
RNA Proximity Labeling: A New Detection Tool for RNA–Protein Interactions

Abstract
:1. Introduction
2. RIP and CLIP
3. RNA Affinity Purification
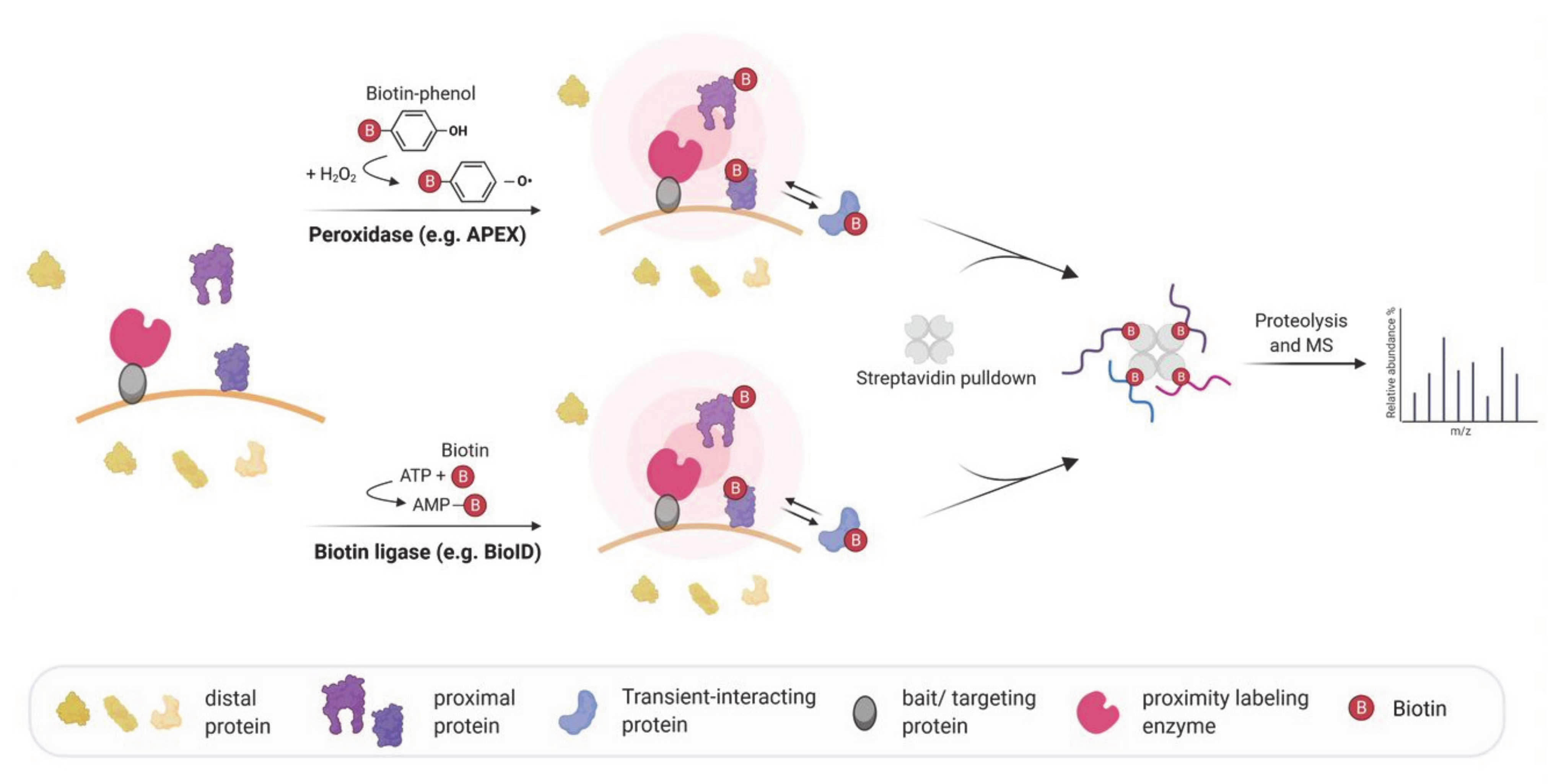
4. Biotin-Based Proximity Labeling Approaches
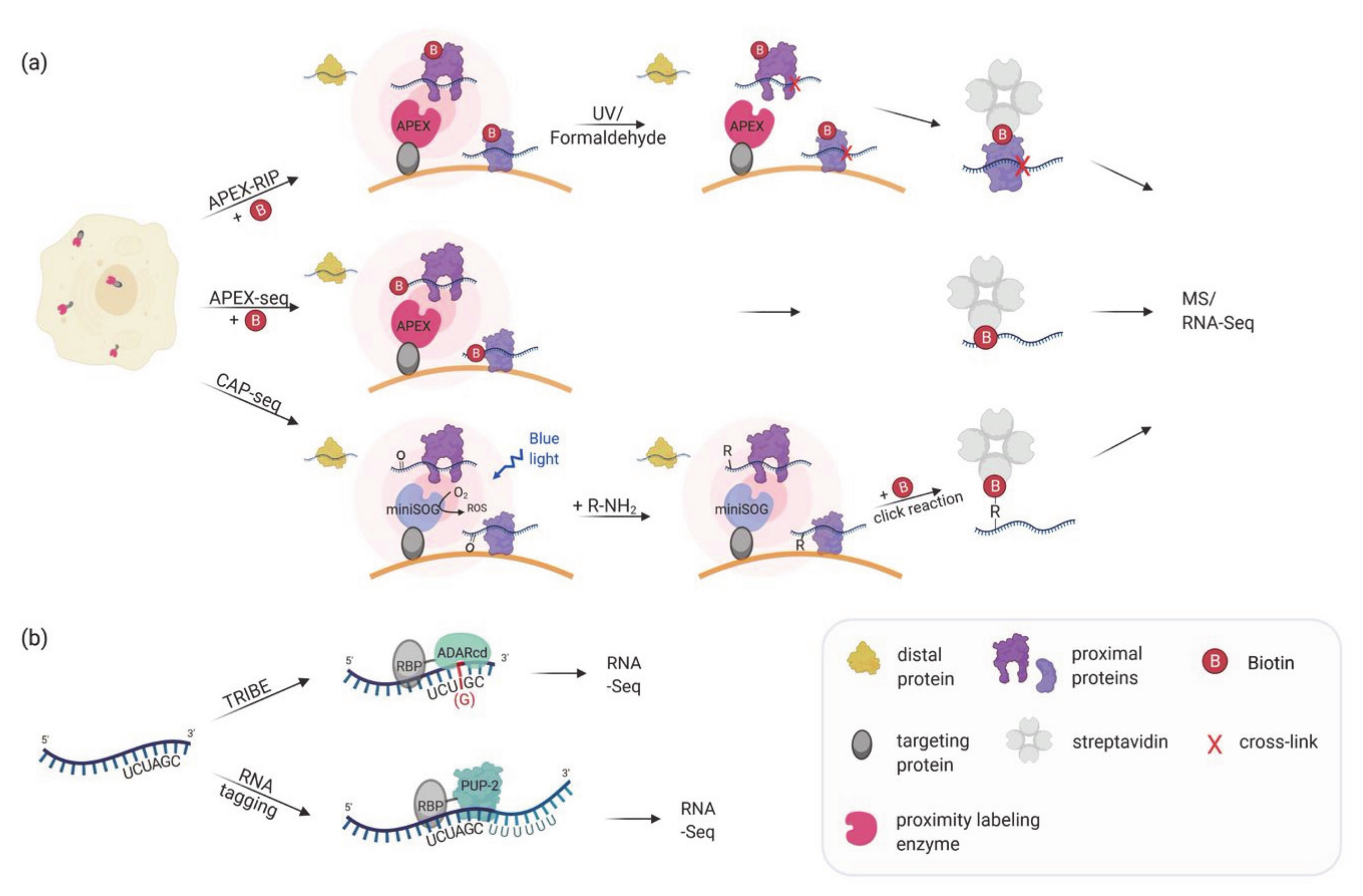
5. Proximity Labeling for Mapping Subcellular Transcriptomes
6. Proximity Labeling of RNA–Protein Interactions: Finding the RNA Partners
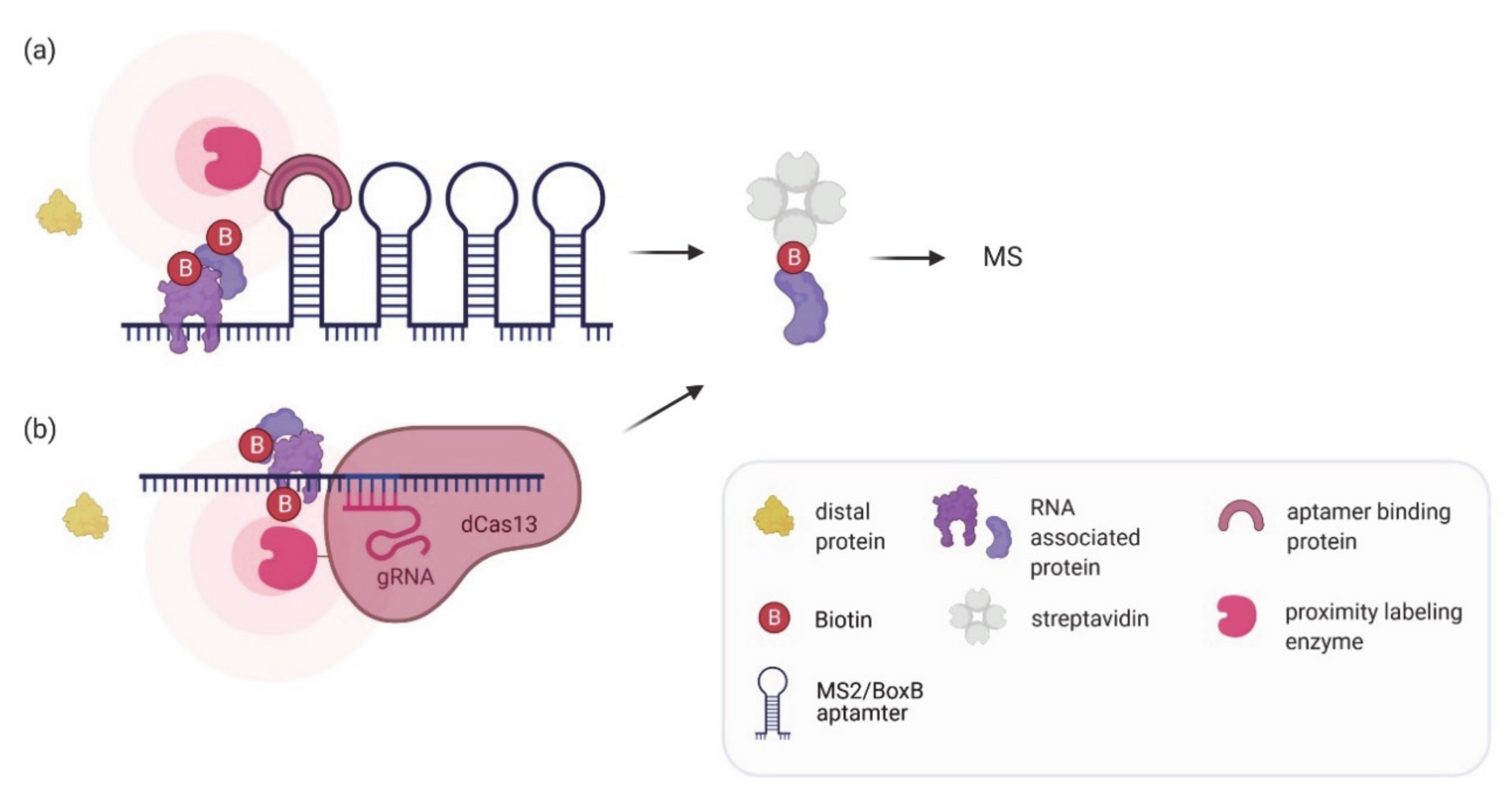
7. Proximity Labeling of RNA–Protein Interactions: Finding the Protein Partners
8. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szeto, R.A.; Tran, T.; Truong, J.; Negraes, P.D.; Trujillo, C.A. RNA processing in neurological tissue: Development, aging and disease. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Halbeisen, R.E.; Galgano, A.; Scherrer, T.; Gerber, A.P. Post-transcriptional gene regulation: From genome-wide studies to principles. Cell. Mol. Life Sci. 2007, 65, 798–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell. Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef]
- Tolino, M.; Köhrmann, M.; Kiebler, M.A. RNA-binding proteins involved in RNA localization and their implications in neuronal diseases. Eur. J. Neurosci. 2012, 35, 1818–1836. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef] [PubMed]
- García-Mauriño, S.M.; Rivero-Rodríguez, F.; Velázquez-Cruz, A.; Hernández-Vellisca, M.; Díaz-Quintana, A.; De La Rosa, M.A.; Díaz-Moreno, I. RNA Binding Protein Regulation and Cross-Talk in the Control of AU-rich mRNA Fate. Front. Mol. Biosci. 2017, 4, 71. [Google Scholar] [CrossRef]
- Keene, J.D.; Lager, P.J. Post-transcriptional operons and regulons co-ordinating gene expression. Chromosom. Res. 2005, 13, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Gilbert, C.; Svejstrup, J.Q. RNA Immunoprecipitation for Determining RNA-Protein Associations In Vivo. Curr. Protoc. Mol. Biol. 2006, 75, 27.4.1–27.4.11. [Google Scholar] [CrossRef]
- Gagliardi, M.; Matarazzo, M.R. RIP: RNA Immunoprecipitation. Methods Mol. Biol. 2016, 1480, 73–86. [Google Scholar] [CrossRef]
- Lambert, N.; Robertson, A.; Jangi, M.; McGeary, S.; Sharp, P.A.; Burge, C.B. RNA Bind-n-Seq: Quantitative Assessment of the Sequence and Structural Binding Specificity of RNA Binding Proteins. Mol. Cell 2014, 54, 887–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, D.; Freese, P.; Alexis, M.S.; Su, A.; Hochman, M.; Palden, T.; Bazile, C.; Lambert, N.J.; Van Nostrand, E.L.; Pratt, G.A.; et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 2018, 70, 854–867.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP Identifies Nova-Regulated RNA Networks in the Brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.C.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Buchbender, A.; Mutter, H.; Sutandy, F.R.; Körtel, N.; Hänel, H.; Busch, A.; Ebersberger, S.; König, J. Improved library preparation with the new iCLIP2 protocol. Methods 2020, 178, 33–48. [Google Scholar] [CrossRef]
