
First Determination of Glycidyl Ester Species in Edible Oils by Reverse-Phase Ultra-Performance Liquid Chromatography Coupled with an Evaporative Light-Scattering Detector


Abstract
:1. Introduction
2. Results and Discussion
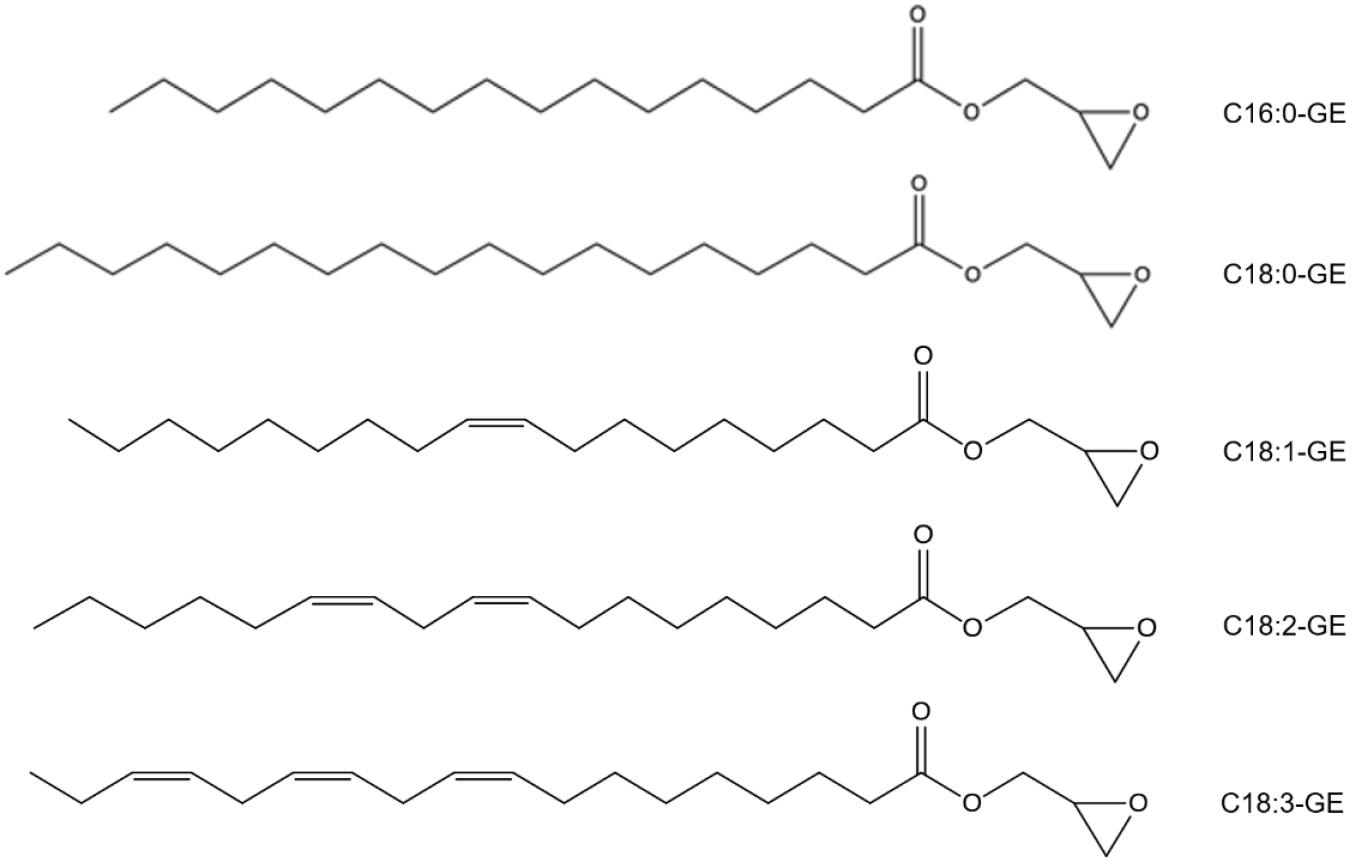
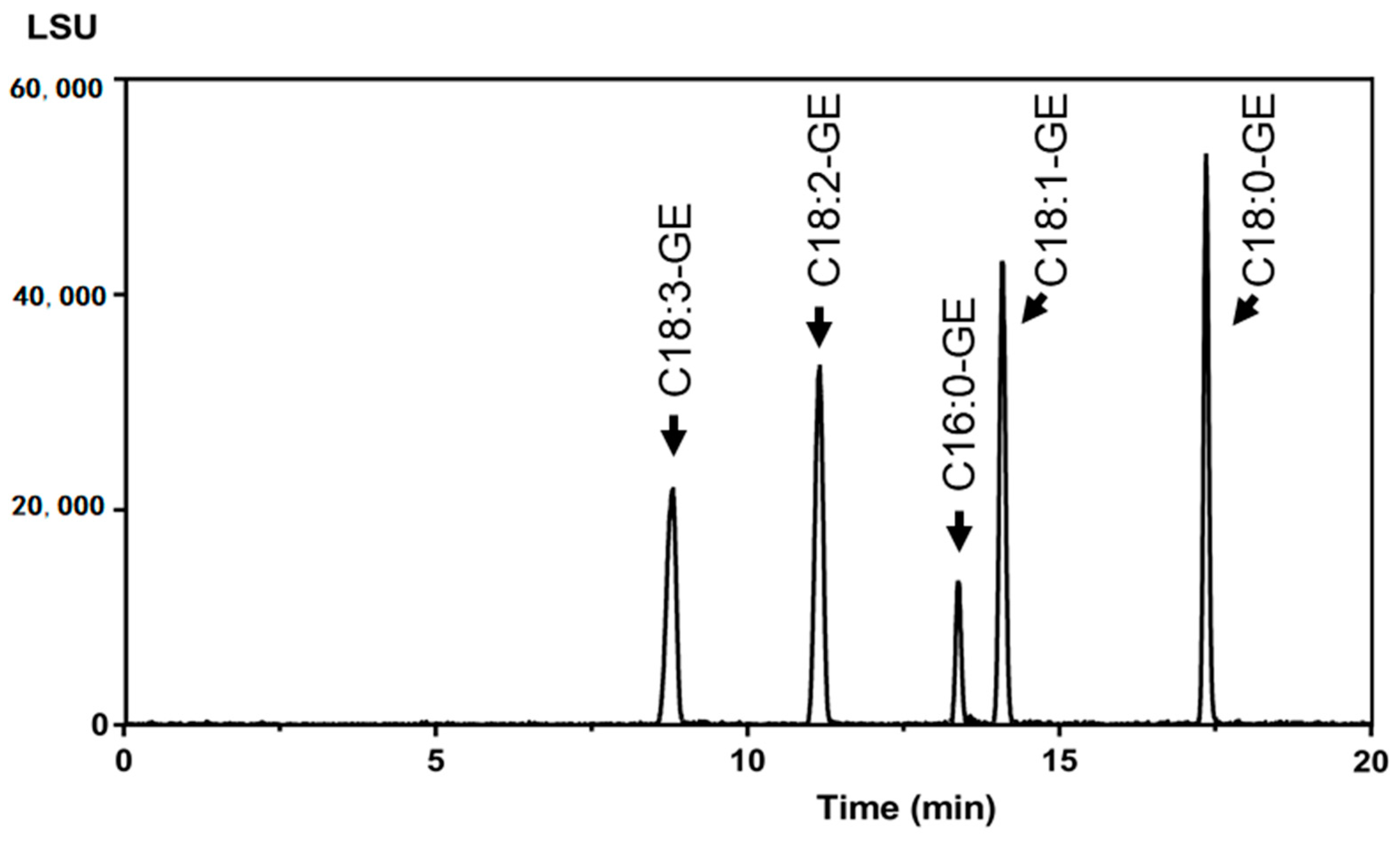
2.1. Development of the UPLC-ELSD Method
2.2. Validation of the UPLC-ELSD Method
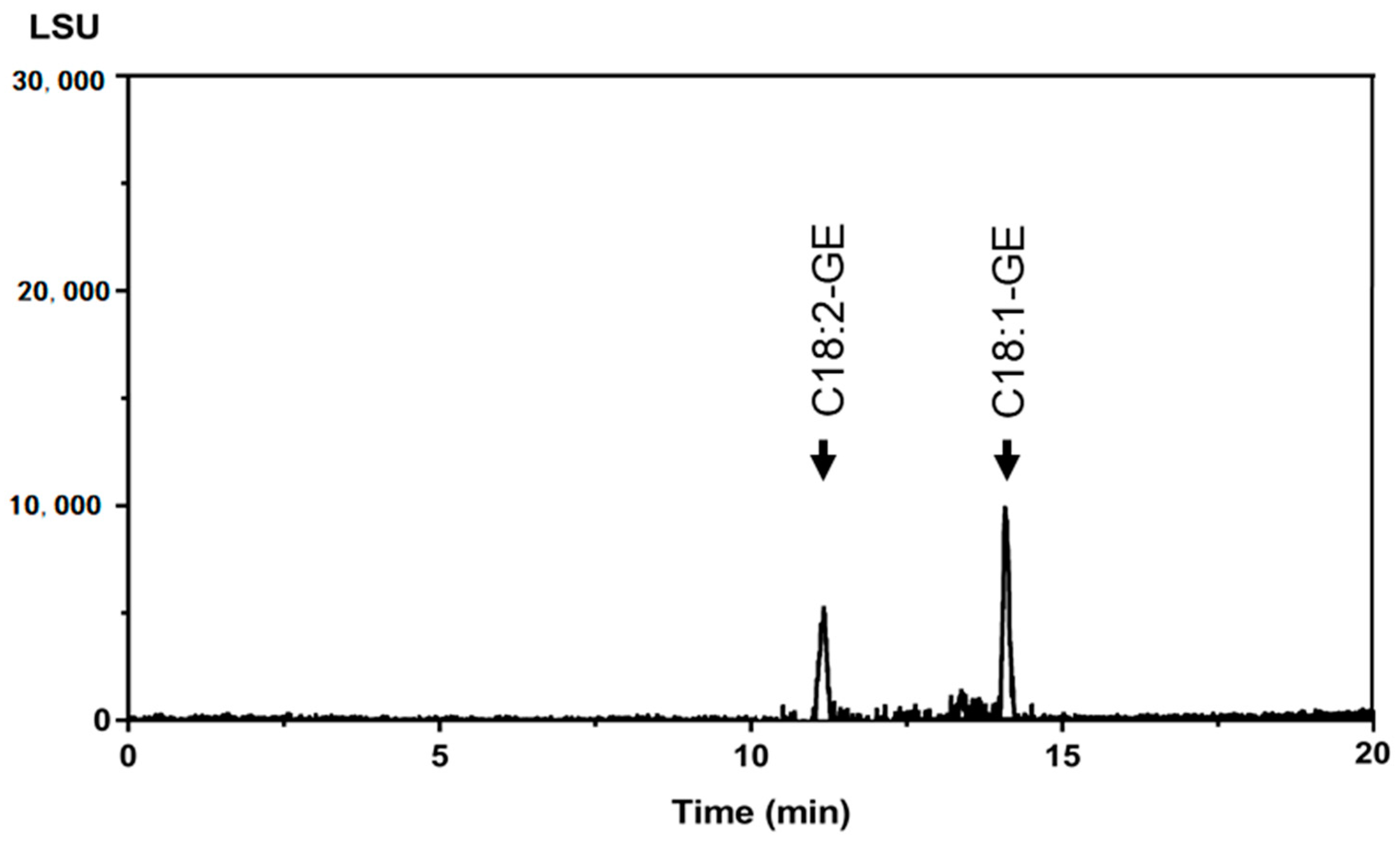
2.3. Sample Analysis by the UPLC-ELSD Method and GC-MS Method
3. Materials and Methods
3.1. Materials and Chemicals
3.2. UPLC Instrumentation and Chromatographic Conditions
3.3. Oil Sample Pretreatment
3.4. Method Validation
3.5. GE Determination by GC-MS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zelinkova, Z.; Svejkovska, B.; Velisek, J.; Dolezal, M. Fatty acid esters of 3-chloropropane-1,2-diol in edible oils. Food Addit. Contam. 2006, 23, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Masukawa, Y.; Shiro, H.; Nakamura, S.; Kondo, N.; Jin, N.; Suzuki, N.; Ooi, N.; Kudo, N. A new analytical method for the quantification of glycidol fatty acid esters in edible oils. J. Oleo Sci. 2010, 59, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisshaar, R.; Perz, R. Fatty acid esters of glycidol in refined fats and oils. Eur. J. Lipid Sci. Technol. 2010, 112, 158–165. [Google Scholar] [CrossRef]
