
A One-Pot Approach to Novel Pyridazine C-Nucleosides


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
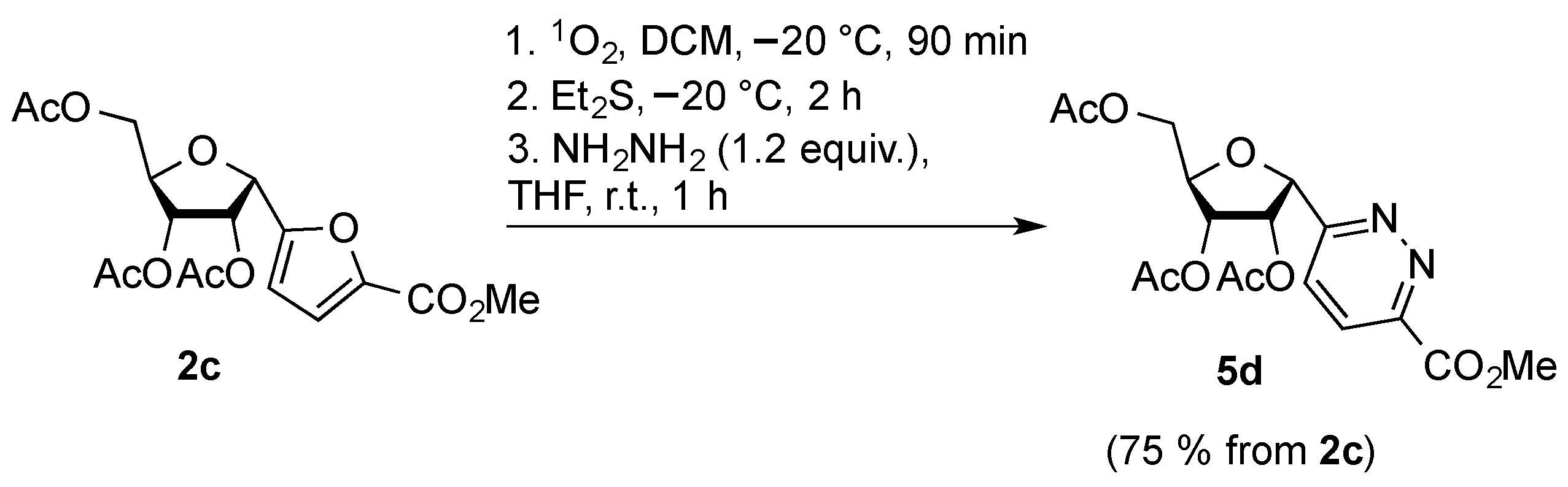
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 2a,b
3.3. Synthesis of Pyridazines: General Procedure
3.4. Aromatization of 5d
3.5. X-ray Crystallography of 5d
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Merino, P. Chemical Synthesis of Nucleoside Analogues; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 978-1-118-00751-8. [Google Scholar]
- Stambaský, J.; Hocek, M.; Kočovský, P. C-Nucleosides: Synthetic Strategies and Biological Applications. Chem. Rev. 2009, 109, 6729–6764. [Google Scholar] [CrossRef] [PubMed]
- Cohn, W.E. Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: Isolation, structure, and chemical characteristics. J. Biol. Chem. 1960, 235, 1488–1498. [Google Scholar] [CrossRef]
- Nishimura, H.; Mayama, M.; Komatsu, Y.; Kato, H.; Shimaoka, N.; Tanaka, Y. Showdomycin, a New Antibiotic from a Streptomyces sp. J. Antibiot. 1964, 17, 148–155. [Google Scholar] [CrossRef]
- Sasaki, K.; Kusakabe, Y.; Esumi, S. The structure of minimycin, a novel carbon-linked nucleoside antibiotic related to β-pseudouridine. J. Antibiot. 1972, 25, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lescrinier, E.; Groaz, E.; Persoons, L.; Daelemans, D.; Herdewijn, P.; De Jonghe, S. Synthesis and Biological Evaluation of Pyrrolo[2,1-f][1,2,4]triazine C-Nucleosides with a Ribose, 2′-Deoxyribose, and 2′,3′-Dideoxyribose Sugar Moiety. ChemMedChem 2018, 13, 97–104. [Google Scholar] [CrossRef]
- Sabat, N.; Migianu-Griffoni, E.; Tudela, T.; Lecouvey, M.; Kellouche, S.; Carreiras, F.; Gallier, F.; Uziel, J.; Lubin-Germain, N. Synthesis and antitumor activities investigation of a C-nucleoside analogue of ribavirin. Eur. J. Med. Chem. 2020, 188. [Google Scholar] [CrossRef]
- Sun, J.; Kang, Y.; Gao, L.; Lu, X.; Ju, H.; Li, X.; Chen, H. Synthesis of tricyclic quinazolinone-iminosugars as potential glycosidase inhibitors via a Mitsunobu reaction. Carbohydr. Res. 2019, 478, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Groaz, E.; Persoons, L.; Daelemans, D.; Herdewijn, P. Synthesis and Antitumor Activity of C-7-Alkynylated and Arylated Pyrrolotriazine C-Ribonucleosides. ACS Med. Chem. Lett. 2020, 11, 1605–1610. [Google Scholar] [CrossRef]
- Postema, M.H.D. C-Glycoside Synthesis; CRC Press: London, UK, 1995; ISBN 0-8493-9150-4. [Google Scholar]
- Keay, B.A.; Hopkins, J.; Dibbie, P.W. Furans and their benzoderivatives. Applications. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C., Scriven, E.F., Eds.; Elsevier: San Diego, CA, USA, 2008; Volume 3, pp. 571–621. ISBN 9780080449920. [Google Scholar]
- Cermola, F.; Iesce, M.; Montella, S. The First Dye-sensitized Photooxygenation of 2-(C-glycosyl)furans. One-Pot Stereoselective Approach to New Carbohydrate Derivatives. Lett. Org. Chem. 2004, 1, 271–275. [Google Scholar] [CrossRef]
- Iesce, M.R.; Cermola, F. Photooxygenation, [2+2] and [4+2]. In CRC Handbook of Organic Photochemistry and Photobiology; Griesbeck, A., Oelgemöller, M., Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 727–764. ISBN 9780080449920. [Google Scholar]
