
Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics


Abstract
:1. Introduction
2. Molecular Docking
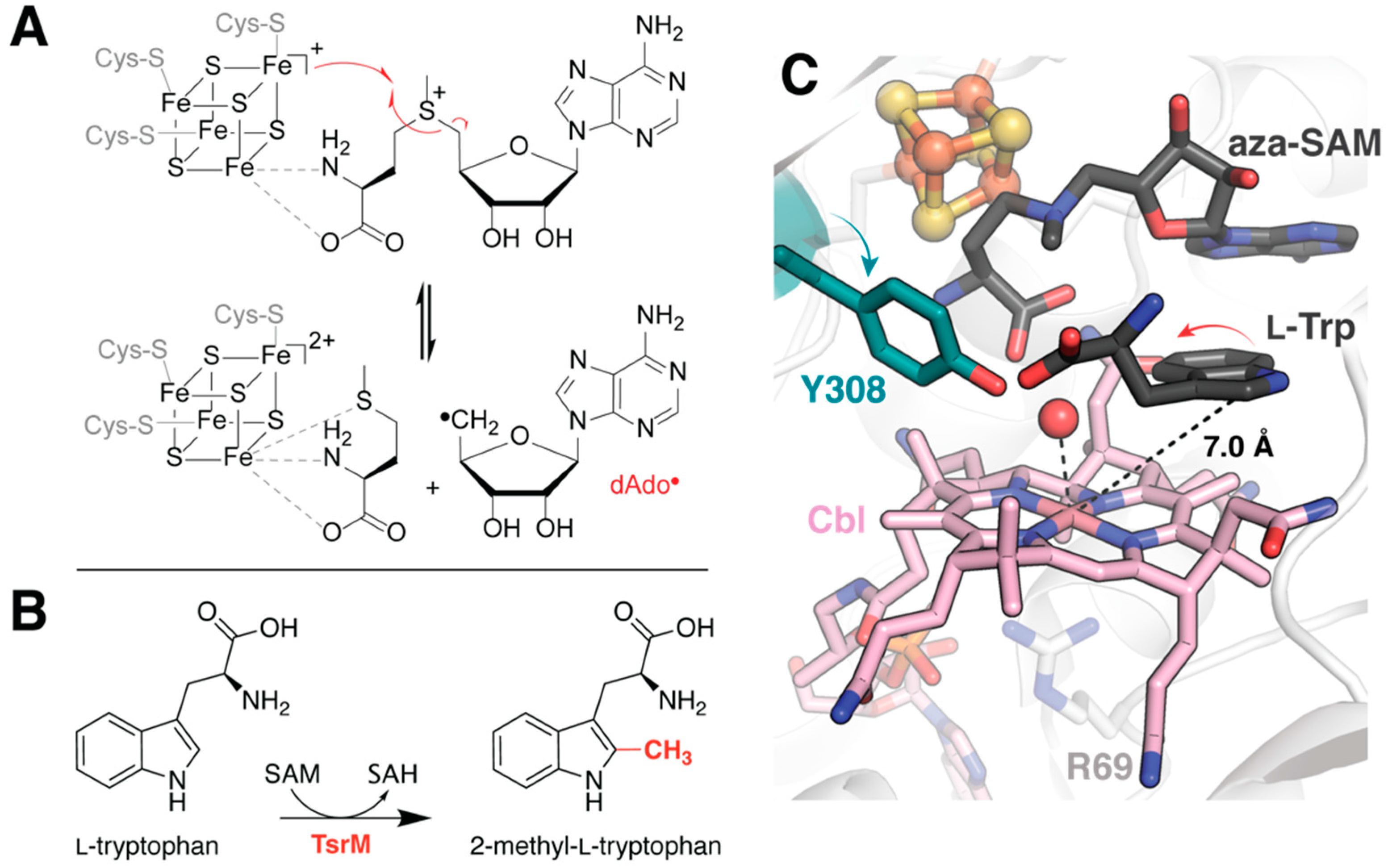
2.1. Correcting for the Wrong Form of Cobalamin in TsrM
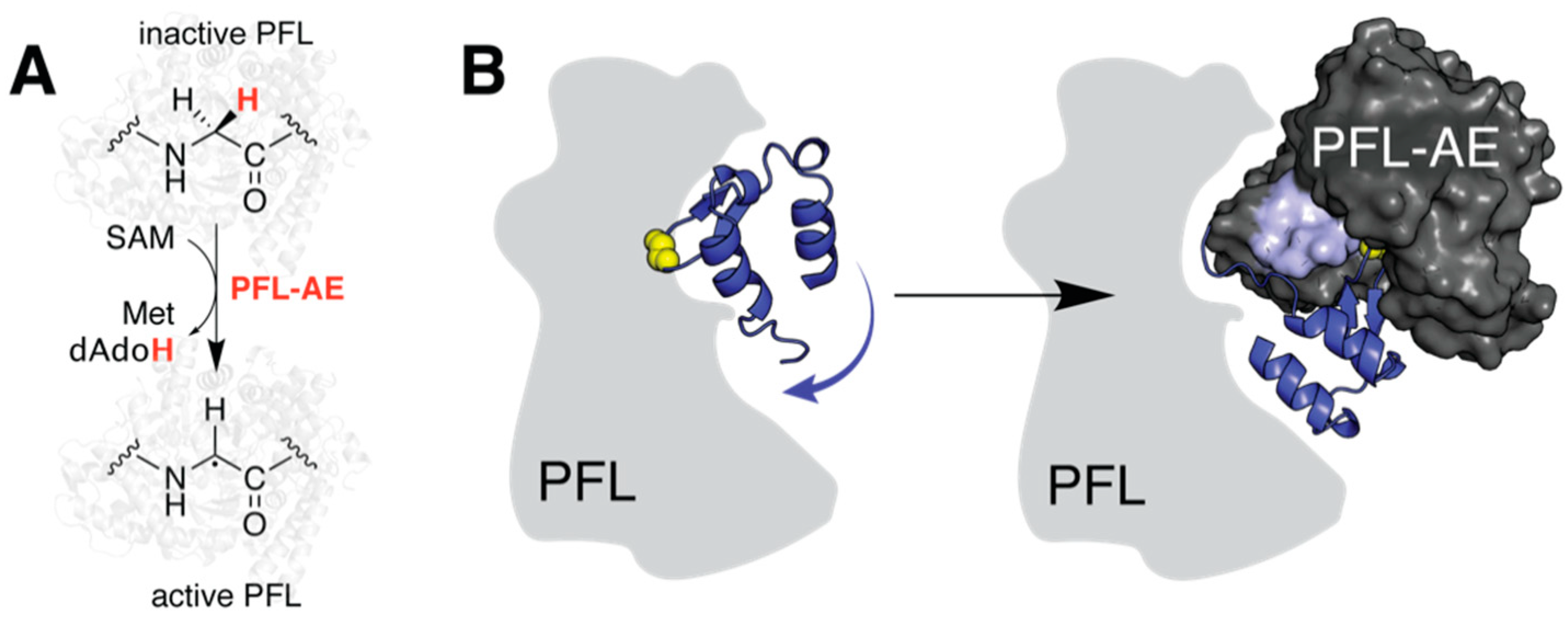
2.2. Predicting a Structural Framework for PFL Activation
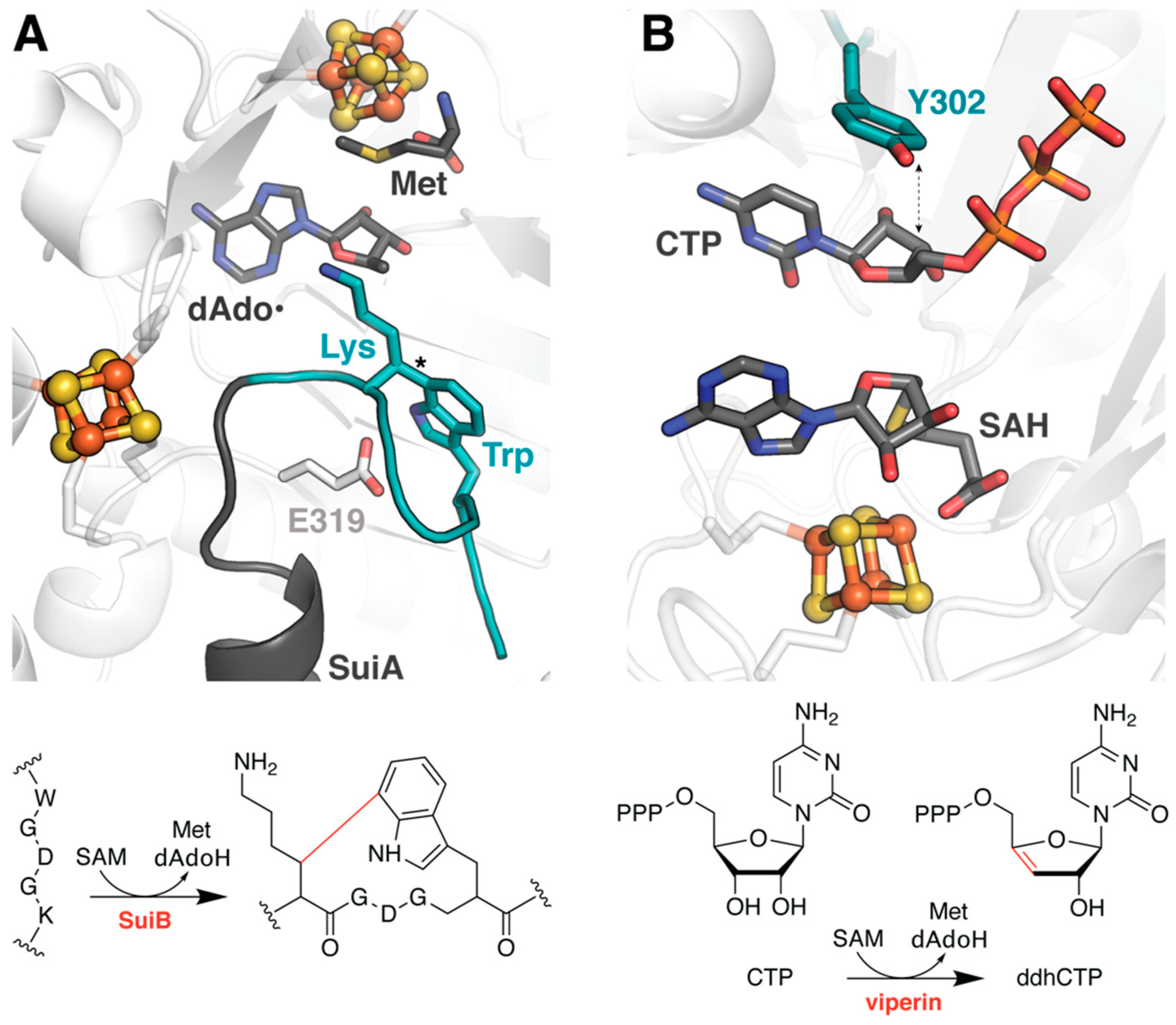
2.3. Elucidating the Transition to Ordered Product in SuiB
2.4. Assessing the Substrate Scope of Viperin and QueE
3. Molecular Dynamics
3.1. The Flexible Binding Pocket of Fungal Viperin
3.2. Substrate Stabilization in QueE
4. Density Functional Theory
4.1. Probing the Basis for SAM Cleavage in PFL-AE
4.2. Analyzing the Effect of Ion Complexation to QueE
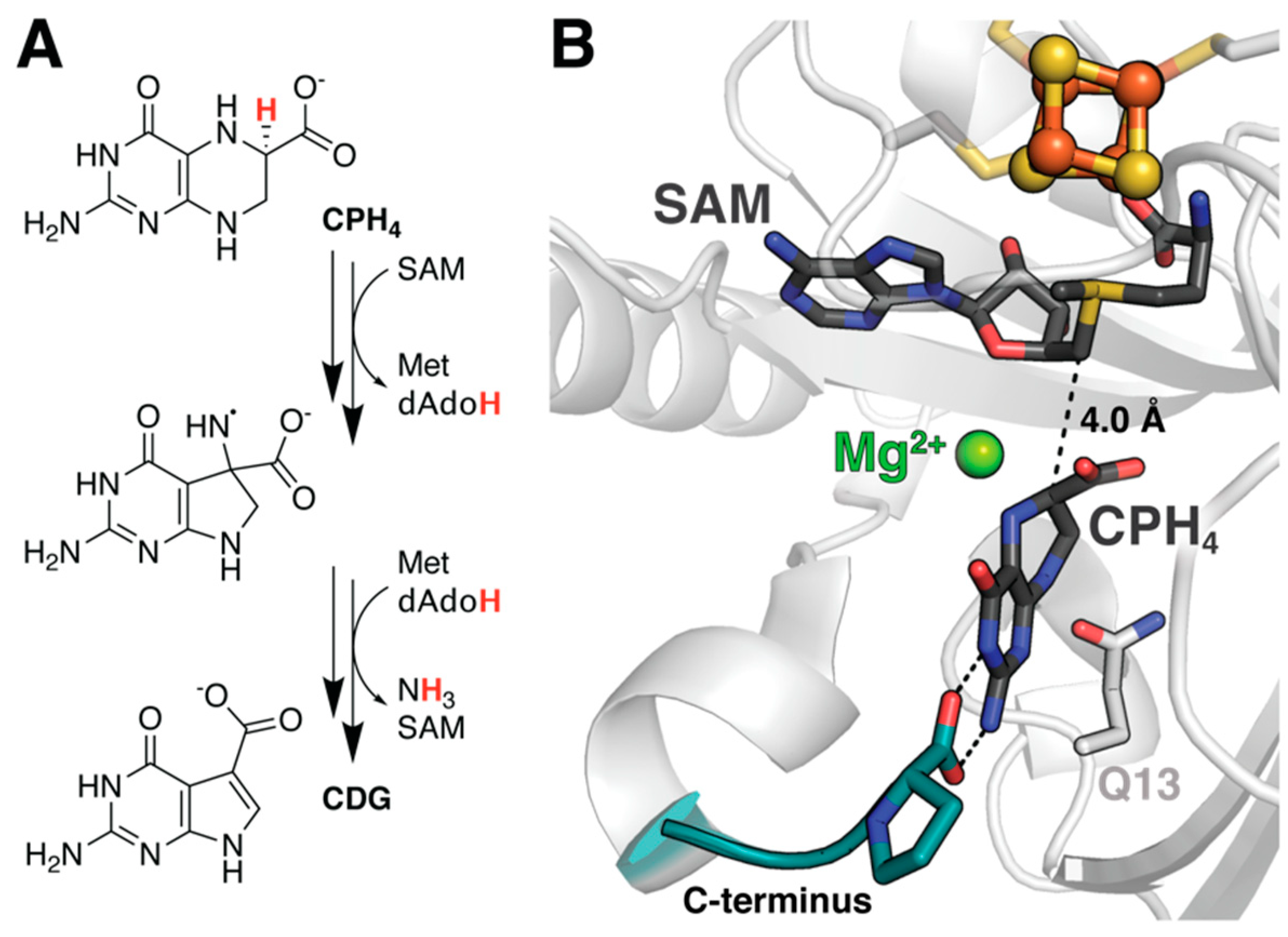
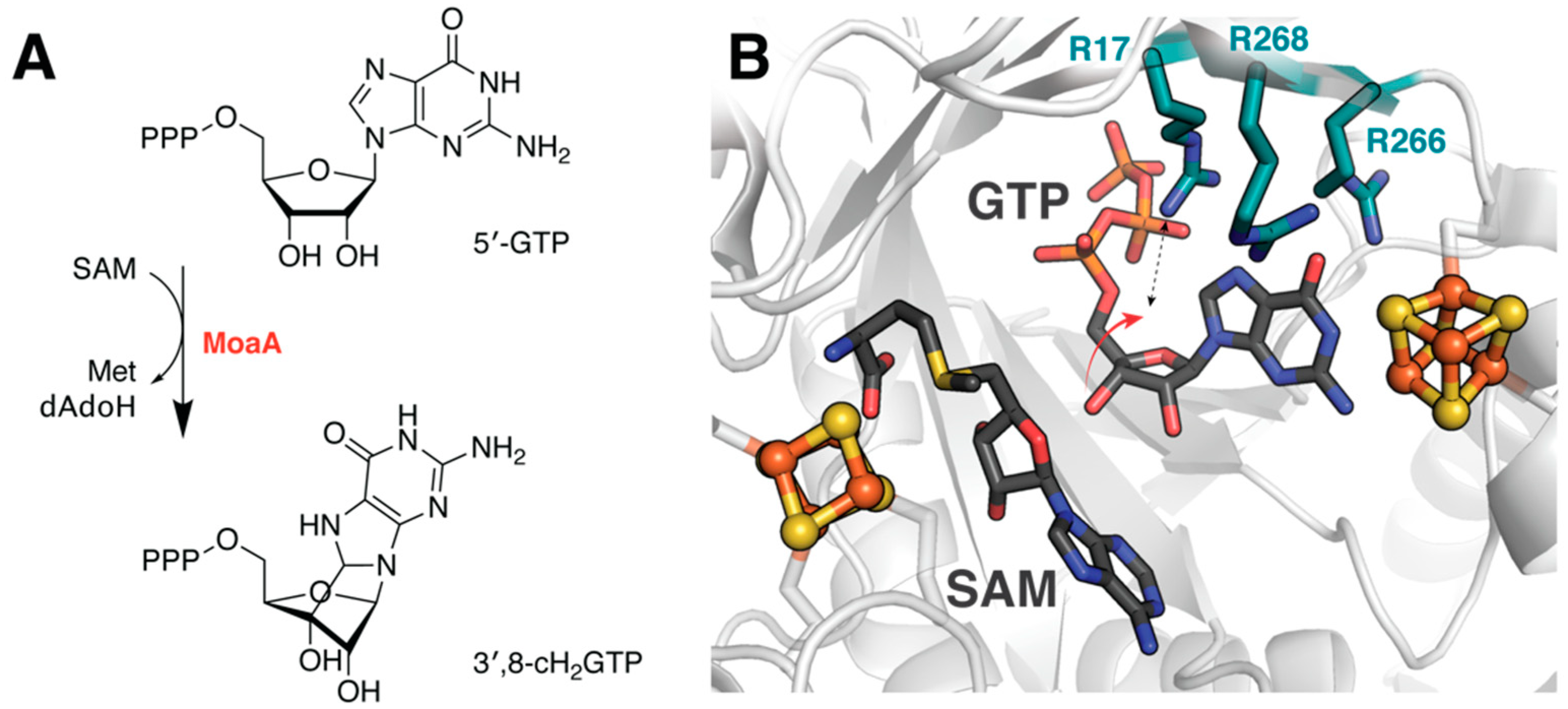
4.3. Determining Transition State Dynamics in MoaA
4.4. Beyond Dynamics
5. Hybrid Methods: Quantum Mechanics/Molecular Mechanics
5.1. Coordinated Changes to the Active Site in QueE
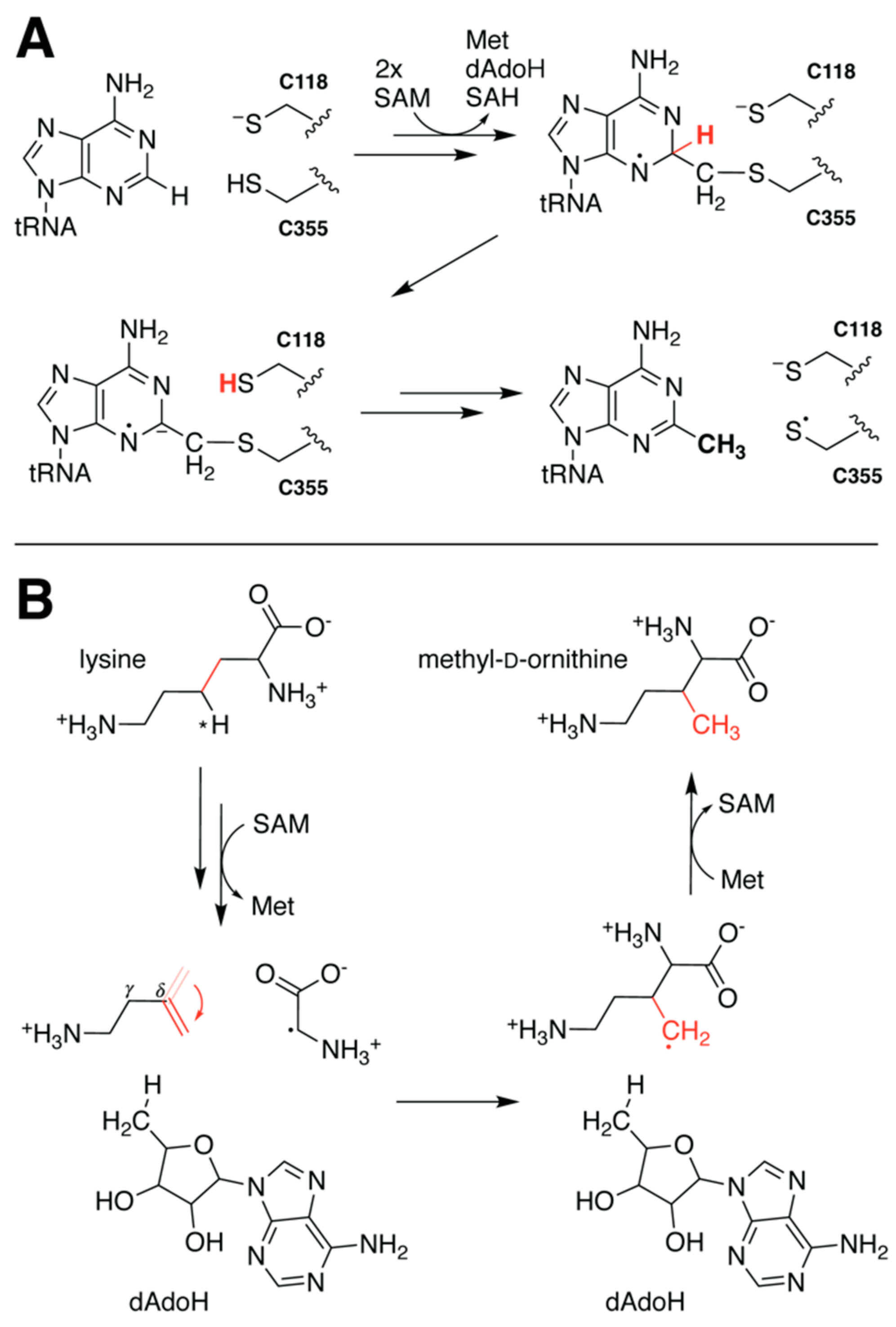
5.2. Proton Transfer Precedes Methyl Transfer in RlmN
5.3. Rotation of Intermediate Crucial for PylB Reactivity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Organism | PDB Code | Computational Method/s |
|---|---|---|---|
| TsrM | Kitasatospora cheerisanensis | 6WTE, 6WTF | D [12] |
| PFL-AE | Escherichia coli | 3C8F, 3CB8 | D [8] DFT [41] |
| SuiB | Streptococcus suis | 5V1Q | D [19] |
| Viperin | Mus musculus Trichoderma virens | 5VSL, 5VSM, 6B4C | D [27,28,29] MD [29,34] |
| QueE | Bacillus multivorans | 4NJI, 4NJK | D, MD, DFT, QM/MM [30,35,36] |
| MoaA | Staphylococcus aureus | 1TV8, 2FB3 | DFT [45] |
| HydE | Thermotoga maritima | 3CIW | DFT [50] |
| RlmN | Escherichia coli | 5HR6 | DFT, QM/MM [56,58] |
| PylB | Methanosarcina barkeri | 3T7V | QM/MM [61] |