- Cheng, W.W.; Liu, G.Q.; Wang, L.Q.; Liu, Z.S. Glycidyl fatty acid esters in refined edible oils: A review on formation, occurrence, analysis, and elimination methods. Compr. Rev. Food Sci. Food Saf. 2017, 16, 263–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.W.; Liu, G.Q.; Liu, X.Q. Formation of glycidyl fatty acid esters both in real edible oils during laboratory-scale refining and in chemical model during high temperature exposure. J. Agric. Food Chem. 2016, 64, 5919–5927. [Google Scholar] [CrossRef] [PubMed]
- Matthaus, B.; Pudel, F.; Fehling, P.; Vosmann, K.; Freudenstein, A. Strategies for the reduction of 3-MCPD esters and related compounds in vegetable oils. Eur. J. Lipid Sci. Technol. 2011, 113, 380–386. [Google Scholar] [CrossRef]
- Craft, B.D.; Nagy, K.; Seefelder, W.; Dubois, M.; Destaillats, F. Glycidyl esters in refined palm (Elaeis guineensis) oil and related fractions. Part II: Practical recommendations for effective mitigation. Food Chem. 2012, 132, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Destaillats, F.; Craft, B.D.; Dubois, M.; Nagy, K. Glycidyl esters in refined palm (Elaeis guineensis) oil and related fractions. Part I: Formation mechanism. Food Chem. 2012, 131, 1391–1398. [Google Scholar] [CrossRef]
- Kamikata, K.; Vicente, E.; Arisseto-Bragotto, A.P.; Miguel, A.M.R.D.; Milani, R.F.; Tfouni, S.A.V. Occurrence of 3-MCPD, 2-MCPD and glycidyl esters in extra virgin olive oils, olive oils and oil blends and correlation with identity and quality parameters. Food Control 2019, 95, 135–141. [Google Scholar] [CrossRef]