- König, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Hinze, F.; Drewe-Boss, P.; Milek, M.; Ohler, U.; Landthaler, M.; Gotthardt, M. Expanding the map of protein-RNA interaction sites via cell fusion followed by PAR-CLIP. RNA Biol. 2018, 15, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogell, B.; Fischer, B.; Rettel, M.; Krijgsveld, J.; Castello, A.; Hentze, M.W. Specific RNP capture with antisense LNA/DNA mixmers. RNA 2017, 23, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic Discovery of Xist RNA Binding Proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nat. Cell Biol. 2015, 521, 232–236. [Google Scholar] [CrossRef]
- Wippich, F.; Ephrussi, A. Transcript specific mRNP capture from Drosophila egg-chambers for proteomic analysis. Methods 2020, 178, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J.; Sproat, B.S.; Ryder, U.; Barabino, S.; Lamond, A.I. Antisense probing of the human U4U6 snRNP with biotinylated 2′-OMe RNA oligonucleotides. Cell 1989, 59, 531–539. [Google Scholar] [CrossRef]
- Castello, A.; Fischer, B.; Frese, C.K.; Horos, R.; Alleaume, A.-M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M.W. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef] [Green Version]
- Conrad, T.; Albrecht, A.-S.; Costa, V.R.D.M.; Sauer, S.; Meierhofer, D.; Ørom, U.A. Serial interactome capture of the human cell nucleus. Nat. Commun. 2016, 7, 11212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matia-González, A.M.; Iadevaia, V.; Gerber, A.P. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 2017, 118-119, 93–100. [Google Scholar] [CrossRef] [Green Version]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The Long Noncoding RNAs NEAT1 and MALAT1 Bind Active Chromatin Sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keryer-Bibens, C.; Barreau, C.; Osborne, H.B. Tethering of proteins to RNAs by bacteriophage proteins. Biol. Cell 2008, 100, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Srisawat, C.; Engelke, D.R. Streptavidin aptamers: Affinity tags for the study of RNAs and ribonucleoproteins. RNA 2001, 7, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butter, F.; Scheibe, M.; Mörl, M.; Mann, M. Unbiased RNA-protein interaction screen by quantitative proteomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10626–10631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2014, 42, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmuth, K.; Urlaub, H.; Vornlocher, H.-P.; Will, C.L.; Gentzel, M.; Wilm, M.; Lührmann, R. Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc. Natl. Acad. Sci. USA 2002, 99, 16719–16724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachler, M.; Schroeder, R.; Von Ahsen, U. StreptoTag: A novel method for the isolation of RNA-binding proteins. RNA 1999, 5, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Bardwell, V.J.; Wickens, M. Purification of RNA and RNA-protein complexes by an R17 coat protein affinity method. Nucleic Acids Res. 1990, 18, 6587–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative Profiling of In Vivo-assembled RNA-Protein Complexes Using a Novel Integrated Proteomic Approach. Mol. Cell. Proteom. 2011, 10, 110–007385. [Google Scholar] [CrossRef] [Green Version]
