
Predicting Accurate Lead Structures for Screening Molecular Libraries: A Quantum Crystallographic Approach


Abstract
:
1. Introduction
2. Results
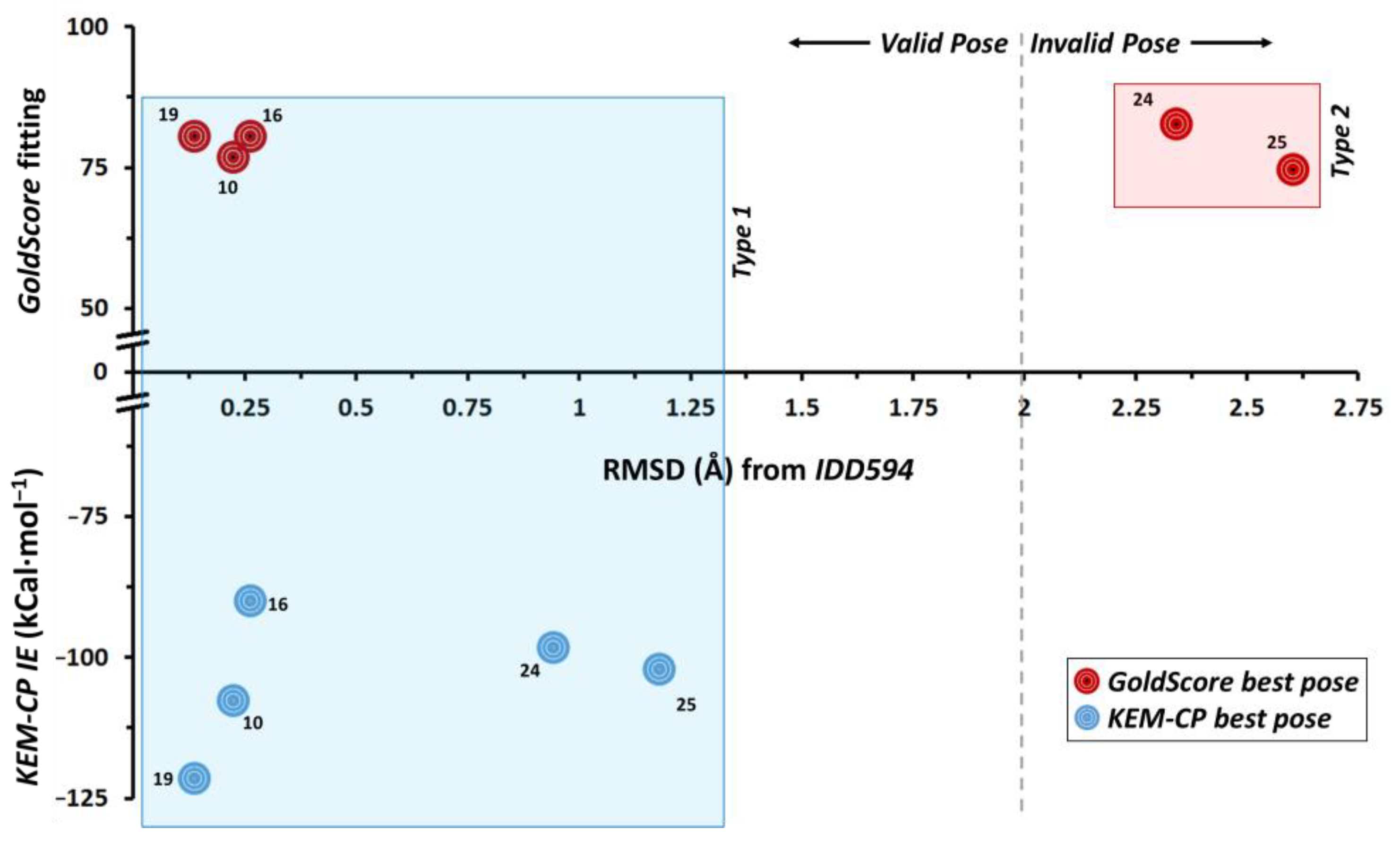
2.1. Case of hAR-IDD594
2.2. Case Study of CDK2-NU6102
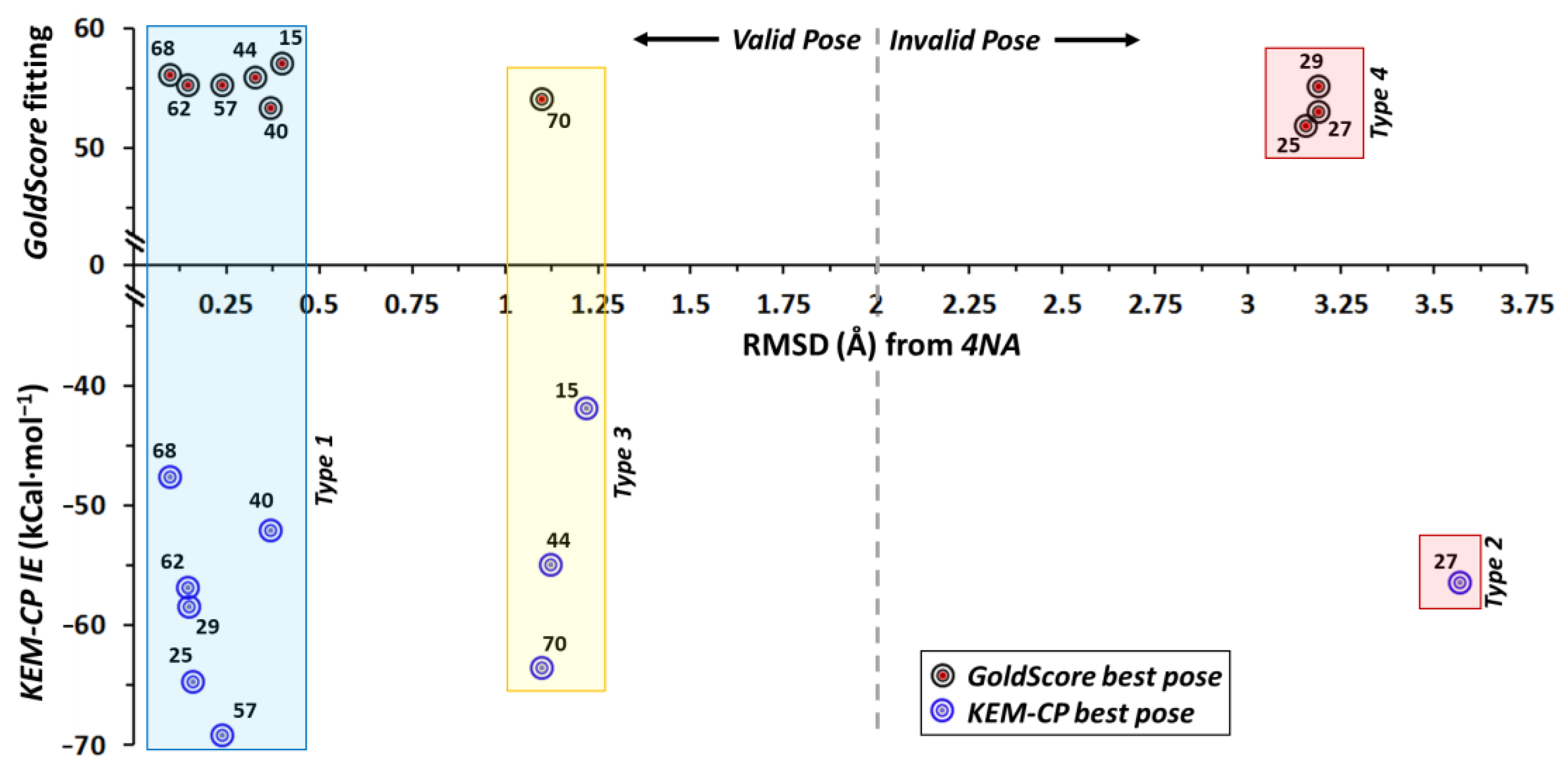
