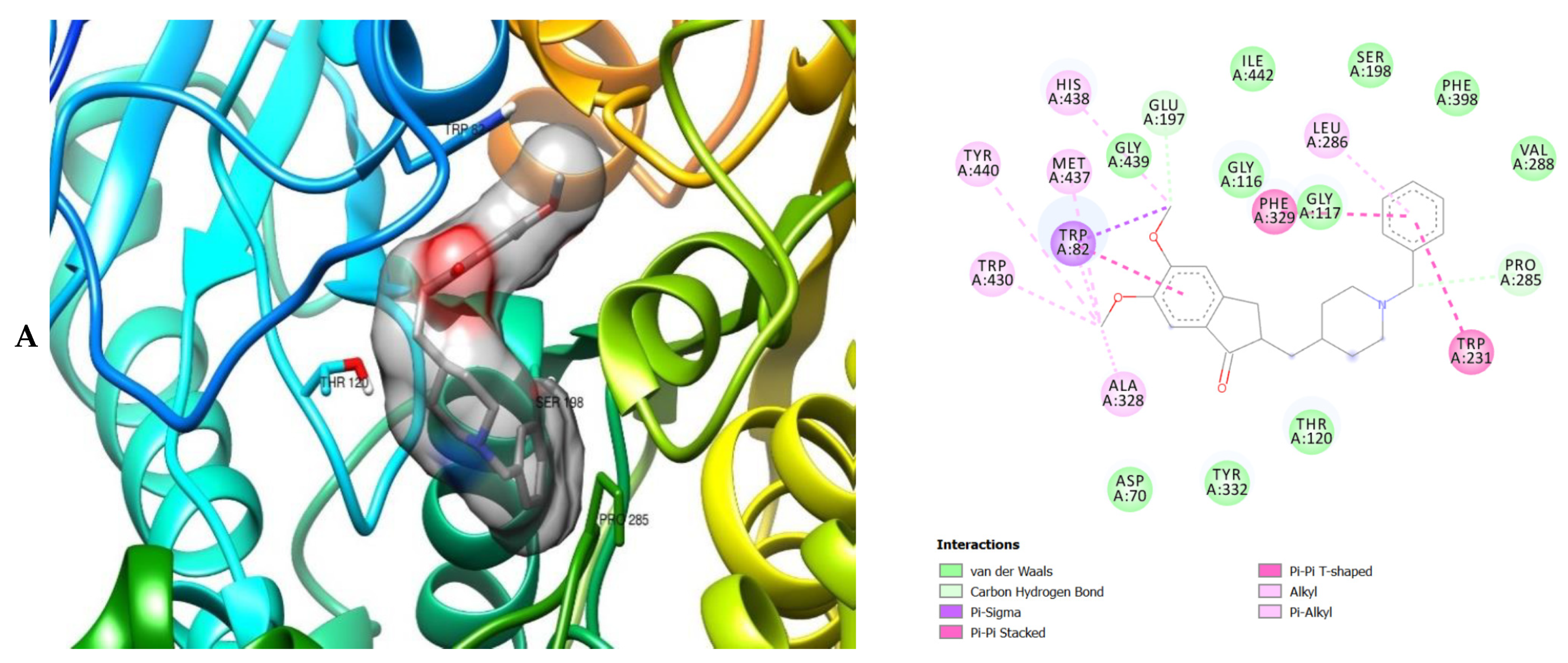




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Phase Hypo Score | EF1% | BEDROC160.9 | ROC | AUAC | Average Outranking Decoys | Total Actives | Ranked Actives | Matches | Excluded Volumes |
|---|---|---|---|---|---|---|---|---|---|---|
| ADRR_1 | 0.83 | 36.76 | 0.58 | 0.45 | 0.69 | 3.2 | 11 | 5 | 4 of 4 | No |
| AARR_1 | 0.82 | 36.76 | 0.58 | 0.45 | 0.66 | 3.4 | 11 | 5 | 4 of 4 | No |
| AADRR_3 | 0.81 | 36.76 | 0.58 | 0.45 | 0.7 | 3 | 11 | 5 | 5 of 5 | No |
| AARR_2 | 0.81 | 36.76 | 0.57 | 0.45 | 0.64 | 5.6 | 11 | 5 | 4 of 4 | No |
| AADRRR_1 | 0.8 | 36.76 | 0.57 | 0.36 | 0.67 | 0 | 11 | 4 | 6 of 6 | No |
| ADDRRR_1 | 0.8 | 36.76 | 0.57 | 0.36 | 0.68 | 0 | 11 | 4 | 6 of 6 | No |
| AAARRR_1 | 0.8 | 36.76 | 0.57 | 0.36 | 0.67 | 0 | 11 | 4 | 6 of 6 | No |
| AADRRR_2 | 0.79 | 36.76 | 0.57 | 0.36 | 0.68 | 0 | 11 | 4 | 6 of 6 | No |
| AAADRR_1 | 0.79 | 36.76 | 0.57 | 0.36 | 0.66 | 0 | 11 | 4 | 6 of 6 | No |
| ADRRR_1 | 0.78 | 36.76 | 0.57 | 0.36 | 0.66 | 0 | 11 | 4 | 5 of 5 | No |
| AAADRR_2 | 0.78 | 36.76 | 0.57 | 0.36 | 0.66 | 0 | 11 | 4 | 6 of 6 | No |
| AADDRR_1 | 0.78 | 36.76 | 0.57 | 0.36 | 0.67 | 0 | 11 | 4 | 6 of 6 | No |
| AADRR_1 | 0.77 | 36.76 | 0.57 | 0.36 | 0.63 | 0 | 11 | 4 | 5 of 5 | No |
| AADRR_2 | 0.77 | 36.76 | 0.57 | 0.36 | 0.64 | 0 | 11 | 4 | 5 of 5 | No |
| ADRRR_2 | 0.77 | 36.76 | 0.57 | 0.36 | 0.64 | 0 | 11 | 4 | 5 of 5 | No |
| AARRR_1 | 0.77 | 36.76 | 0.57 | 0.36 | 0.64 | 0 | 11 | 4 | 5 of 5 | No |
| DDRRR_1 | 0.77 | 36.76 | 0.57 | 0.36 | 0.67 | 0 | 11 | 4 | 5 of 5 | No |
| AARRR_2 | 0.77 | 36.76 | 0.57 | 0.36 | 0.63 | 0 | 11 | 4 | 5 of 5 | No |
| AADR_1 | 0.77 | 36.76 | 0.57 | 0.45 | 0.66 | 15.6 | 11 | 5 | 4 of 4 | No |
| ADRRR_3 | 0.77 | 36.76 | 0.57 | 0.36 | 0.66 | 0 | 11 | 4 | 5 of 5 | No |
| AARRR_3 | 0.77 | 36.76 | 0.57 | 0.36 | 0.64 | 0 | 11 | 4 | 5 of 5 | No |
| ADRR_2 | 0.76 | 36.76 | 0.57 | 0.36 | 0.61 | 0 | 11 | 4 | 4 of 4 | No |
| DRRR_1 | 0.76 | 36.76 | 0.57 | 0.36 | 0.61 | 0 | 11 | 4 | 4 of 4 | No |
| ARRR_1 | 0.75 | 36.76 | 0.57 | 0.36 | 0.59 | 0 | 11 | 4 | 4 of 4 | No |
| ADRR_3 | 0.75 | 36.76 | 0.57 | 0.36 | 0.55 | 0 | 11 | 4 | 4 of 4 | No |
| ARRR_2 | 0.75 | 36.76 | 0.57 | 0.36 | 0.59 | 0 | 11 | 4 | 4 of 4 | No |
| ARRR_3 | 0.75 | 36.76 | 0.57 | 0.36 | 0.59 | 0 | 11 | 4 | 4 of 4 | No |
| Compounds | 6u3p_AChE | 3o9m_BChE | 2bk5_MAO |
|---|---|---|---|
