
A New Approach for Controlling Mesoporosity in Activated Carbon by the Consecutive Process of Air Oxidation, Thermal Destruction of Surface Functional Groups, and Carbon Activation (the OTA Method)



Abstract
:1. Introduction
2. Results and Discussion
2.1. Thermal Decomposition Behavior of Longan Seed Precursor
2.2. Proximate and Ultimate Analyses of Longan Seed Precursor and Activated Carbon
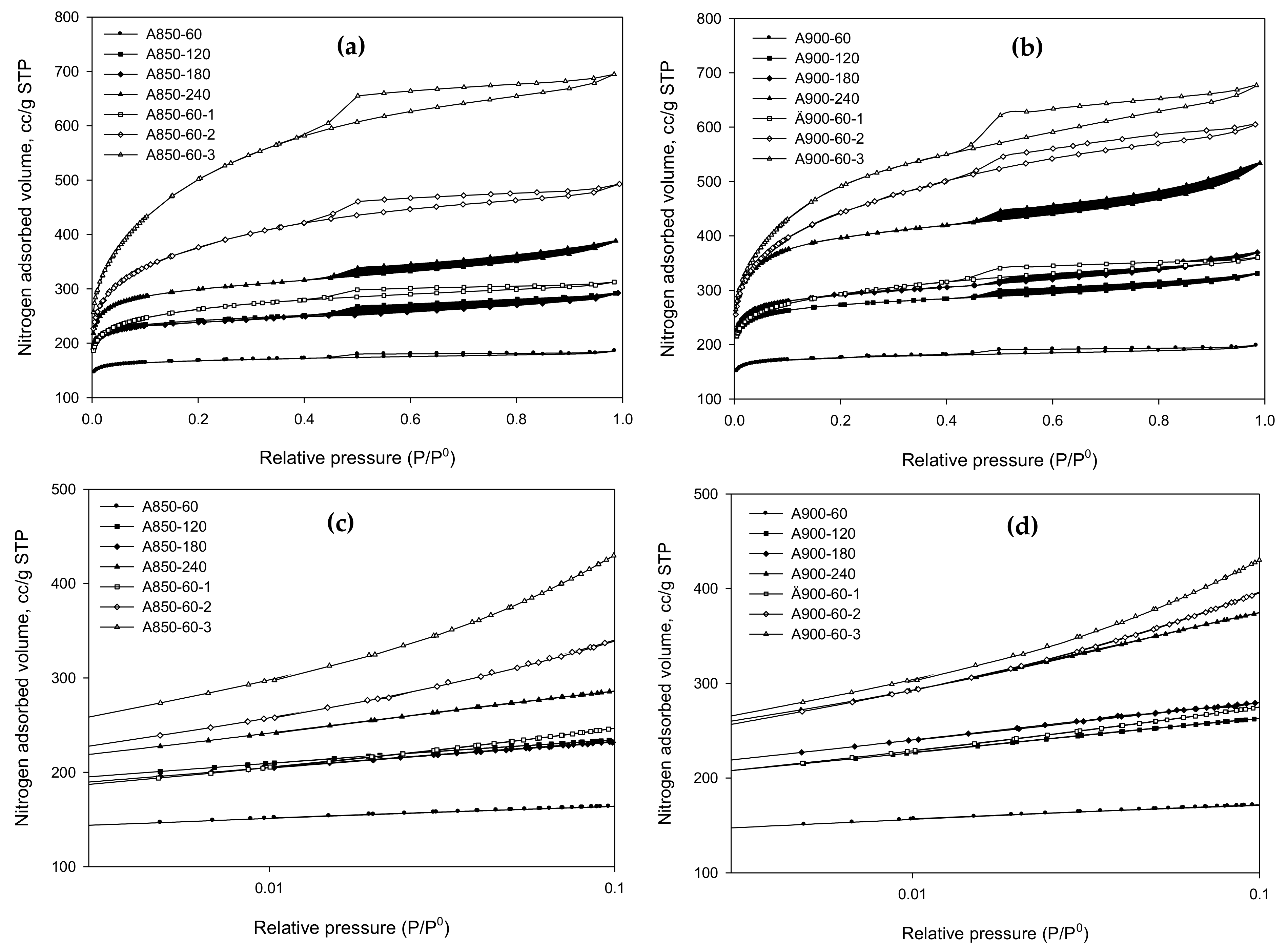
2.3. N2 Isotherms of Prepared Activated Carbons
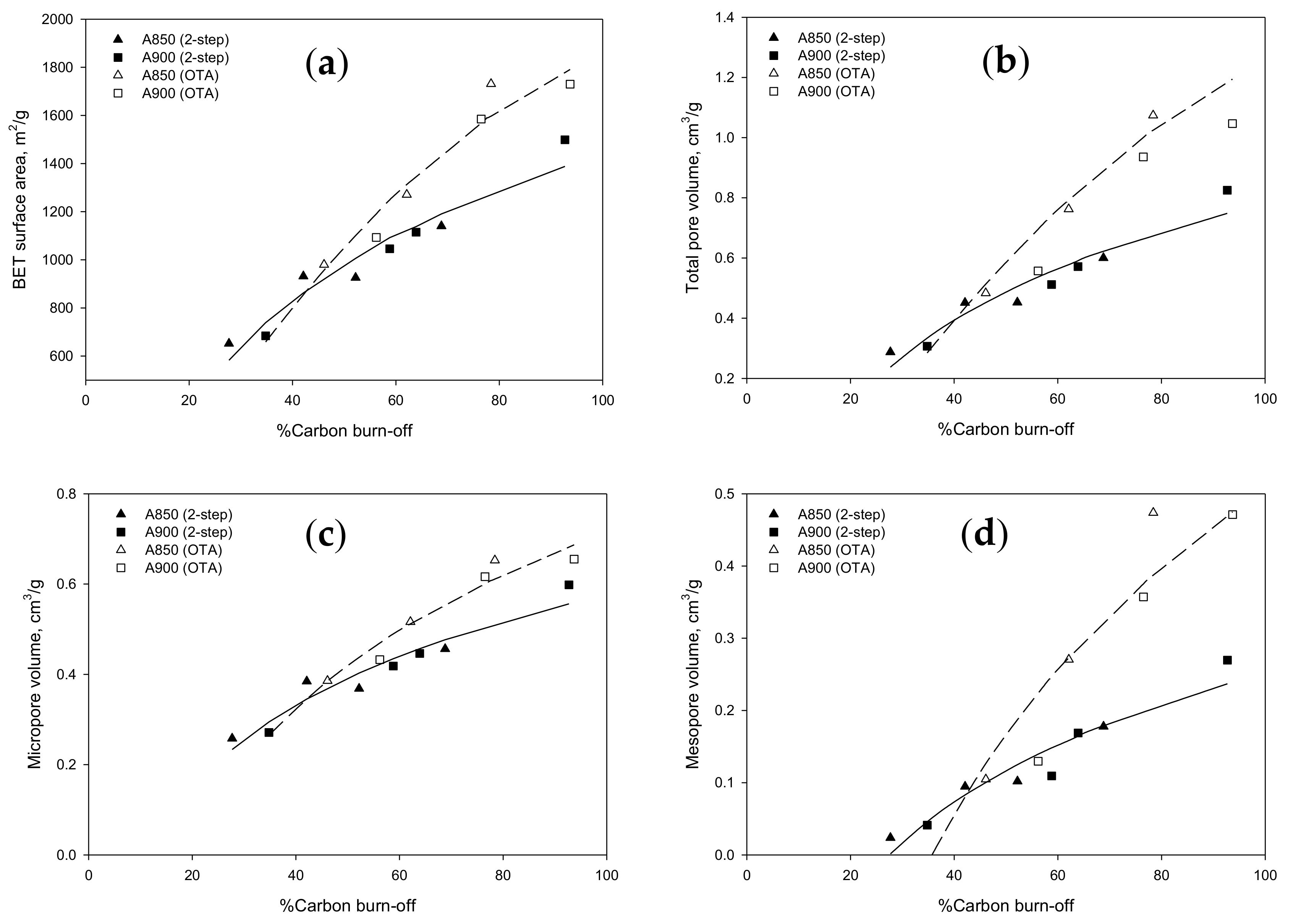
2.4. Porous Properties of Prepared Activated Carbons
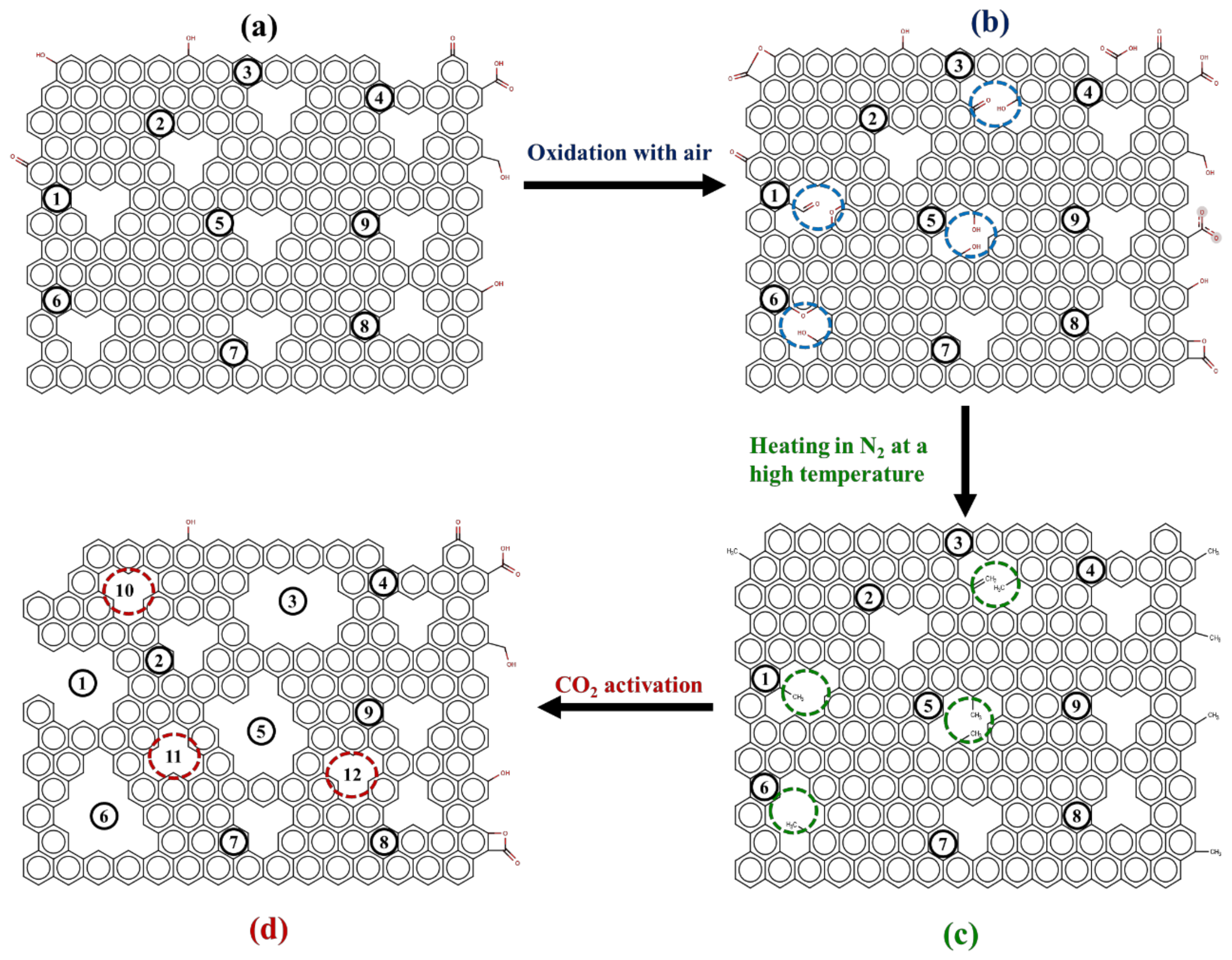
2.5. On Mesopore Development by the OTA Method