2.3. Case of ERβ-4NA
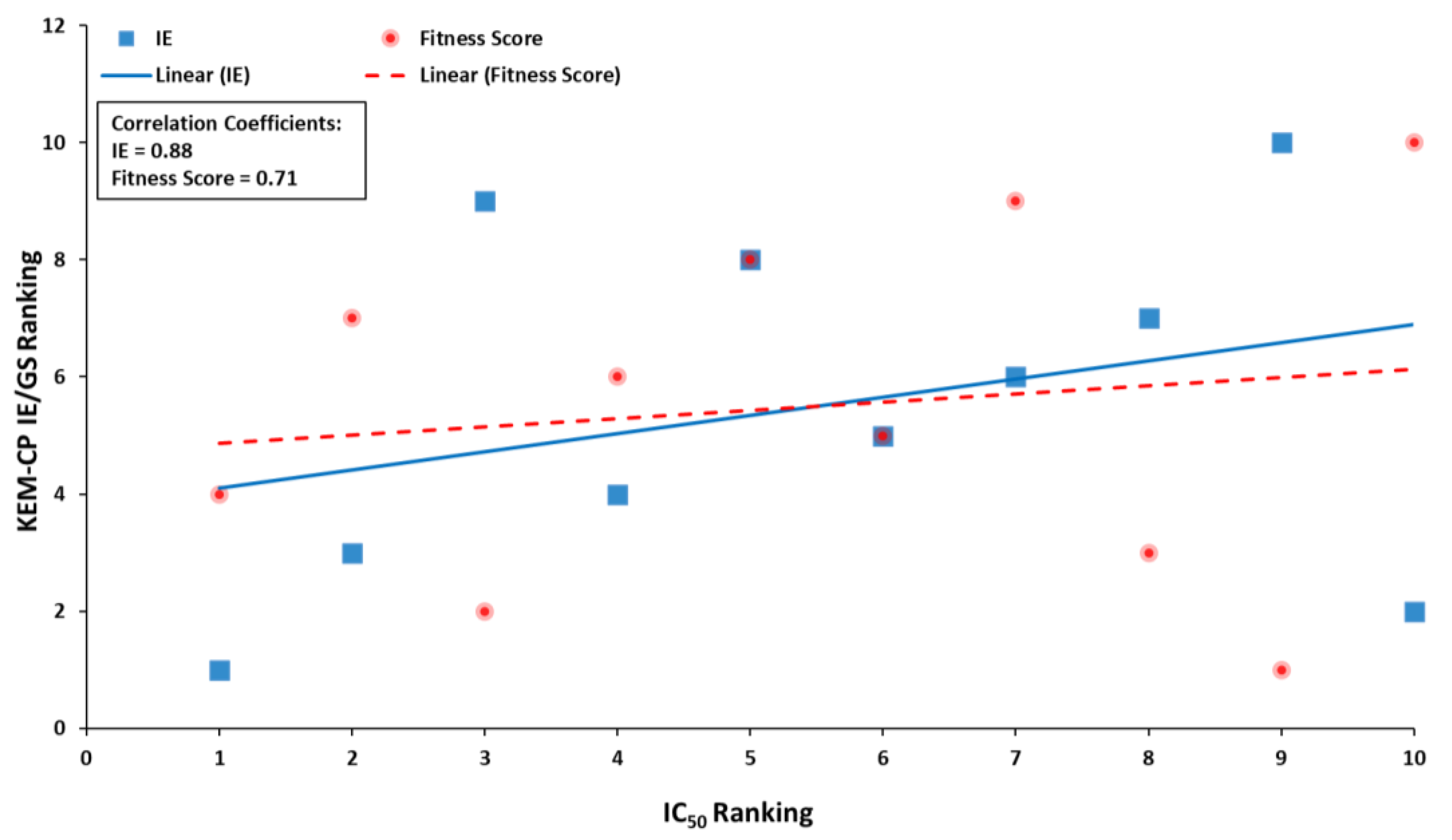
3. Discussion
4. Materials and Methods
4.1. Selection of Complex Structures and Preparation of the Targets and Their Ligands
- (1)
- hAR-IDD594: The ultra-high resolution (0.66 Å) complex structure was previously considered by some of us for studying protein-ligand interactions and for benchmarking KEM-CP approach against MOE scoring function [43]. Here, once again, we consider this complex structure with moderately hydrophobic active site environment for benchmarking KEM-CP approach against GoldScore.