| Apigenin | −10.2 | −9.4 | −9.2 |
| Caffeic Acid | −7.2 | −6.7 | −7.8 |
| Chlorogenic acid | −9.6 | −8.6 | −9.9 |
| (R)-Donepezil | −10.7 | −9.7 | −10.8 |
| Ellagic acid | −9.8 | −9.9 | −8.9 |
| Galantamine | −7.5 | −8.6 | −6.1 |
| Gallic acid | −6.5 | −6.1 | −6.3 |
| Kaempferol | −9.6 | −9.4 | −8.4 |
| Luteolin | −10.4 | −9.7 | −9.3 |
| p-Coumaric acid | −7.1 | −6.6 | −7 |
| Quercetin | −9.4 | −9.6 | −8.8 |
| Compounds | iLOGP | XLOGP3 | WLOGP | MLOGP | Silicos-IT Log P | Consensus Log P |
|---|---|---|---|---|---|---|
| Apigenin | 1.89 | 3.02 | 2.58 | 0.52 | 2.52 | 2.11 |
| Caffeic Acid | 0.97 | 1.15 | 1.09 | 0.7 | 0.75 | 0.93 |
| Chlorogenic acid | 0.87 | −0.42 | −0.75 | −1.05 | −0.61 | −0.39 |
| (R)-Donepezil | 3.92 | 4.28 | 3.83 | 3.06 | 4.91 | 4 |
| Ellagic acid | 0.79 | 1.1 | 1.31 | 0.14 | 1.67 | 1 |
| Galanthamine | 2.67 | 1.84 | 1.32 | 1.74 | 2.03 | 1.92 |
| Gallic acid | 0.21 | 0.7 | 0.5 | −0.16 | −0.2 | 0.21 |
| Kaempferol | 1.7 | 1.9 | 2.28 | −0.03 | 2.03 | 1.58 |
| Luteolin | 1.86 | 2.53 | 2.28 | −0.03 | 2.03 | 1.73 |
| p-Coumaric acid | 0.95 | 1.46 | 1.38 | 1.28 | 1.22 | 1.26 |
| Quercetin | 1.63 | 1.54 | 1.99 | −0.56 | 1.54 | 1.23 |
| Compounds | ESOL Log S | ESOL Solubility (mg/mL) | ESOL Class | Ali Log S | Ali Solubility (mg/mL) | Ali Class | Silicos-IT LogSw | Silicos-IT Solubility (mg/mL) | Silicos-IT Class | Bio-Availability Score |
|---|---|---|---|---|---|---|---|---|---|---|
| Apigenin | −3.94 | 3.07 × 10−2 | Soluble | −4.59 | 6.88 × 10−3 | Moderately soluble | −4.4 | 1.07 × 10−2 | Moderately soluble | 0.55 |
| Caffeic Acid | −1.89 | 2.32 × 100 | Very soluble | −2.38 | 7.55 × 10−1 | Soluble | −0.71 | 3.51 × 101 | Soluble | 0.55 |
| Chlorogenic acid | −1.62 | 8.50 × 100 | Very soluble | −2.58 | 9.42 × 10−1 | Soluble | 0.4 | 8.94 × 102 | Soluble | 0.55 |
| (R)- Donepezil | −1.481 | 5.87 × 10−3 | Soluble | −4.81 | 5.92 × 10−3 | Moderately soluble | −6.9 | 4.78 × 10−5 | Poorly soluble | 0.56 |
| Ellagic acid | −2.94 | 3.43 × 10−1 | Soluble | −3.66 | 6.60 × 10−2 | Soluble | −3.35 | 1.36 × 10−1 | soluble | 0.55 |
| Galanthamine | −2.93 | 3.41 × 10−1 | Soluble | −2.34 | 1.31 × 100 | Soluble | −2.96 | 3.17 × 10−1 | soluble | 0.55 |
| Gallic acid | −1.64 | 3.90 × 100 | Very soluble | −2.34 | 7.86 × 10−1 | Soluble | −0.04 | 1.55 × 102 | Soluble | 0.56 |
| Kaempferol | −3.31 | 1.40 × 10−1 | Soluble | −3.86 | 3.98 × 10−2 | soluble | −3.82 | 4.29 × 10−2 | Soluble | 0.55 |