- MacMahon, S.; Mazzola, E.; Begley, T.H.; Diachenko, G.W. Analysis of processing contaminants in edible oils. Part 1. Liquid chromatography−tandem mass spectrometry method for the direct detection of 3-monochloropropanediol monoesters and glycidyl esters. J. Agric. Food Chem. 2013, 61, 4737–4747. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; Glycidol: LYON, France, 2000; pp. 469–486. [Google Scholar]
- Scholz, G.; Schilter, B. Toxicological Properties of Glycidyl Esters. In Processing Contaminants in Edible Oils. MCPD and Glycidyl Esters; MacMahon, S., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Chapter 6; pp. 169–182. [Google Scholar]
- European Union (EU). Commission Regulation (EU) 2018/290 of 26 February 2018. Off. J. Eur. Union 2018, L55, 27–29. [Google Scholar]
- Albuquerque, T.G.; Costa, H.S.; Silva, M.A. Are chloropropanols and glycidyl fatty acid esters a matter of concern in palm oil? Trends Food Sci. Technol. 2019, 105, 494–514. [Google Scholar] [CrossRef]
- American Oil Chemists’ Society. Official Methods and Recommended Practices of the AOCS, 7th ed.; American Oil Chemists’ Society: Urbana, IL, USA, 2017. [Google Scholar]
- Hori, K.; Koriyama, N.; Omori, H.; Kuriyama, M.; Arishima, T.; Tsumura, K. Simultaneous determination of 3-MCPD fatty acid esters and glycidol fatty acid esters in edible oils using liquid chromatography time-of-flight mass spectrometry. LWT-Food Sci. Technol. 2012, 48, 204–208. [Google Scholar] [CrossRef]