- (2)
- CDK2-NU6102: This standard resolution (2.0 Å) complex structure has a hydrophilic environment in its active site and previous report [28] suggests that GoldScore provides better results for such systems. Therefore, we select this system to check the superiority of KEM-CP over GoldScore.
- (3)
- ERβ-4NA: This complex structure consists of a hydrophobic (or lyophilic) active site and reported at a low resolution of 2.7 Å. As reported earlier, GoldScore fails to rank potent ligand accurately for proteins with hydrophobic environments [28] (Table 1) and hence IE study on such a system provides us an opportunity to test the potentiality of KEM-CP approach.
- For hAR, we select five ligands (including IDD594) with similar scaffolds (Scheme S1, Supplementary Materials) as reported by Ferrari et al. [7].
- For CDK2, seven ligands (Scheme S2, Supplementary Materials) with best experimental IC50 values are chosen from the study by Hardcastle et al. [48]. Despite having lower IC50 value the ligand 33 is retained in the list because it is an isomer of NU-6102 (with sulphonamide substitution on phenyl ring at the meta position instead of the ortho position). This provides an additional opportunity to explore the applicability of KEM-CP approach for the isomers.
- For ERβ, although Mewshaw et al. [49] have studied ~70 ligands with IC50 values ranging from 2.0 μM–0.5 nM, an IC50 cut-off of 3.0 nM resulted in 24 ligands, out of which we select 10 ligands to include various kinds of functional groups (Scheme S3, Supplementary Materials) in our analysis.
4.2. Scoring Function and Docking Studies
4.3. KEM-CP Interaction Energy Calculation and Kernel Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gohlke, H.; Klebe, G. Approaches to the Description and Prediction of the Binding Affinity of Small-Molecule Ligands to Macromolecular Receptors. Angew. Chem. Int. Ed. 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Gilson, M.K.; Zhou, H.-X. Calculation of Protein-Ligand Binding Affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, C.R.W.; Boger, A.D.L.; Jorgensen, W.L. Elucidation of Fatty Acid Amide Hydrolase Inhibition by Potent α-Ketoheterocycle Derivatives from Monte Carlo Simulations. J. Am. Chem. Soc. 2005, 127, 17377–17384. [Google Scholar] [CrossRef] [PubMed]
- Simonson, T.; Archontis, G.; Karplus, M. Free Energy Simulations Come of Age: Protein−Ligand Recognition. Acc. Chem. Res. 2002, 35, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães, C.R.W.; Cardozo, M. MM-GB/SA Rescoring of Docking Poses in Structure-Based Lead Optimization. J. Chem. Inf. Model. 2008, 48, 958–970. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Degliesposti, G.; Sgobba, M.; Rastelli, G. Validation of an Automated Procedure for the Prediction of Relative Free Energies of Binding on a Set of Aldose Reductase Inhibitors. Bioorganic Med. Chem. 2007, 15, 7865–7877. [Google Scholar] [CrossRef]
- Barreiro, G.; Guimarães, C.R.W.; Tubert-Brohman, I.; Lyons, T.M.; Tirado-Rives, J.; Jorgensen, W.L. Search for Non-Nucleoside Inhibitors of HIV-1 Reverse Transcriptase Using Chemical Similarity, Molecular Docking, and MM-GB/SA Scoring. J. Chem. Inf. Model. 2007, 47, 2416–2428. [Google Scholar] [CrossRef] [Green Version]