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmidt, M. Time-resolved macromolecular crystallography at pulsed X-ray sources. Int. J. Mol. Sci. 2019, 20, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, H.N. Structure determination using X-ray free-electron laser pulses. Methods Mol. Biol. 2017, 1607, 295–324. [Google Scholar] [CrossRef]
- Holcomb, J.; Spellmon, N.; Zhang, Y.; Doughan, M.; Li, C.; Yang, Z. Protein crystallization: Eluding the bottleneck of X-ray crystallography. AIMS Biophys. 2017, 4, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Akiva, E.; Brown, S.; Almonacid, D.E.; Barber, A.E., 2nd; Custer, A.F.; Hicks, M.A.; Huang, C.C.; Lauck, F.; Mashiyama, S.T.; Meng, E.C.; et al. The Structure–Function Linkage Database. Nucleic Acids Res. 2013, 42, D521–D530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, J.B.; Duffus, B.R.; Duschene, K.S.; Shepard, E.M. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. [Google Scholar] [CrossRef] [PubMed]
- Frey, P.A.; Booker, S.J. Radical mechanisms of S-adenosylmethionine-dependent enzymes. Adv. Protein Chem. 2001, 58, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Sofia, H.J.; Chen, G.; Hetzler, B.G.; Reyes-Spindola, J.F.; Miller, N.E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: Functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vey, J.L.; Yang, J.; Li, M.; Broderick, W.E.; Broderick, J.B.; Drennan, C.L. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 16137–16141. [Google Scholar] [CrossRef] [Green Version]
- Dowling, D.P.; Vey, J.L.; Croft, A.K.; Drennan, C.L. Structural diversity in the AdoMet radical enzyme superfamily. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2012, 1824, 1178–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duschene, K.S.; Veneziano, S.E.; Silver, S.C.; Broderick, J.B. Control of radical chemistry in the AdoMet radical enzymes. Curr. Opin. Chem. Biol. 2009, 13, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Knox, H.L.; Chen, P.Y.