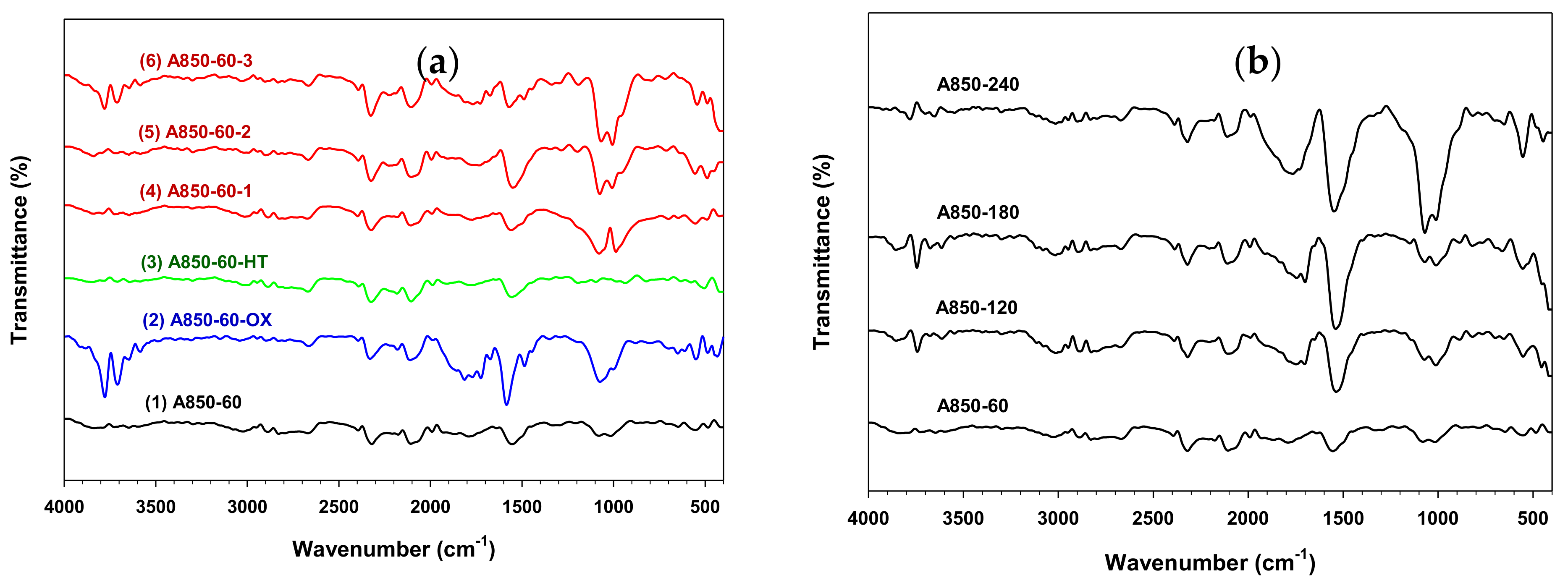
2.6. Surface Functional Groups of Prepared Activated Carbon
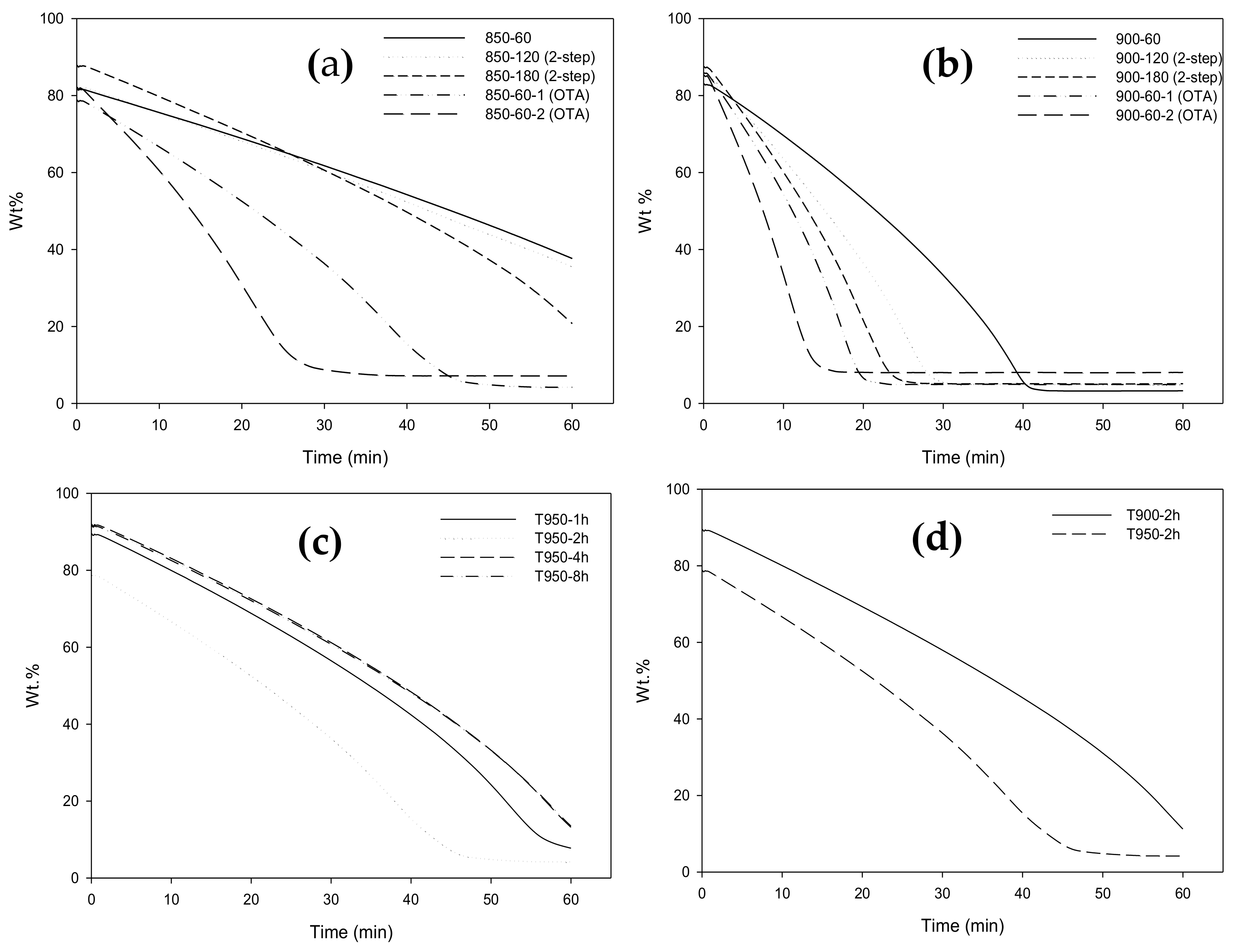
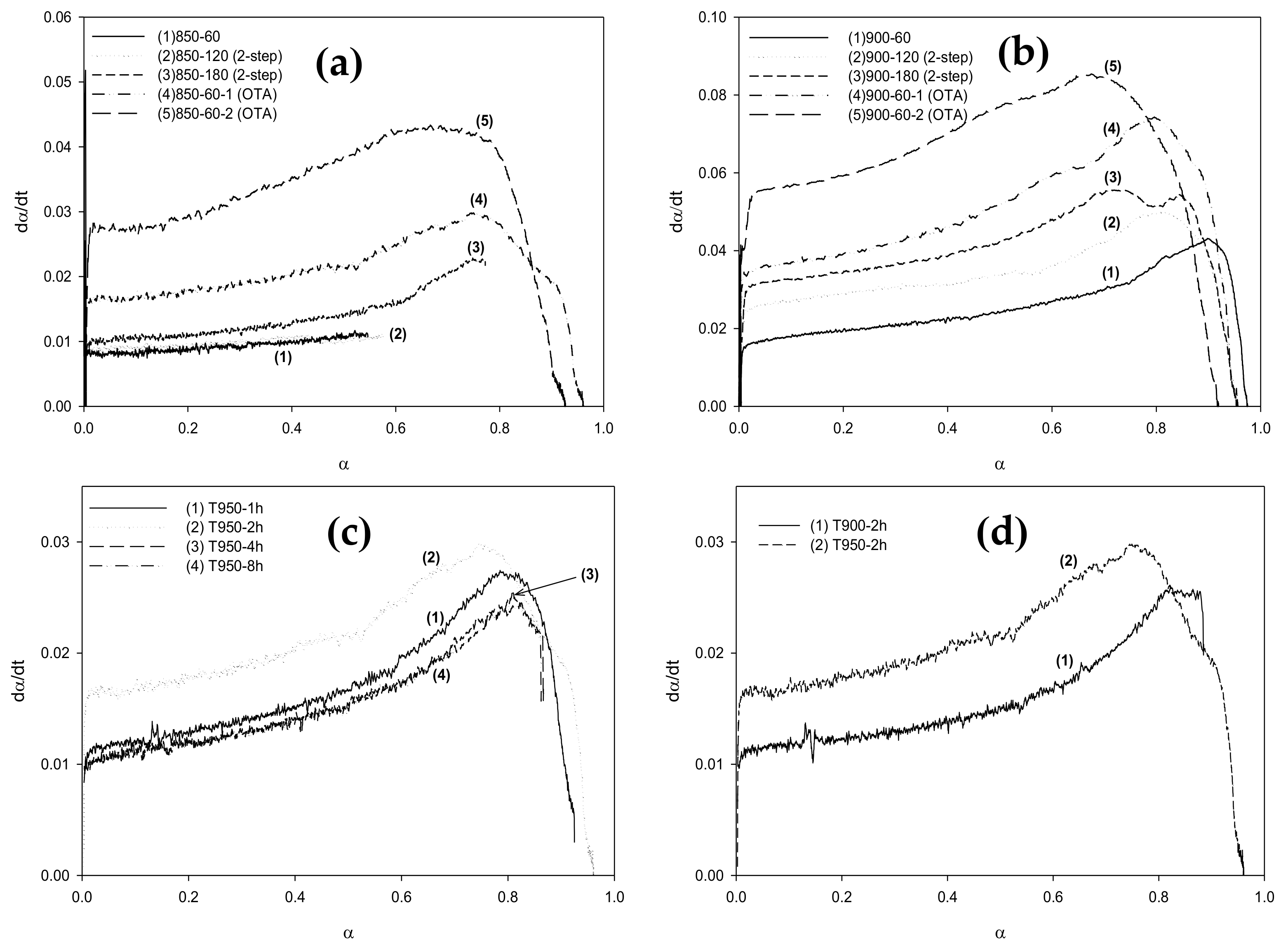
2.7. Carbon Reactivity towards CO2 Gasification
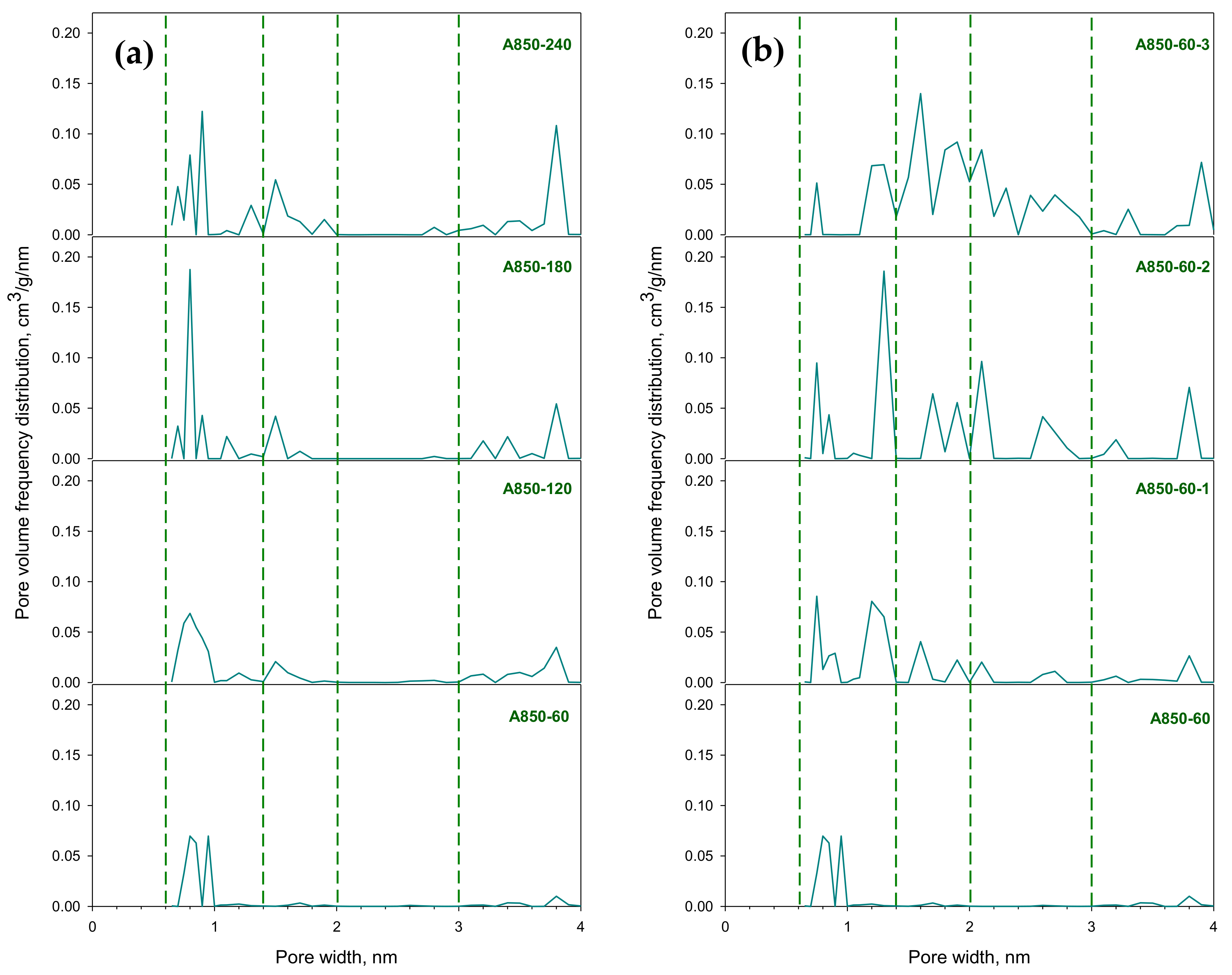
2.8. Pore Size Distributions and Surface Morphology of Prepared Activated Carbons
3. Materials and Methods
3.1. Raw Materials
3.2. Preparation of Char and Microporous Activated Carbons
3.3. Preparation of Mesoporous Activated Carbon by the OTA Method
3.4. Porous Properties of the Prepared Activated Carbons
3.5. Surface Reactivity of Activated Carbon towards CO2 Gasification Reaction