- Hogg, J.R.; Collins, K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 2007, 13, 868–880. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Srikantan, S.; Gorospe, M. MS2-TRAP (MS2-tagged RNA affinity purification): Tagging RNA to identify associated miRNAs. Methods 2012, 58, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Graindorge, A.; Pinheiro, I.; Nawrocka, A.; Mallory, A.C.; Tsvetkov, P.; Gil, N.; Carolis, C.; Buchholz, F.; Ulitsky, I.; Heard, E.; et al. In-cell identification and measurement of RNA-protein interactions. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.F.; Parker, R. MS2 coat proteins bound to yeast mRNAs block 5’ to 3’ degradation and trap mRNA decay products: Implications for the localization of mRNAs by MS2-MCP system. RNA 2015, 21, 1393–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haimovich, G.; Zabezhinsky, D.; Haas, B.; Slobodin, B.; Purushothaman, P.; Fan, L.; Levin, J.Z.; Nusbaum, C.; Gerst, J.E. Use of the MS2 aptamer and coat protein for RNA localization in yeast: A response to “MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: Implications for the localization of mRNAs by MS2-MCP system”. RNA 2016, 22, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.E.; Dertinger, D.; Lecuyer, K.A.; Behlen, L.S.; Greef, C.H.; Uhlenbeck, O.C. A thermodynamic analysis of the sequence-specific binding of RNA by bacteriophage MS2 coat protein. Proc. Natl. Acad. Sci. USA 1998, 95, 9244–9249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobodin, B.; Gerst, J.E. A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA 2010, 16, 2277–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pederson, T. Half a Century of “The Nuclear Matrix”. Mol. Biol. Cell 2000, 11, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, E.C.; Van Nostrand, E.L.; Yeo, G.W. Advances and challenges in the detection of transcriptome-wide protein-RNA interactions. Wiley Interdiscip. Rev. RNA 2017, 9, e1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, R.D.; Sanjeev, M.; Woodward, L.A.; Mabin, J.W.; Bundschuh, R.; Singh, G. Chemical crosslinking enhances RNA immunoprecipitation for efficient identification of binding sites of proteins that photo-crosslink poorly with RNA. RNA 2020, 26, 1216–1233. [Google Scholar] [CrossRef]
- Mili, S. Evidence for reassociation of RNA-binding proteins after cell lysis: Implications for the interpretation of immunoprecipitation analyses. RNA 2004, 10, 1692–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Martell, J.D.; Deerinck, T.J.; Sancak, Y.; Poulos, T.L.; Mootha, V.K.; Sosinsky, G.E.; Ellisman, M.H.; Ting, A.Y. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 2021, 18, 133–143. [Google Scholar] [CrossRef]
- Kim, D.I.; Kc, B.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, K.J.; Kim, D.I.; Burke, B.; May, D.G. BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 2018, 91, 19.23.1–19.23.15. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; MacLean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firat-Karalar, E.N.; Stearns, T. Probing mammalian centrosome structure using BioID proximity-dependent biotinylation. Methods Cell Biol. 2015, 129, 153–170. [Google Scholar] [CrossRef] [Green Version]