- Blumhorst, M.R.; Collison, M.W.; Cantrill, R.; Shiro, H.; Masukawa, Y.; Kawai, S.; Yasunaga, K. Collaborative study for the analysis of glycidyl fatty acid esters in edible oils using LC-MS. J. Am. Oil Chem. Soc. 2013, 90, 493–500. [Google Scholar] [CrossRef]
- Aniolowska, M.; Kita, A. The effect of frying on glycidyl esters content in palm oil. Food Chem. 2016, 203, 95–103. [Google Scholar] [CrossRef]
- Becalski, A.; Feng, S.; Lau, B.P.Y.; Zhao, T. A pilot survey of 2-and 3-monochloropropanediol and glycidol fatty acid esters in foods on the Canadian market 2011–2013. J. Food Compos. Anal. 2015, 37, 58–66. [Google Scholar] [CrossRef]
- Haines, T.D.; Adlaf, K.J.; Pierceall, R.M.; Lee, I.; Venkitasubramanian, P.; Collison, M.W. Direct determination of MCPD fatty acid esters and glycidyl fatty acid esters in vegetable oils by LC-TOFMS. J. Am. Oil Chem. Soc. 2011, 88, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hrncirik, K.; Zelinkova, Z.; Ermacora, A. Critical factors of indirect determination of 3-chloropropane-1,2-diol esters. Eur. J. Lipid Sci. Technol. 2011, 113, 361–367. [Google Scholar] [CrossRef]
- Varache, M.; Ciancone, M.; Couffin, A.C. Development and validation of a novel UPLC-ELSD method for the assessment of lipid composition of nanomedicine formulation. Int. J. Pharmaceut. 2019, 566, 11–23. [Google Scholar] [CrossRef]