-T.; Blaszczyk, A.J.; Mukherjee, A.; Grove, T.L.; Schwalm, E.L.; Wang, B.; Drennan, C.L.; Booker, S.J. Structural basis for non-radical catalysis by TsrM, a radical SAM methylase. Nat. Chem. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, T.; Zhou, P.; Floss, H.G. Formation of 2-methyltryptophan in the biosynthesis of thiostrepton: Isolation of S-adenosylmethionine: Tryptophan 2-methyltransferase. Arch. Biochem. Biophys. 1990, 278, 35–40. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, P.J.; Grove, T.L.; Sites, L.A.; McLaughlin, M.I.; Booker, S.J.; Drennan, C.L. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc. Natl. Acad. Sci. USA 2013, 201302417. [Google Scholar] [CrossRef] [Green Version]
- Knappe, J.; Schmitt, T. A novel reaction of S-adenosyl-L-methionine correlated with the activation of pyruvate formate-lyase. Biochem. Biophys. Res. Commun. 1976, 71, 1110–1117. [Google Scholar] [CrossRef]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.M.; Schramma, K.R.; Hansen, W.A.; Bacik, J.P.; Khare, S.D.; Seyedsayamdost, M.R.; Ando, N. Structures of the peptide-modifying radical SAM enzyme SuiB elucidate the basis of substrate recognition. Proc. Natl. Acad. Sci. USA 2017, 114, 10420–10425. [Google Scholar] [CrossRef] [Green Version]
- Schramma, K.R.; Seyedsayamdost, M.R. Lysine-tryptophan-crosslinked peptides produced by radical SAM enzymes in pathogenic streptococci. ACS Chem. Biol. 2017, 12, 922–927. [Google Scholar] [CrossRef]
- Zanghellini, A.; Jiang, L.; Wollacott, A.M.; Cheng, G.; Meiler, J.; Althoff, E.A.; Röthlisberger, D.; Baker, D. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006, 15, 2785–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatib, F.; Cooper, S.; Tyka, M.D.; Xu, K.; Makedon, I.; Popović, Z.; Baker, D.; Players, F. Algorithm discovery by protein folding game players. Proc. Natl. Acad. Sci. USA 2011, 108, 18949–18953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramma, K.R.; Forneris, C.C.; Caruso, A.; Seyedsayamdost, M.R. Mechanistic