- Van Itallie, C.M.; Tietgens, A.J.; Aponte, A.; Fredriksson, K.; Fanning, A.S.; Gucek, M.; Anderson, J.M. Biotin ligase tagging identifies proteins proximal to E-cadherin, including lipoma preferred partner, a regulator of epithelial cell–cell and cell–substrate adhesion. J. Cell Sci. 2014, 127, 885–895. [Google Scholar] [CrossRef] [Green Version]
- McAllaster, M.R.; Ikeda, K.N.; Lozano-Núñez, A.; Anrather, D.; Unterwurzacher, V.; Gossenreiter, T.; Perry, J.A.; Crickley, R.; Mercadante, C.J.; Vaughan, S.; et al. Proteomic identification of novel cytoskeletal proteins associated with TbPLK, an essential regulator of cell morphogenesis in Trypanosoma brucei. Mol. Biol. Cell 2015, 26, 3013–3029. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, K.; Van Itallie, C.M.; Aponte, A.; Gucek, M.; Tietgens, A.J.; Anderson, J.M. Proteomic Analysis of Proteins Surrounding Occludin and Claudin-4 Reveals Their Proximity to Signaling and Trafficking Networks. PLoS ONE 2015, 10, e0117074. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.L.; Kim, E.W.; Toh, J.Y.; Vashisht, A.A.; Rashoff, A.Q.; Van, C.; Huang, A.S.; Moon, A.S.; Bell, H.N.; Bentolila, L.A.; et al. Novel Components of the Toxoplasma Inner Membrane Complex Revealed by BioID. mBio 2015, 6, e02357-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, J.; Hermesh, O.; Eliscovich, C.; Nalpas, N.; Franz-Wachtel, M.; Maček, B.; Jansen, R.-P. β-Actin mRNA interactome mapping by proximity biotinylation. Proc. Natl. Acad. Sci. USA 2019, 116, 12863–12872. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Ramanathan, M.; Majzoub, K.; Rao, D.S.; Neela, P.H.; Zarnegar, B.J.; Mondal, S.; Roth, J.G.; Gai, H.; Kovalski, J.R.; Siprashvili, Z.; et al. RNA–protein interaction detection in living cells. Nat. Methods 2018, 15, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- LaRochelle, M.; Bergeron, D.; Arcand, B.; Bachand, F. Proximity-dependent biotinylation mediated by TurboID to identify protein–protein interaction networks in yeast. J. Cell Sci. 2019, 132, jcs232249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, A.; Xu, S.-L.; Branon, T.C.; Ting, A.Y.; Bergmann, D.C. Proximity labeling of protein complexes and cell-type-specific organellar proteomes in Arabidopsis enabled by TurboID. eLife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, G.; Lal, N.K.; Nagalakshmi, U.; Li, Y.; Zheng, W.; Huang, P.-J.; Branon, T.C.; Ting, A.Y.; Walley, J.W.; et al. TurboID-based proximity labeling reveals that UBR7 is a regulator of N NLR immune receptor-mediated immunity. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, J.S.; Philip, J.A.; Perrett, S.; Lilley, K.S.; Jackson, A.P. Selective Proteomic Proximity Labeling Assay Using Tyramide (SPPLAT): A Quantitative Method for the Proteomic Analysis of Localized Membrane-Bound Protein Clusters. Curr. Protoc. Protein Sci. 2015, 80, 19.27.1–19.27.18. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.-M.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Lobingier, B.T.; Hüttenhain, R.; Eichel, K.; Miller, K.B.; Ting, A.Y.; von Zastrow, M.; Krogan, N.J. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 2017, 169, 350–360.