- Fidelak, J.; Juraszek, J.; Branduardi, D.; Bianciotto, M.; Gervasio, F.L. Free-Energy-Based Methods for Binding Profile Determination in a Congeneric Series of CDK2 Inhibitors. J. Phys. Chem. B 2010, 114, 9516–9524. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-Based Scoring Function to Predict Protein-Ligand Interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. An Iterative Knowledge-Based Scoring Function to Predict Protein–Ligand Interactions: II. Validation of the Scoring Function. J. Comput. Chem. 2006, 27, 1876–1882. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B.O. A Machine Learning Approach to Predicting Protein–Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Olson, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Leaver-Fay, A.; Tyka, M.D.; Davis, I.W.; Cooper, S.; Treuille, A.; Mandell, D.J.; Richter, F.; Ban, Y.-E.A.; Fleishman, S.J.; Corn, J.E.; et al. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [CrossRef] [Green Version]
- Davis, I.W.; Baker, D. Rosetta Ligand Docking with Full Ligand and Receptor Flexibility. J. Mol. Biol. 2009, 385, 381–392. [Google Scholar] [CrossRef]
- Sotriffer, C.A. Accounting for Induced-Fit Effects in Docking: What is Possible and What is Not? Curr. Top. Med. Chem. 2011, 11, 179–191. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring Functions and their Evaluation Methods for Protein-Ligand Docking: Recent Advances and Future Directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Tiikkainen, P.; Markt, P.; Wolber, G.; Kirchmair, J.; Distinto, S.; Poso, A.; Kallioniemi, O. Critical Comparison of Virtual Screening Methods against the MUV Data Set. J. Chem. Inf. Model. 2009, 49, 2168–2178. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef]
- Morrone, J.A.; Weber, J.K.; Huynh, T.; Luo, H.; Cornell, W.D. Combining Docking Pose Rank and Structure with Deep Learning Improves Protein–Ligand Binding Mode Prediction over a Baseline Docking Approach. J. Chem. Inf. Model. 2020, 60, 4170–4179. [Google Scholar] [CrossRef] [Green Version]
- Gomes, P.D.S.F.C.; Da Silva, F.; Bret, G.; Rognan, D. Ranking Docking Poses by Graph Matching of Protein-Ligand Interactions: Lessons Learned from the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2017, 32, 75–87. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Cuzzolin, A.; Bolcato, G.; Sturlese, M.; Moro, S. A Deep-Learning Approach toward Rational Molecular Docking Protocol Selection. Molecules 2020, 25, 2487. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Hu, Y.; Wang, Z.; Zhang, X.; Pang, J.; Wang, G.; Zhong, H.; Xu, L.; Cao, D.; Hou, T. Beware of the Generic Machine Learning-Based Scoring Functions in Structure-Based Virtual Screening. Brief. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative Evaluation of 11 Scoring Functions for Molecular Docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef]
- Xu, W.; Lucke, A.J.; Fairlie, D.P. Comparing Sixteen Scoring Functions for Predicting Biological Activities of Ligands for Protein Targets. J. Mol. Graph. Model. 2015, 57, 76–88. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Massa, L.; Karle, J. Kernel Energy Method Illustrated with Peptides. Int. J. Quantum Chem. 2005, 103, 808–817. [Google Scholar] [CrossRef]