investigations of lysine–tryptophan cross-link formation catalyzed by streptococcal radical S-adenosylmethionine enzymes. Biochemistry 2018, 57, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Bernheim, A.; Millman, A.; Ofir, G.; Meitav, G.; Avraham, C.; Shomar, H.; Rosenberg, M.M.; Tal, N.; Melamed, S.; Amitai, G.; et al. Prokaryotic viperins produce diverse antiviral molecules. Nature 2021, 589, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Serrano, E.E.; Gizzi, A.S.; Arnold, J.J.; Grove, T.L.; Almo, S.C.; Cameron, C.E. Viperin reveals its true function. Annu. Rev. Virol. 2020, 7, 421–446. [Google Scholar] [CrossRef]
- Gizzi, A.S.; Grove, T.L.; Arnold, J.J.; Jose, J.; Jangra, R.K.; Garforth, S.J.; Du, Q.; Cahill, S.M.; Dulyaninova, N.G.; Love, J.D.; et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558, 610–614. [Google Scholar] [CrossRef]
- Mikulecky, P.; Andreeva, E.; Amara, P.; Weissenhorn, W.; Nicolet, Y.; Macheboeuf, P. Human viperin catalyzes the modification of GPP and FPP potentially affecting cholesterol synthesis. FEBS Lett. 2018, 592, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honarmand Ebrahimi, K.; Carr, S.B.; McCullagh, J.; Wickens, J.; Rees, N.H.; Cantley, J.; Armstrong, F.A. The radical-SAM enzyme Viperin catalyzes reductive addition of a 5′-deoxyadenosyl radical to UDP-glucose in vitro. FEBS Lett. 2017, 591, 2394–2405. [Google Scholar] [CrossRef] [Green Version]
- Honarmand Ebrahimi, K.; Rowbotham, J.S.; McCullagh, J.; James, W.S. Mechanism of diol dehydration by a promiscuous radical-SAM enzyme homologue of the antiviral enzyme Viperin (RSAD2). ChemBioChem 2020, 21, 1605–1612. [Google Scholar] [CrossRef]
- Suess, C.J.; Martins, F.L.; Croft, A.K.; Jäger, C.M. Radical stabilization energies for enzyme engineering: Tackling the substrate scope of the radical enzyme QueE. J. Chem. Inf. Model. 2019, 59, 5111–5125. [Google Scholar] [CrossRef]