3.6. Surface Functional Groups of Activated Carbons
3.7. GCMC Simulation for Determining Pore Size Distributions in Activated Carbon
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pallarés, J.; González-Cencerrado, A.; Arauzo, I. Production and characterization of activated carbon from barley straw by physical activation with carbon dioxide and steam. Biomass Bioenergy 2018, 115, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Nor, N.M.; Lau, L.C.; Lee, K.T.; Mohamed, A.R. Synthesis of activated carbon from lignocellulosic biomass and its applications in air pollution control—a review. J. Environ. Chem. Eng. 2013, 1, 658–666. [Google Scholar] [CrossRef]
- González-García, P. Activated carbon from lignocellulosics precursors: A review of the synthesis methods, characterization techniques and applications. Renew. Sustain. Energy Rev. 2018, 82, 1393–1414. [Google Scholar] [CrossRef]
- Román, S.; González, J.; González-García, C.; Zamora, F. Control of pore development during CO2 and steam activation of olive stones. Fuel Process. Technol. 2008, 89, 715–720. [Google Scholar] [CrossRef]
- Carrott, P.; Carrott, M.R.; Mourão, P. Pore size control in activated carbons obtained by pyrolysis under different conditions of chemically impregnated cork. J. Anal. Appl. Pyrolysis 2006, 75, 120–127. [Google Scholar] [CrossRef]