- Řezáč, J.; Salahub, D.R. Multilevel Fragment-Based Approach (MFBA): A Novel Hybrid Computational Method for the Study of Large Molecules. J. Chem. Theory Comput. 2009, 6, 91–99. [Google Scholar] [CrossRef]
- Massa, L.; Matta, C.F. Quantum Crystallography: A Perspective. J. Comput. Chem. 2017, 39, 1021–1028. [Google Scholar] [CrossRef]
- Huang, L.; Massa, L.; Karle, J. Kernel Energy Method: Application to DNA. Biochemistry 2005, 44, 16747–16752. [Google Scholar] [CrossRef]
- Huang, L.; Massa, L.; Karle, J. The Kernel Energy Method: Application to a tRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 1233–1237. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Massa, L.; Karle, J. Kernel Energy Method: Application to Insulin. Proc. Natl. Acad. Sci. USA 2005, 102, 12690–12693. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Massa, L. Quantum Kernel Applications in Medicinal Chemistry. Future Med. Chem. 2012, 4, 1479–1494. [Google Scholar] [CrossRef]
- Huang, L.; Massa, L.; Karle, J. Drug Target Interaction Energies by the Kernel Energy Method in Aminoglycoside Drugs and Ribosomal A Site RNA Targets. Proc. Natl. Acad. Sci. USA 2007, 104, 4261–4266. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A Direct MP2 Gradient Method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 Energy Evaluation by Direct Methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does Basis Set Superposition Error Change the Potential Surfaces for Hydrogen-Bonded Dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
- Halkier, A.; Klopper, W.; Helgaker, T.; Jorgensen, P.; Taylor, P.R. Basis Set Convergence of the Interaction Energy of Hydrogen-Bonded Complexes. J. Chem. Phys. 1999, 111, 9157–9167. [Google Scholar] [CrossRef]
- Brauer, B.; Kesharwani, M.K.; Martin, J.M.L. Some Observations on Counterpoise Corrections for Explicitly Correlated Calculations on Noncovalent Interactions. J. Chem. Theory Comput. 2014, 10, 3791–3799. [Google Scholar] [CrossRef]
- Mandal, S.K.; Saha, P.; Munshi, P.; Sukumar, N. Exploring Potent Ligand for Proteins: Insights from Knowledge-Based Scoring Functions and Molecular Interaction Energies. Struct. Chem. 2017, 28, 1537–1552. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical Scoring Functions: I. The Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible Docking using Tabu Search and an Empirical Estimate of Binding Affinity. Proteins Struct. Funct. Bioinform. 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Hardcastle, I.R.; Arris, C.E.; Jewsbury, P.; Menyerol, J.; Mesguiche, V.; Newell, D.R.; Noble, M.E.M.; Pratt, D.J.; Wang, A.L.-Z.; Whitfield†, H.J.; et al. N2-SubstitutedO6-Cyclohexylmethylguanine Derivatives: Potent Inhibitors of Cyclin-Dependent Kinases 1 and 2. J. Med. Chem. 2004, 47, 3710–3722. [Google Scholar] [CrossRef]
- Mewshaw, R.E.; Edsall, R.J.; Yang, C.; Manas, E.S.; Xu, Z.B.; Henderson, R.A.; Keith, J.C.; Harris, H.A. ERβ Ligands. 3. Exploiting Two Binding Orientations of the 2-Phenylnaphthalene Scaffold to Achieve ERβ Selectivity. J. Med. Chem. 2005, 48, 3953–3979. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of theCCP4 Suite and Current Developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Complexes | hAR-IDD594 | CDK2-NU6102 | ERβ-4NA |
|---|---|---|---|
| PDB ID | 1US0 | 1H1S | 1YY4 |