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, S.; Nguyen, N.; Richter, J.; Setayesh, J.; Lonquich, B.; Oon, C.H.; Wozniak, J.M.; Barahona, R.; Kamei, C.N.; Houston, J.; et al. EGFR is required for Wnt9a–Fzd9b signalling specificity in haematopoietic stem cells. Nat. Cell Biol. 2019, 21, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Kohli, P.; Höhne, M.; Jüngst, C.; Bertsch, S.; Ebert, L.K.; Schauss, A.C.; Benzing, T.; Rinschen, M.M.; Schermer, B. The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep. 2017, 18, 1521–1535. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Peterson, C.W.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 97–112.e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zheng, J.; Sun, W.; Huo, Y.; Zhang, L.; Hao, P.; Wang, H.; Zhuang, M. A proximity-tagging system to identify membrane protein–protein interactions. Nat. Methods 2018, 15, 715–722. [Google Scholar] [CrossRef] [PubMed]
- De Munter, S.; Goernemann, J.; Derua, R.; Lesage, B.; Qian, J.; Heroes, E.; Waelkens, E.; Van Eynde, A.; Beullens, M.; Bollen, M. Split-BioID: A proximity biotinylation assay for dimerization-dependent protein interactions. FEBS Lett. 2017, 591, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Schopp, I.M.; Ramirez, C.C.A.; Debeljak, J.; Kreibich, E.; Skribbe, M.; Wild, K.; Béthune, J. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat. Commun. 2017, 8, 15690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Branon, T.C.; Martell, J.D.; Boassa, D.; Shechner, D.; Ellisman, M.H.; Ting, A. Directed Evolution of Split APEX2 Peroxidase. ACS Chem. Biol. 2019, 14, 619–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.-W.; Lee, I.-K.; Carr, S.A.; et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12143–12154. [Google Scholar] [CrossRef] [PubMed]
- Kaewsapsak, P.; Shechner, D.M.; Mallard, W.; Rinn, J.L.; Ting, A.Y. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 2017, 6, e29224. [Google Scholar] [CrossRef] [PubMed]
- Benhalevy, D.; Anastasakis, D.G.; Hafner, M. Proximity-CLIP provides a snapshot of protein-occupied RNA elements in subcellular compartments. Nat. Methods 2018, 15, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, G.; Wang, P.; Li, Z.; Yue, T.; Wang, J.; Zou, P. Expanding APEX2 Substrates for Proximity-Dependent Labeling of Nucleic Acids and Proteins in Living Cells. Angew. Chem. Int. Ed. 2019, 58, 11763–11767. [Google Scholar] [CrossRef] [PubMed]
- Padrón, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA Labeling by APEX-Seq Reveals the Organization of Translation Initiation Complexes and Repressive RNA Granules. Mol. Cell 2019, 75, 875–887.e5. [Google Scholar] [CrossRef] [PubMed]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490.e26. [Google Scholar] [CrossRef]