- Hadoun, H.; Sadaoui, Z.; Souami, N.; Sahel, D.; Toumert, I. Characterization of mesoporous carbon prepared from date stems by H3PO4 chemical activation. Appl. Surf. Sci. 2013, 280, 1–7. [Google Scholar] [CrossRef]
- Sayğılı, H.; Güzel, F. High surface area mesoporous activated carbon from tomato processing solid waste by zinc chloride activation: Process optimization, characterization and dyes adsorption. J. Clean. Prod. 2016, 113, 995–1004. [Google Scholar] [CrossRef]
- Marrakchi, F.; Ahmed, M.; Khanday, W.; Asif, M.; Hameed, B. Mesoporous-activated carbon prepared from chitosan flakes via single-step sodium hydroxide activation for the adsorption of methylene blue. Int. J. Biol. Macromol. 2017, 98, 233–239. [Google Scholar] [CrossRef]
- Jawad, A.H.; Rashid, R.A.; Ismail, K.; Sabar, S. High surface area mesoporous activated carbon developed from coconut leaf by chemical activation with H3PO4 for adsorption of methylene blue. Desalination Water Treat. 2017, 74, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Nasrullah, A.; Saad, B.; Bhat, A.; Khan, A.S.; Danish, M.; Isa, M.H.; Naeem, A. Mangosteen peel waste as a sustainable precursor for high surface area mesoporous activated carbon: Characterization and application for methylene blue removal. J. Clean. Prod. 2019, 211, 1190–1200. [Google Scholar] [CrossRef]