| Resolution | 0.66 Å | 2.0 Å | 2.7 Å |
| SiteMap Score [47] * | |||
| Hydrophobic | 3.0 | 1.4 | 4.4 |
| Hydrophilic | 0.7 | 1.0 | 0.3 |
| Balance ** | 4.2 | 1.4 | 13.3 |
| Ligand # | Experimental IC50 (nM) | Pose Type 1 | Pose Type 2 | ||||
|---|---|---|---|---|---|---|---|
| Avg. IE (kCal·mol−1) | GoldScore | Avg. IE (kCal·mol−1) | GoldScore | ||||
| Fitness Score | Rank | Fitness Score | Rank | ||||
| 10 | 176 | −107.67 | 76.93 | 1 | 2.35 | 58.51 | 19 |
| 16 | 44 | −89.93 | 80.56 | 1 | −18.68 | 72.97 | 4 |
| 19 | 30 | −121.11 | 87.08 | 1 | 18.24 | 76.64 | 2 |
| 24 | 7 | −98.39 | 77.90 | 2 | −33.42 | 82.73 | 1 |
| 25 | 6 | −100.45 | 73.76 | 2 | −19.97 | 74.75 | 1 |
| Ligand # | Pose Type 1 | Pose Type 2 | ||
|---|---|---|---|---|
| Avg. IE (kCal·mol−1) | RMSD Crystal Geometry (Å) | Avg. IE (kCal·mol−1) | RMSD Crystal Geometry (Å) | |
| Predicted pose superimposed on crystal geometry (grey) |  |  | ||
| 10 | −107.67 | 0.223 | 2.35 | 2.042 |
| 16 | −89.93 | 0.263 | −18.68 | 2.563 |
| 19 | −121.11 | 0.137 | 18.24 | 2.128 |
| 24 | −98.39 | 0.943 | −33.42 | 2.342 |
| 25 | −100.45 | 1.180 | −19.97 | 2.603 |
| Ligand # | Experimental IC50 (nM) | Pose Type 1 | Pose Type 2 | Pose Type 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Avg. IE (kCal·mol−1) | GoldScore | Avg. IE (kCal·mol−1) | GoldScore | Avg. IE (kCal·mol−1) | GoldScore | |||||
| Fitness Score | Rank | Fitness Score | Rank | Fitness Score | Rank | |||||
| 3 | 5.4 ± 1.0 | −434.26 | 67.87 | 1 | −422.08 | 63.07 | 2 | −299.76 | 60.32 | 3 |
| 25 | 69 ± 1 | −304.97 | 62.98 | 1 | −258.26 | 59.74 | 2 | −219.28 | 55.47 | 3 |
| 28 * | 7.0 ± 0.1 | −433.67 | 68.88 | 1 | - | - | - | −334.16 | 56.93 | 10 |
| 29 | 56 ± 20 | −434.11 | 73.24 | 1 | −376.89 | 59.73 | 4 | −376.57 | 55.92 | 11 |
| 30 | 63 ± 7 | −398.53 | 66.33 | 1 | −498.35 | 66.16 | 3 | −362.61 | 54.99 | 12 |
| 33 | 210 ± 40 | −432.88 | 66.71 | 1 | −414.07 | 63.00 | 4 | −317.21 | 56.66 | 14 |
| 34 | 64 ± 33 | −418.00 | 64.60 | 1 | −334.12 | 59.39 | 2 | Failed | 55.37 | 6 |
| Ligand # | Pose Type 1 | Pose Type 2 | Pose Type 3 | |||
|---|---|---|---|---|---|---|
| Avg. IE (kCal·mol−1) | RMSD with Crystal Geometry (Å) | Avg. IE (kCal·mol−1) | RMSD with Crystal Geometry (Å) | Avg. IE (kCal·mol−1) | RMSD with Crystal Geometry (Å) | |
| Predicted pose superimposed on crystal geometry (grey) |  |  |  | |||
| 3 | −434.26 | 0.832 | −422.08 | 4.072 | −299.76 | 3.212 |
| 25 | −304.97 | 0.944 | −258.26 | 4.379 | −219.28 | 2.947 |
| 28 | −433.67 | 0.844 | - | - | −334.16 | 3.221 |
| 29 | −434.11 | 0.798 | −376.89 | 3.797 | −376.57 | 2.752 |
| 30 | −398.53 | 0.811 | −498.35 | 3.454 | −362.61 | 2.950 |
| 33 | −432.88 | 0.567 | −414.07 | 3.680 | −317.21 | 2.908 |
| 34 | −418.00 | 0.975 | −334.12 | 3.330 | Failed | 2.875 |
| Ligand # | Experimental IC50 (nM) | Pose Type 1 | Pose Type 2 | Pose Type 3 | Pose Type 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Avg. IE (kCal·mol−1) | GoldScore | Avg. IE (kCal·mol−1) | GoldScore | Avg. IE (kCal·mol−1) | GoldScore | Avg. IE (kCal·mol−1) | GoldScore | ||||||
| Fitness Score | Rank | Fitness Score | Rank | Fitness Score | Rank | Fitness Score | Rank | ||||||
| 15 | 2.52 ± 1.3 | −40.33 | 57.10 | 1 | −30.44 | 52.58 | 3 | −41.90 | 55.11 | 2 | −25.86 | 51.85 | 4 |