- McCarty, R.M.; Somogyi, Á.; Lin, G.; Jacobsen, N.E.; Bandarian, V. The deazapurine biosynthetic pathway revealed: In vitro enzymatic synthesis of PreQ0 from guanosine 5′-triphosphate in four steps. Biochemistry 2009, 48, 3847–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Margreitter, C.; Oostenbrink, C. MDplot: Visualise Molecular Dynamics. R J. 2017, 9, 164–186. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Selvadurai, K.; Shahoei, R.; Lee, H.; Fatma, S.; Tajkhorshid, E.; Huang, R.H. Reconstitution and substrate specificity for isopentenyl pyrophosphate of the antiviral radical SAM enzyme viperin. J. Biol. Chem. 2018, 293, 14122–14133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Liu, Y. Ring contraction catalyzed by the metal-dependent radical SAM enzyme: 7-Carboxy-7-deazaguanine synthase from B. multivorans. Theoretical insights into the reaction mechanism and the influence of metal ions. ACS Catal. 2015, 5, 3953–3965. [Google Scholar] [CrossRef]
- Jäger, C.M.; Croft, A.K. Radical reaction control in the AdoMet radical enzyme CDG synthase (QueE): Consolidate, destabilize, accelerate. Chem. Eur. J. 2017, 23, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Burke, K. Perspective on density functional theory. Int. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. RMP 2015, 87, 897–923. [Google Scholar] [CrossRef] [Green Version]
- Walsby, C.J.; Ortillo, D.; Broderick, W.E.; Broderick, J.B.; Hoffman, B.M. An anchoring role for FeS clusters: Chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe− 4S] cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 2002, 124, 11270–11271. [Google Scholar] [CrossRef]
- Walsby, C.J.; Ortillo, D.; Yang, J.; Nnyepi, M.R.; Broderick, W.E.; Hoffman, B.M.; Broderick, J.B. Spectroscopic approaches to elucidating novel iron−sulfur chemistry in the “radical-SAM” protein superfamily. Inorg. Chem. 2005, 44, 727–741. [Google Scholar] [CrossRef]