- Cermola, F.; Iesce, M.R. Dye-sensitized photooxygenation of sugar-furans as synthetic strategy for novel C-nucleosides and functionalized exo-glycals. Tetrahedron 2006, 62, 10694–10699. [Google Scholar] [CrossRef]
- Cermola, F.; Sferruzza, R.; Iesce, M.R. Spiroketals of monosaccharides by dye-sensitized photooxygenation of furyl ketoses. Tetrahedron Lett. 2014, 55, 737–740. [Google Scholar] [CrossRef]
- Cermola, F.; Iesce, M.R.; Buonerba, G. Dye-Sensitized Photooxygenation of Furanosyl Furans: Synthesis of a New Pyridazine C-Nucleoside. J. Org. Chem. 2005, 70, 6503–6505. [Google Scholar] [CrossRef] [PubMed]
- Astarita, A.; Cermola, F.; Rosaria Iesce, M.; Previtera, L. Dye-sensitized photooxygenation of sugar furans: Novel bis-epoxide and spirocyclic C-nucleosides. Tetrahedron 2008, 64, 6744–6748. [Google Scholar] [CrossRef]
- Maeba, I.; Iwata, K.; Usami, F.; Furukawa, H. C-Nucleosides. 1. Synthesis of 3-(β-d-Ribofuranosyl)pyridazines. J. Org. Chem. 1983, 48, 2998–3002. [Google Scholar] [CrossRef]
- Elnagdi, M.H.; Al-Awadi, N.A.; Abdelhamid, I.A. Recent Developments in Pyridazine and Condensed Pyridazine Synthesis. In Advances Heterocyclic Chemistry; Katritzky, A.R., Ed.; Elsevier: Oxford, UK, 2009; pp. 1–43. ISBN 9780080449920. [Google Scholar]
- Bel Abed, H.; Mammoliti, O.; Bande, O.; Van Lommen, G.; Herdewijn, P. Strategy for the synthesis of pyridazine heterocycles and their derivatives. J. Org. Chem. 2013, 78, 7845–7858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Zhu, Y.; Lian, M.; Liu, M.; Yuan, J.; Yin, G.; Wu, A. Unexpected C-C bond cleavage: A route to 3,6-diarylpyridazines and 6-arylpyridazin-3-ones from 1,3-dicarbonyl compounds and methyl ketones. J. Org. Chem. 2012, 77, 9865–9870. [Google Scholar] [CrossRef]
- Guo, Y.Q.; Zhao, M.N.; Ren, Z.H.; Guan, Z.H. Copper-Promoted 6- endo-trig Cyclization of β,γ-Unsaturated Hydrazones for the Synthesis of 1,6-Dihydropyridazines. Org. Lett. 2018, 20, 3337–3340. [Google Scholar] [CrossRef]
- Czernecki, S.; Ville, G. Stereospecific C-Glycosides.7. Stereospecific C-Glycosylation of Aromatic and Heterocyclic Rings. J. Org. Chem. 1989, 987, 610–612. [Google Scholar] [CrossRef]
- Macdonald, S.J.F.; Huizinga, W.B.; McKenzie, T.C. Retention of configuration in the coupling of aluminated heterocycles with glycopyranosyl fluorides. J. Org. Chem. 1988, 53, 3371–3373. [Google Scholar] [CrossRef]
- Guo, J.; Ye, X.S. Protecting groups in carbohydrate chemistry: Influence on stereoselectivity of glycosylations. Molecules 2010, 15, 7235–7265. [Google Scholar] [CrossRef]
- Spadafora, M.; Mehiri, M.; Burger, A.; Benhida, R. Friedel-Crafts and modified Vorbrüggen ribosylation. A short synthesis of aryl and heteroaryl-C-nucleosides. Tetrahedron Lett. 2008, 49, 3967–3971. [Google Scholar] [CrossRef]