- Mayr, C. Regulation by 3′-Untranslated Regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Tang, W.; Li, Z.; Zou, Z.; Zhou, Y.; Li, R.; Xiong, T.; Wang, J.; Zou, P. Mapping spatial transcriptome with light-activated proximity-dependent RNA labeling. Nat. Chem. Biol. 2019, 15, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.C.; Rahman, R.; Jin, H.; Shen, J.L.; Fieldsend, A.; Luo, W.; Rosbash, M. TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 2016, 165, 742–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Rahman, R.; Rosbash, M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA 2017, 24, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, C.P.; Wilinski, D.; Saunders, H.A.J.; Wickens, M. Protein-RNA networks revealed through covalent RNA marks. Nat. Methods 2015, 12, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, C.P.; Preston, M.A.; Wilinski, D.; Saunders, H.A.J.; Campbell, Z.T.; Wickens, M. Architecture and dynamics of overlapped RNA regulatory networks. RNA 2017, 23, 1636–1647. [Google Scholar] [CrossRef] [Green Version]
- Medina-Munoz, H.C.; Lapointe, C.P.; Porter, D.F.; Wickens, M. Records of RNA locations in living yeast revealed through covalent marks. Proc. Natl. Acad. Sci. USA 2020, 117, 23539–23547. [Google Scholar] [CrossRef]
- Han, S.; Zhao, B.S.; Myers, S.A.; Carr, S.A.; He, C.; Ting, A.Y. RNA–protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc. Natl. Acad. Sci. USA 2020, 117, 22068–22079. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR–Cas13. Nat. Cell Biol. 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Sun, W.; Shi, T.; Lu, P.; Zhuang, M.; Liu, J.-L. Capturing RNA–protein interaction via CRUIS. Nucleic Acids Res. 2020, 48, e52. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Li, J.; Zhu, X.; Wang, X.; Fan, L.; Sun, W.; Liao, L.; Zhang, J.; Li, X.; Ye, J.; et al. CRISPR-assisted detection of RNA–protein interactions in living cells. Nat. Methods 2020, 17, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Fonseca, M.A.S.; Corona, R.I.; Lawrenson, K. In vivo discovery of RNA proximal proteins in human cells via proximity-dependent biotinylation. BioRxiv 2020. Preprint. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, K.A.; Bass, B.L. Double-Stranded RNA Adenosine Deaminases ADAR1 and ADAR2 Have Overlapping Specificities. Biochemistry 2000, 39, 12875–12884. [Google Scholar] [CrossRef] [PubMed]
- Eggington, J.M.; Greene, T.; Bass, B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011, 2, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutucci, E.; Livingston, N.M.; Singer, R.H.; Wu, B. Imaging mRNA In Vivo, from Birth to Death. Annu. Rev. Biophys. 2018, 47, 85–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Wei, W. Proximity labeling to detect RNA–protein interactions in live cells. FEBS Open Biol. 2019, 9, 1860–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionnet, T.; Czaplinski, K.; Darzacq, X.; Shav-Tal, Y.; Wells, A.L.; Chao, J.A.; Park, H.Y.; De Turris, V.; Lopez-Jones, M.; Singer, R.H. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat. Methods 2011, 8, 165–170. [Google Scholar] [CrossRef]