- Dey, A.; Peng, Y.; Broderick, W.E.; Hedman, B.; Hodgson, K.O.; Broderick, J.B.; Solomon, E.I. S K-edge XAS and DFT calculations on SAM dependent pyruvate formate-lyase activating enzyme: Nature of interaction between the Fe4S4 cluster and SAM and its role in reactivity. J. Am. Chem. Soc. 2011, 133, 18656–18662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolet, Y.; Amara, P.; Mouesca, J.-M.; Fontecilla-Camps, J.C. Unexpected electron transfer mechanism upon AdoMet cleavage in radical SAM proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 14867–14871. [Google Scholar] [CrossRef] [Green Version]
- Impano, S.; Yang, H.; Jodts, R.J.; Pagnier, A.; Swimley, R.; McDaniel, E.C.; Shepard, E.M.; Broderick, W.E.; Broderick, J.B.; Hoffman, B.M. Active-site controlled, Jahn–Teller enabled regioselectivity in reductive S–C bond cleavage of S-adenosylmethionine in radical SAM enzymes. J. Am. Chem. Soc. 2021, 143, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Hover, B.M.; Loksztejn, A.; Ribeiro, A.A.; Yokoyama, K. Identification of a cyclic nucleotide as a cryptic intermediate in molybdenum cofactor biosynthesis. J. Am. Chem. Soc. 2013, 135, 7019–7032. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Lilla, E.A.; Zhang, P.; Zhang, D.; Shields, T.P.; Scott, L.G.; Yang, W.; Yokoyama, K. Mechanism of rate acceleration of radical C–C bond formation reaction by a radical SAM GTP 3′,8-cyclase. J. Am. Chem. Soc. 2020, 142, 9314–9326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Y.; Chen, D.; Yu, Y.; Duan, L.; Shen, B.; Liu, W. Radical-mediated enzymatic carbon chain fragmentation-recombination. Nat. Chem. Biol. 2011, 7, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Qianzhu, H.; Ji, W.; Ji, X.; Chu, L.; Guo, C.; Lu, W.; Ding, W.; Gao, J.; Zhang, Q. Reactivity of the nitrogen-centered tryptophanyl radical in the catalysis by the radical SAM enzyme NosL. Chem. Commun. 2017, 53, 344–347. [Google Scholar] [CrossRef]