- Lee, W.J.; Su, N.W.; Lee, M.H.; Lin, J.T. Assessment of authenticity of sesame oil by modified Villavecchia Test and HPLC-ELSD analysis of triacylglycerol profile. Food Res. Int. 2013, 53, 195–202. [Google Scholar] [CrossRef]
- Lee, W.J.; Weng, S.H.; Su, N.W. Individual phosphatidylcholine species analysis by RP-HPLC-ELSD for determination of polyenylphosphatidylcholine in lecithins. J. Agric. Food Chem. 2015, 63, 3851–3858. [Google Scholar] [CrossRef]
- Rombaut, R.; De Clercq, N.; Foubert, I.; Dewettinck, K. Triacylglycerol analysis of fats and oils by evaporative light scattering detection. J. Am. Oil Chem. Soc. 2009, 86, 19–25. [Google Scholar] [CrossRef]
- Costa, P.P.K.G.; Mendes, T.D.; Salum, T.F.C.; Pacheco, T.F.; Braga, S.C.; de Almeida, J.R.M.; Goncalves, S.B.; Damaso, M.C.T.; Rodrigues, C.M. Development and validation of HILIC-UHPLC-ELSD methods for determination of sugar alcohols stereoisomers and its application for bioconversion processes of crude glycerin. J. Chromatogr. A 2019, 1589, 56–64. [Google Scholar] [CrossRef]
- Taiwan Food and Drug Administration (TFDA). Guidelines for the Validation of Chemical Methods for Foods. 2013. Available online: https://www.fda.gov.tw/tc/includes/GetFile.ashx?id=f636921436165222394 (accessed on 20 June 2019).
- Taiwan Food and Drug Administration (TFDA). Fatty Acid Composition of Edible Oils and Fats. 2011. Available online: https://www.fda.gov.tw/upload/133/2017012016382977055.xls (accessed on 30 June 2019).