- Spille, J.-H.; Hecht, M.; Grube, V.; Cho, W.-K.; Lee, C.; Cissé, I.I. A CRISPR/Cas9 platform for MS2-labelling of single mRNA in live stem cells. Methods 2019, 153, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Tutucci, E.; Vera, M.; Biswas, J.; Garcia, J.; Parker, R.; Singer, R.H. An improved MS2 system for accurate reporting of the mRNA life cycle. Nat. Methods 2018, 15, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Laprade, H.; Querido, E.; Smith, M.J.; Guérit, D.; Crimmins, H.; Conomos, D.; Pourret, E.; Chartrand, P.; Sfeir, A. Single-Molecule Imaging of Telomerase RNA Reveals a Recruitment-Retention Model for Telomere Elongation. Mol. Cell 2020, 79, 115–126.e6. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.M.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Bandaru, S.; Tsuji, M.H.; Shimizu, Y.; Usami, K.; Lee, S.; Takei, N.K.; Yoshitome, K.; Nishimura, Y.; Otsuki, T.; Ito, T. Structure-based design of gRNA for Cas13. Sci. Rep. 2020, 10, 11610. [Google Scholar] [CrossRef] [PubMed]
- Minajigi, A.; Froberg, J.E.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349, aab2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined proximity labeling and affinity purification−mass spectrometry workflow for mapping and visualizing protein interaction networks. Nat. Protoc. 2020, 15, 1–30. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Proximity Labeling for Mapping Subcellular Transcriptomes | ||||||
|---|---|---|---|---|---|---|
| Method | Description | Model Organism/ Cell Type | Achievements | Strengths | Weaknesses | Ref. |
| APEX-RIP, Proximity-CLIP | Targeting of a labeling enzyme (e.g., APEX2) to subcellular compartments in order to biotinylate proximal proteins. After crosslinking proteins and RNA, biotinylated proteins are enriched by streptavidin beads and bound RNAs are identified via RNA-Seq. | HEK293T | Identification of compartment specific RNAs, e.g., in the nucleus, cytoplasm, mitochondrial matrix and at the ER membrane. | Proximity-Clip allows identification of RBP-protected regions of RNA targets. Short labeling time. No need of specific antibody. | Limited detection of RNAs in non-membrane bound cellular regions. | [81,82] |
| APEX-seq, CAP-seq | Targeting of a labeling enzyme (e.g., APEX2, miniSOG) to subcellular compartments or complexes to directly label RNAs. Biotinylated RNAs are enriched by streptavidin beads and identified using RNA-Seq. | HEK293T | Identification of RNAs localized to various locations, including nucleolus, nuclear lamina, nuclear pore, the outer mitochondrial membrane, the mitochondrial matrix, the ER lumen, the ER cytosolic interface and RNA granules. | No crosslinking required. Can identify proximal RNAs in insoluble and open cellular regions. Short labeling time. | Interactomes of individual RBPs cannot be assessed. | [83,84,85] |
| Proximity Labeling of RNA–Protein Interactions: Finding the RNA Partners | ||||||
| Method | Description | Model Organism/ Cell Type | Achievements | Strengths | Weaknesses | Ref. |
| TRIBE | An RBP of interest is fused to the catalytic domain of the RNA editing enzyme ADAR. ADAR edits target RNAs (A-to-I editing) bound by the RBP, which can be identified by RNA-Seq. | Drosophila S2 cells and neurons | Identification of RNAs bound to Drosophila RBPs: Hrp48, dFMR1 and NonA. | No crosslinking required. No specific substrate required for labeling. Can be used to identify the RNA region close to the RBP binding site. | The edited sequence is biased due to the binding and editing preference of ADARcd. ADAR can also edit RNAs in the vicinity but not bound by the RBP. Cannot be used to detect dynamic interactions. | [88,89] |