- Molle, T.; Moreau, Y.; Clemancey, M.; Forouhar, F.; Ravanat, J.-L.; Duraffourg, N.; Fourmond, V.; Latour, J.-M.; Gambarelli, S.; Mulliez, E.; et al. Redox behavior of the S-adenosylmethionine (SAM)-binding Fe–S cluster in methylthiotransferase RimO, toward understanding dual SAM activity. Biochemistry 2016, 55, 5798–5808. [Google Scholar] [CrossRef]
- Tao, L.; Pattenaude, S.A.; Joshi, S.; Begley, T.P.; Rauchfuss, T.B.; Britt, R.D. Radical SAM enzyme HydE generates adenosylated Fe(I) intermediates en route to the [FeFe]-hydrogenase catalytic H-cluster. J. Am. Chem. Soc. 2020, 142, 10841–10848. [Google Scholar] [CrossRef]
- Hioe, J.; Zipse, H. Hydrogen transfer in SAM-mediated enzymatic radical reactions. Chem. Eur. J. 2012, 18, 16463–16472. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- Bruender, N.A.; Grell, T.A.J.; Dowling, D.P.; McCarty, R.M.; Drennan, C.L.; Bandarian, V. 7-Carboxy-7-deazaguanine synthase: A radical S-adenosyl-L-methionine enzyme with polar tendencies. J. Am. Chem. Soc. 2017, 139, 1912–1920. [Google Scholar] [CrossRef]
- Yan, F.; LaMarre, J.M.; Röhrich, R.; Wiesner, J.; Jomaa, H.; Mankin, A.S.; Fujimori, D.G. RlmN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J. Am. Chem. Soc. 2010, 132, 3953–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grove, T.L.; Benner, J.S.; Radle, M.I.; Ahlum, J.H.; Landgraf, B.J.; Krebs, C.; Booker, S.J. A radically different mechanism for S-adenosylmethionine–dependent methyltransferases. Science 2011, 332, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, X.; Wang, E.; Li, B. Quantum chemistry studies of adenosine 2503 methylation by S-adenosylmethionine-dependent enzymes. Int. J. Quantum Chem. 2013, 113, 1409–1415. [Google Scholar] [CrossRef]