| Luteolin | −3.71 | 5.63 × 10−2 | Soluble | −4.51 | 8.84 × 10−3 | Moderately soluble | −3.82 | 4.29 × 10−2 | Soluble | 0.55 |
| p-Coumaric acid | −2.02 | 1.58 × 100 | Soluble | −2.27 | 8.73 × 10−1 | Soluble | −1.28 | 8.67 × 100 | Soluble | 0.56 |
| Quercetin | −3.16 | 2.11 × 10−1 | Soluble | −3.91 | 3.74 × 10−2 | Soluble | −3.24 | 1.73 × 10−1 | soluble | 0.55 |
| Apigenin | Caffeic Acid | Chlorogenic Acid | (R)-Donepezil | Ellagic Acid | Galanthamine | Gallic Acid | Kaempferol | Luteolin | p-Coumaric Acid | Quercetin | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GI absorption | High | High | Low | High | High | High | High | High | High | High | High |
| BBB permeant | No | No | No | Yes | Yes | No | No | No | No | Yes | No |
| Pgp substrate | No | No | No | Yes | Yes | No | No | Yes | Yes | No | Yes |
| CYP1A2 inhibitor | Yes | No | No | No | Yes | No | No | Yes | Yes | No | Yes |
| CYP2C19 inhibitor | No | No | No | No | No | No | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No | No | No | No | No | No | No |
| CYP2D6 inhibitor | Yes | No | No | Yes | No | Yes | No | Yes | Yes | No | Yes |
| CYP3A4 | Yes | No | No | Yes | No | No | Yes | Yes | Yes | No | Yes |
| Skin permeability logKp (cm/s) | −5.8 | −6.58 | −8.76 | −5.58 | −7.36 | −6.75 | −6.84 | −6.7 | −6.25 | −6.26 | −7.05 |
| Apigenin | Caffeic Acid | Chlorogenic Acid | (R)- Donepezil | Ellagic Acid | Galanthamine | Gallic Acid | Kaempferol | Luteolin | p-Coumaric Acid | Quercetin | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | 270.24 | 180.16 | 354.31 | 379.49 | 302.19 | 287.35 | 170.12 | 286.24 | 286.24 | 164.16 | 302.24 |
| #Heavy atoms | 20 | 13 | 25 | 28 | 22 | 21 | 12 | 21 | 21 | 12 | 22 |
| #Aromatic heavy atoms | 16 | 6 | 6 | 12 | 16 | 6 | 6 | 16 | 16 | 6 | 16 |
| Fraction Csp3 | 0 | 0 | 0.38 | 0.46 | 0 | 0.53 | 0 | 0 | 0 | 0 | 0 |
| #Rotatable bonds | 1 | 2 | 5 | 6 | 0 | 4 | 1 | 4 | 4 | 4 | 2 |
| #H-bond donors | 3 | 3 | 6 | 0 | 4 | 1 | 4 | 4 | 4 | 2 | 5 |
| MR | 73.99 | 47.16 | 83.5 | 115.31 | 75.31 | 84.05 | 39.47 | 76.01 | 76.01 | 45.13 | 78.04 |
| TPSA | 90.9 | 77.76 | 164.75 | 38.77 | 141.34 | 41.93 | 97.99 | 111.13 | 111.13 | 57.53 | 131.36 |