| RNA tagging | An RBP of interest is fused to the uridine polymerase PUP-2. PUP-2 attaches an uracil tail to RNAs bound by the RBP which allow their identification by RNA-Seq. | S. cerevisiae | Identification of RNA targets of yeast pumilio proteins as well as RNAs localized to ER and mitochondrial surfaces. | No crosslinking required. Counting of added uracil residues allows differentiation of true and false interactors. | Might miss proteins that interact close to the 5′ of the RNA. Can stress cells. Cannot be used to detect dynamic interactions. | [90,91,92] |
| Proximity Labeling of RNA–Protein Interactions: Finding the Protein Partners | ||||||
| Method | Description | Model Organism/ Cell Type | Achievements | Strengths | Weaknesses | Ref. |
| RaPID, RNA-BioID | An RNA sequence of interest is tagged with either BoxB or MS2 aptamers. The aptamers recruit a viral coat protein fused to a labeling enzyme (BirA*, BASU, APEX2) which biotinylates associated proteins. | HEK293T, huh7, mouse embryonic fibroblasts | Identification of proteins binding various RNA motifs, the UTR of the Zika virus RNA genome, human telomerase RNA, or β-actin mRNA. | Allows identification of weak or transient interactions. High specificity and affinity of MCP or λ N-peptide for their corresponding aptamer. No crosslinking required. | Aptamer insertion might affect RNA function or regulation. Technically challenging to genomically integrate the aptamer cassette at correct location. | [62,64,93,94] |
| CARPID, dCas13d-dsRBD-APEX2, RPL | Catalytically inactive Cas13 fused to a labeling enzyme (BioID2, BASU, APEX, APEX2) is targeted to an RNA of interest using guide RNAs. | HEK293T | Identification of proteins binding to Xist, MALAT1, DANCR, hTR and U1 snRNA. | No need for changes in target RNA. Probing of different endogenous RNAs can easily be achieved by changing the gRNA. Can be used to probe a specific region on the RNA. No crosslinking required. | Background biotinylation from off-target gRNAs or unbound Cas13-labeling complex possible. Thorough optimization of the Cas13-labeling enzyme construct required. | [93,95,96,97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weissinger, R.; Heinold, L.; Akram, S.; Jansen, R.-P.; Hermesh, O. RNA Proximity Labeling: A New Detection Tool for RNA–Protein Interactions. Molecules 2021, 26, 2270. https://doi.org/10.3390/molecules26082270
Weissinger R, Heinold L, Akram S, Jansen R-P, Hermesh O. RNA Proximity Labeling: A New Detection Tool for RNA–Protein Interactions. Molecules. 2021; 26(8):2270. https://doi.org/10.3390/molecules26082270
Chicago/Turabian StyleWeissinger, Ronja, Lisa Heinold, Saira Akram, Ralf-Peter Jansen, and Orit Hermesh. 2021. "RNA Proximity Labeling: A New Detection Tool for RNA–Protein Interactions" Molecules 26, no. 8: 2270. https://doi.org/10.3390/molecules26082270
APA StyleWeissinger, R., Heinold, L., Akram, S., Jansen, R.-P., & Hermesh, O. (2021). RNA Proximity Labeling: A New Detection Tool for RNA–Protein Interactions. Molecules, 26(8), 2270. https://doi.org/10.3390/molecules26082270