{kind=link}
{kind=link}
{kind=link}
| Condition | 1 | 2 | 3 | 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stationary phase | CSH C18 column 2.1 × 100 mm, 1.7 µm | BEH C18 column 2.1 × 100 mm, 1.7 µm | BEH C18 column 2.1 × 150 mm, 1.7 μm | BEH C18 column 2.1 × 150 mm, 1.7 μm | ||||||||
| Mobile phase | Time (min) | A: ACN | B: IPA | Time (min) | A: ACN | B: IPA | Time (min) | A: 85% MeOH | B: 97.5% MeOH | Time (min) | A: 85% MeOH | B: 2.5% MeOH |
| 0 | 89% | 11% | 0 | 89% | 11% | 0 | 15% | 85% | 0 | 10% | 90% | |
| 5.5 | 62.5% | 37.5% | 5.5 | 62.5% | 37.5% | 9.6 | 10% | 90% | 9.6 | 12.5% | 87.5% | |
| 5.51 | 10% | 90% | 5.51 | 10% | 90% | 19.2 | 0% | 100% | 19.2 | 15% | 85% | |
| 6.51 | 89% | 11% | 6.51 | 89% | 11% | 22 | 0% | 100% | 22 | 15% | 85% | |
| 7.5 | 89% | 11% | 7.5 | 89% | 11% | 25 | 15% | 85% | 25 | 10% | 90% | |
| GE Species | Retention Time (min) | |||
|---|---|---|---|---|
| Condition 1 | Condition 2 | Condition 3 | Condition 4 | |
| C16:0-GE | 1.77 | 2.59 | 4.9 | 12.7 |
| C18:0-GE | 2.19 | 3.24 | 6.9 | 16.6 |
| C18:1-GE | 1.73 | 2.56 | 5.1 | 13.3 |
| C18:2-GE | 1.45 | 2.14 | 4.0 | 10.4 |
| C18:3-GE | 1.26 | 1.87 | 3.3 | 8.3 |
| GE Species | Equation of Regression Curve | R2 | Instrumental LOD (µg/mL) | Instrumental LOQ (µg/mL) |
|---|---|---|---|---|
| C16:0-GE | y = 2.52x2 − 102.06x + 939.78 | 0.9999 | 2.4 | 8.0 |
| C18:0-GE | y = 3.43x2 − 61.48x + 523.09 | 1.0000 | 1.7 | 5.0 |
| C18:1-GE | y = 4.02x2 − 85.29x + 612.33 | 1.0000 | 1.7 | 5.0 |
| C18:2-GE | y = 4.53x2 − 114.32x + 912.71 | 0.9999 | 1.7 | 5.0 |
| C18:3-GE | y = 3.41x2 − 84.44x + 653.53 | 0.9999 | 1.7 | 5.0 |
| Accuracy | Nominal Concentration (µg/mL) | Analyte | ||||
|---|---|---|---|---|---|---|
| C16:0-GE | C18:0-GE | C18:1-GE | C18:2-GE | C18:3-GE | ||
| Day 1a | 80 | 99.5 | 104.4 | 102.2 | 101.2 | 101.7 |
| 50 | 98.1 | 104.9 | 102.1 | 101.7 | 101.9 | |
| 40 | 102.5 | 107.1 | 104.6 | 105.3 | 106.2 | |
| Day 1b | 80 | 100.2 | 104.9 | 102.3 | 101.1 | 101.3 |
| 50 | 97.2 | 105.5 | 102.0 | 101.5 | 102.2 | |
| 40 | 102.7 | 107.3 | 104.6 | 105.4 | 106.0 | |
| Intra-day precision | Mean, µg/mL, n = 6 | 100.0 | 105.7 | 102.9 | 102.7 | 103.2 |
| CV, %, n = 6 | 2.1 | 1.1 | 1.1 | 1.8 | 2.0 | |
| Day 2 | 80 | 103.5 | 94.3 | 84.2 | 102.1 | 102.2 |
| 50 | 99.9 | 91.8 | 81.3 | 99.2 | 98.7 | |
| 40 | 102.1 | 93.4 | 82.8 | 100.7 | 100.9 | |
| Day 3 | 80 | 93.4 | 93.4 | 97.7 | 96.8 | 96.5 |
| 50 | 91.4 | 91.4 | 97.4 | 96.9 | 96.6 | |
| 40 | 92.8 | 92.8 | 95.6 | 95.8 | 95.7 | |
| Inter-day precision | Mean, µg/mL, n = 12 | 98.6 | 99.3 | 96.4 | 100.7 | 100.8 |
| CV, %, n = 12 | 4.0 | 6.6 | 8.6 | 2.9 | 3.3 | |
| Intermediate Accuracy | Nominal Concentration (µg/mL) | Analyte | ||||