- Chavelas-Hernández, L.; Valdéz-Camacho, J.R.; Hernández-Vázquez, L.G.; Dominguez-Mendoza, B.E.; Vasquez-Ríos, M.G.; Escalante, J. A New Approach Using Aromatic-Solvent-Induced Shifts in NMR Spectroscopy to Analyze β-Lactams with Various Substitution Patterns. Synlett 2020, 31, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Goodyear, M.D.; Hill, M.L.; West, J.P.; Whitehead, A.J. Practical enantioselective synthesis of lamivudine (3TCTM) via a dynamic kinetic resolution. Tetrahedron Lett. 2005, 46, 8535–8538. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS (Version 2.03). Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cacarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Bourguignon, J.J.; Oumouch, S.; Schmitt, M. Use of Polyfunctionalized Pyridazines as Reactive Species for Building Chemical Diversity. Curr. Org. Chem. 2006, 10, 277–295. [Google Scholar] [CrossRef]
- Ritchie, T.J.; MacDonald, S.J.F.; Peace, S.; Pickett, S.D.; Luscombe, C.N. The developability of heteroaromatic and heteroaliphatic rings—Do some have a better pedigree as potential drug molecules than others? Medchemcomm 2012, 3, 1062–1069. [Google Scholar] [CrossRef]
- Wermuth, C.G. Are pyridazines privileged structures? Medchemcomm 2011, 2, 935–941. [Google Scholar] [CrossRef]
- Jaballah, M.Y.; Serya, R.T.; Abouzid, K. Pyridazine Based Scaffolds as Privileged Structures in anti-Cancer Therapy. Drug Res. 2017, 67, 138–148. [Google Scholar] [CrossRef]
- Kilic, B.; Gulcan, H.O.; Aksakal, F.; Ercetin, T.; Oruklu, N.; Umit Bagriacik, E.; Dogruer, D.S. Design and synthesis of some new carboxamide and propanamide derivatives bearing phenylpyridazine as a core ring and the investigation of their inhibitory potential on in-vitro acetylcholinesterase and butyrylcholinesterase. Bioorg. Chem. 2018, 79, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Sabt, A.; Eldehna, W.M.; Al-Warhi, T.; Alotaibi, O.J.; Elaasser, M.M.; Suliman, H.; Abdel-Aziz, H.A. Discovery of 3,6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: Synthesis, biological evaluation and in silico insights. J. Enzyme Inhib. Med. Chem. 2020, 35, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Hassan, M.S.A.; El-Malah, A.A.; Kassab, A.E. New pyridazine derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents; design, synthesis and biological evaluation. Bioorg. Chem. 2020, 95, 103497. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cermola, F.; Vella, S.; DellaGreca, M.; Tuzi, A.; Iesce, M.R. A One-Pot Approach to Novel Pyridazine C-Nucleosides. Molecules 2021, 26, 2341. https://doi.org/10.3390/molecules26082341
Cermola F, Vella S, DellaGreca M, Tuzi A, Iesce MR. A One-Pot Approach to Novel Pyridazine C-Nucleosides. Molecules. 2021; 26(8):2341. https://doi.org/10.3390/molecules26082341
Chicago/Turabian StyleCermola, Flavio, Serena Vella, Marina DellaGreca, Angela Tuzi, and Maria Rosaria Iesce. 2021. "A One-Pot Approach to Novel Pyridazine C-Nucleosides" Molecules 26, no. 8: 2341. https://doi.org/10.3390/molecules26082341
APA StyleCermola, F., Vella, S., DellaGreca, M., Tuzi, A., & Iesce, M. R. (2021). A One-Pot Approach to Novel Pyridazine C-Nucleosides. Molecules, 26(8), 2341. https://doi.org/10.3390/molecules26082341