| 25 | 2.8 ± 0.1 | −64.69 | 50.76 | 3 | −54.59 | 49.80 | 6 | −57.96 | 50.73 | 4 | −48.19 | 51.87 | 1 |
| 27 | 2.3 ± 0.1 | −49.90 | 49.68 | 7 | −56.42 | 50.17 | 3 | −43.79 | 50.02 | 5 | −50.99 | 53.05 | 1 |
| 29 | 1.4 ± 0.6 | −58.45 | 52.28 | 3 | −37.37 | 51.88 | 5 | −42.65 | 51.99 | 4 | −50.74 | 55.15 | 1 |
| 40 | 1.6 ± 0.7 | −52.07 | 53.38 | 1 | −52.03 | 49.54 | 7 | −49.17 | 52.11 | 3 | −41.80 | 52.68 | 2 |
| 44 | 2.3 ± 1.7 | −53.64 | 55.86 | 1 | −42.58 | 49.53 | 5 | −54.89 | 54.53 | 3 | −24.37 | 52.34 | 4 |
| 57 | 0.5 ± 0.5 | −69.15 | 55.29 | 1 | −36.44 | 47.02 | 9 | −51.64 | 52.72 | 3 | −21.68 | 49.41 | 5 |
| 62 | 2.1 ± 0.9 | −56.79 | 55.28 | 1 | −26.68 | 53.51 | 5 | −40.83 | 55.12 | 3 | −38.80 | 49.52 | 11 |
| 68 | 1.2 ± 0.7 | −47.56 | 56.13 | 1 | −2.31 | 47.19 | 9 | −39.92 | 55.18 | 3 | −19.58 | 45.38 | 11 |
| 70 | 1.1 ± 1.6 | −47.88 | 53.93 | 2 | −38.95 | 51.50 | 7 | −63.55 | 54.13 | 1 | −28.36 | 50.32 | 10 |
| Ligand # | Pose Type 1 | Pose Type 2 | Pose Type 3 | Pose Type 4 | ||||
|---|---|---|---|---|---|---|---|---|
| Avg. IE (kCal·mol−1) | RMSD (Å) | Avg. IE (kCal·mol−1) | RMSD (Å) | Avg. IE (kCal·mol−1) | RMSD (Å) | Avg. IE (kCal·mol−1) | RMSD (Å) | |
| Predicted pose superimposed on crystal geometry (grey) |  |  |  |  | ||||
| 15 | −40.33 | 0.399 | −30.44 | 3.526 | −41.90 | 1.219 | −25.86 | 3.185 |
| 25 | −64.69 | 0.161 | −54.59 | 3.430 | −57.96 | 1.100 | −48.19 | 3.155 |
| 27 | −49.90 | 0.276 | −56.42 | 3.570 | −43.79 | 1.027 | −50.99 | 3.189 |
| 29 | −58.45 | 0.149 | −37.37 | 3.390 | −42.65 | 0.978 | −50.74 | 3.190 |
| 40 | −52.07 | 0.371 | −52.03 | 3.452 | −49.17 | 1.044 | −41.80 | 3.193 |
| 44 | −53.64 | 0.329 | −42.58 | 3.515 | −54.89 | 1.122 | −24.37 | 3.221 |
| 57 | −69.15 | 0.239 | −36.44 | 3.499 | −51.64 | 1.117 | −21.68 | 3.227 |
| 62 | −56.79 | 0.147 | −26.68 | 3.420 | −40.83 | 1.040 | −38.80 | 3.380 |
| 68 | −47.56 | 0.099 | −2.31 | 3.393 | −39.92 | 0.992 | −19.58 | 3.255 |
| 70 | −47.88 | 0.153 | −38.95 | 3.147 | −63.55 | 1.100 | −28.36 | 3.434 |
| Protein | Active Site Environment | No. of Ligands | No. of Ligands Predicted Correctly By | % of Ligands Predicted Correctly By | ||
|---|---|---|---|---|---|---|
| GoldScore | KEM-CP | GoldScore | KEM-CP | |||
| hAR | Moderately hydrophobic | 5 | 3 | 5 | 60 | 100 |
| CDK2 | Highly hydrophilic | 7 | 7 | 6 | 100 | 86 |
| ERβ | Highly hydrophobic | 10 | 7 | 9 | 70 | 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, S.K.; Munshi, P. Predicting Accurate Lead Structures for Screening Molecular Libraries: A Quantum Crystallographic Approach. Molecules 2021, 26, 2605. https://doi.org/10.3390/molecules26092605
Mandal SK, Munshi P. Predicting Accurate Lead Structures for Screening Molecular Libraries: A Quantum Crystallographic Approach. Molecules. 2021; 26(9):2605. https://doi.org/10.3390/molecules26092605
Chicago/Turabian StyleMandal, Suman Kumar, and Parthapratim Munshi. 2021. "Predicting Accurate Lead Structures for Screening Molecular Libraries: A Quantum Crystallographic Approach" Molecules 26, no. 9: 2605. https://doi.org/10.3390/molecules26092605
APA StyleMandal, S. K., & Munshi, P. (2021). Predicting Accurate Lead Structures for Screening Molecular Libraries: A Quantum Crystallographic Approach. Molecules, 26(9), 2605. https://doi.org/10.3390/molecules26092605