| Lipinski #violations | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ghose #violations | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| Veber #violations | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Egan #violations | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Muegge #violations | 0 | 1 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| PAINS #alerts | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 |
| Brenk #alerts | 0 | 2 | 2 | 0 | 3 | 1 | 1 | 0 | 1 | 1 | 1 |
| Leadlikeness #violations | 0 | 1 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Synthetic Accessibility | 2.96 | 1.81 | 4.16 | 3.62 | 3.17 | 4.57 | 1.22 | 3.14 | 3.02 | 1.61 | 3.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojo, .A.; Ojo, A.B.; Okolie, C.; Nwakama, M.-A.C.; Iyobhebhe, M.; Evbuomwan, I.O.; Nwonuma, C.O.; Maimako, R.F.; Adegboyega, A.E.; Taiwo, O.A.; et al. Deciphering the Interactions of Bioactive Compounds in Selected Traditional Medicinal Plants against Alzheimer’s Diseases via Pharmacophore Modeling, Auto-QSAR, and Molecular Docking Approaches. Molecules 2021, 26, 1996. https://doi.org/10.3390/molecules26071996
Ojo A, Ojo AB, Okolie C, Nwakama M-AC, Iyobhebhe M, Evbuomwan IO, Nwonuma CO, Maimako RF, Adegboyega AE, Taiwo OA, et al. Deciphering the Interactions of Bioactive Compounds in Selected Traditional Medicinal Plants against Alzheimer’s Diseases via Pharmacophore Modeling, Auto-QSAR, and Molecular Docking Approaches. Molecules. 2021; 26(7):1996. https://doi.org/10.3390/molecules26071996
Chicago/Turabian StyleOjo, Oluwafemi Adeleke, Adebola Busola Ojo, Charles Okolie, Mary-Ann Chinyere Nwakama, Matthew Iyobhebhe, Ikponmwosa Owen Evbuomwan, Charles Obiora Nwonuma, Rotdelmwa Filibus Maimako, Abayomi Emmanuel Adegboyega, Odunayo Anthonia Taiwo, and et al. 2021. "Deciphering the Interactions of Bioactive Compounds in Selected Traditional Medicinal Plants against Alzheimer’s Diseases via Pharmacophore Modeling, Auto-QSAR, and Molecular Docking Approaches" Molecules 26, no. 7: 1996. https://doi.org/10.3390/molecules26071996
APA StyleOjo, . A., Ojo, A. B., Okolie, C., Nwakama, M.-A. C., Iyobhebhe, M., Evbuomwan, I. O., Nwonuma, C. O., Maimako, R. F., Adegboyega, A. E., Taiwo, O. A., Alsharif, K. F., & Batiha, G. E.-S. (2021). Deciphering the Interactions of Bioactive Compounds in Selected Traditional Medicinal Plants against Alzheimer’s Diseases via Pharmacophore Modeling, Auto-QSAR, and Molecular Docking Approaches. Molecules, 26(7), 1996. https://doi.org/10.3390/molecules26071996