|---|---|---|---|---|---|---|
| C16:0-GE | C18:0-GE | C18:1-GE | C18:2-GE | C18:3-GE | ||
| 40 | 91.6 ± 11.7 | 90.9 ± 10.9 | 88.3 ± 10.3 | 93.0 ± 5.2 | 93.6 ± 6.3 | |
| Intermediate precision | CV, %, n = 5 | 12.4 | 12.0 | 11.6 | 5.6 | 6.7 |
| 20 | 94.2 ± 8.6 | 107.8 ± 4.3 | 103.3 ± 1.5 | 106.8 ± 2.3 | 101.2 ± 3.1 | |
| Intermediate precision | CV, %, n = 5 | 9.1 | 4.0 | 1.5 | 2.1 | 3.0 |
| 1 Repeatability | CV, %, n = 10 | 2.1 | 2.0 | 2.3 | 2.6 | 2.5 |
| Oil Sample | GC-MS Method | UPLC-ELSD Method | Relative Percent Difference (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Glycidol Equivalent (µg/g) | C16:0-GE (µg/g) | C18:0-GE (µg/g) | C18:1-GE (µg/g) | C18:2-GE (µg/g) | C18:3-GE (µg/g) | Glycidol Equivalent (µg/g) | ||
| Palm oil (a) | 1.63 ± 0.14 | 5.87 ± 0.33 | <LOQ | <LOQ | <LOQ | <LOQ | 1.41 ± 0.08 | 14.47 |
| Palm oil (b) | 5.71 ± 0.94 | 17.42 ± 2.01 | <LOQ | 3.93 ± 0.68 | <LOQ | <LOQ | 5.03 ± 0.53 | 12.66 |
| Rice bran oil | 2.96 ± 0.13 | <LOQ | <LOQ | 7.85 ± 0.11 | 6.07 ± 0.07 | <LOQ | 2.64 ± 0.25 | 11.42 |
| Sunflower oil | 0.12 ± 0.02 | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | NA | NA |
| Safflower oil | 0.36 ± 0.02 | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | NA | NA |
| Canola oil | 0.29 ± 0.03 | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | NA | NA |
| Extra virgin olive oil | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | <LOQ | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, P.-Y.; Chen, H.; Su, N.-W.; Chiou, T.-Y.; Lee, W.-J. First Determination of Glycidyl Ester Species in Edible Oils by Reverse-Phase Ultra-Performance Liquid Chromatography Coupled with an Evaporative Light-Scattering Detector. Molecules 2021, 26, 2702. https://doi.org/10.3390/molecules26092702
Wu P-Y, Chen H, Su N-W, Chiou T-Y, Lee W-J. First Determination of Glycidyl Ester Species in Edible Oils by Reverse-Phase Ultra-Performance Liquid Chromatography Coupled with an Evaporative Light-Scattering Detector. Molecules. 2021; 26(9):2702. https://doi.org/10.3390/molecules26092702
Chicago/Turabian StyleWu, Ping-Yi, Hsuan Chen, Nan-Wei Su, Tai-Ying Chiou, and Wei-Ju Lee. 2021. "First Determination of Glycidyl Ester Species in Edible Oils by Reverse-Phase Ultra-Performance Liquid Chromatography Coupled with an Evaporative Light-Scattering Detector" Molecules 26, no. 9: 2702. https://doi.org/10.3390/molecules26092702
APA StyleWu, P.-Y., Chen, H., Su, N.-W., Chiou, T.-Y., & Lee, W.-J. (2021). First Determination of Glycidyl Ester Species in Edible Oils by Reverse-Phase Ultra-Performance Liquid Chromatography Coupled with an Evaporative Light-Scattering Detector. Molecules, 26(9), 2702. https://doi.org/10.3390/molecules26092702