- Schwalm, E.L.; Grove, T.L.; Booker, S.J.; Boal, A.K. Crystallographic capture of a radical S-adenosylmethionine enzyme in the act of modifying tRNA. Science 2016, 352, 309–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Dong, L.; Liu, Y. A QM/MM study of the catalytic mechanism of SAM methyltransferase RlmN from Escherichia coli. Proteins Struct. Funct. Bioinform. 2017, 85, 1967–1974. [Google Scholar] [CrossRef]
- Gaston, M.A.; Zhang, L.; Green-Church, K.B.; Krzycki, J.A. The complete biosynthesis of the genetically encoded amino acid pyrrolysine from lysine. Nature 2011, 471, 647–650. [Google Scholar] [CrossRef] [Green Version]
- Quitterer, F.; List, A.; Eisenreich, W.; Bacher, A.; Groll, M. Crystal structure of methylornithine synthase (PylB): Insights into the pyrrolysine biosynthesis. Angew. Chem. Int. Ed. 2012, 51, 1339–1342. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, Y.; Zhang, R. QM/MM study of the conversion mechanism of lysine to methylornithine catalyzed by methylornithine synthase (PylB). Theor. Chem. Acc. 2013, 132, 1–8. [Google Scholar] [CrossRef]
- Holliday, G.L.; Akiva, E.; Meng, E.C.; Brown, S.D.; Calhoun, S.; Pieper, U.; Sali, A.; Booker, S.J.; Babbitt, P.C. Atlas of the radical SAM superfamily: Divergent evolution of function using a “plug and play” domain. Methods Enzymol. 2018, 606, 1–71. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blue, T.C.; Davis, K.M. Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics. Molecules 2021, 26, 2590. https://doi.org/10.3390/molecules26092590
Blue TC, Davis KM. Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics. Molecules. 2021; 26(9):2590. https://doi.org/10.3390/molecules26092590
Chicago/Turabian StyleBlue, Tamra C., and Katherine M. Davis. 2021. "Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics" Molecules 26, no. 9: 2590. https://doi.org/10.3390/molecules26092590
APA StyleBlue, T. C., & Davis, K. M. (2021). Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics. Molecules, 26(9), 2590. https://doi.org/10.3390/molecules26092590