- Borhan, A.; Yusup, S.; Lim, J.W.; Show, P.L. Characterization and Modelling Studies of Activated Carbon Produced from Rubber-Seed Shell Using KOH for CO2 Adsorption. Processes 2019, 7, 855. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-H.; Lee, H.-M.; Chung, D.; Kim, B.-J. Effect of Mesopore Development on Butane Working Capacity of Biomass-Derived Activated Carbon for Automobile Canister. Nanomaterials 2021, 11, 673. [Google Scholar] [CrossRef]
- Tseng, R.-L. Mesopore control of high surface area NaOH-activated carbon. J. Colloid Interface Sci. 2006, 303, 494–502. [Google Scholar] [CrossRef]
- Tseng, R.-L.; Tseng, S.-K. Pore structure and adsorption performance of the KOH-activated carbons prepared from corncob. J. Colloid Interface Sci. 2005, 287, 428–437. [Google Scholar] [CrossRef]
- Muniandy, L.; Adam, F.; Mohamed, A.R.; Ng, E.-P. The synthesis and characterization of high purity mixed microporous/mesoporous activated carbon from rice husk using chemical activation with NaOH and KOH. Microporous Mesoporous Mater. 2014, 197, 316–323. [Google Scholar] [CrossRef]
- Khamkeaw, A.; Asavamongkolkul, T.; Perngyai, T.; Jongsomjit, B.; Phisalaphong, M. Interconnected Micro, Meso, and Macro Porous Activated Carbon from Bacterial Nanocellulose for Superior Adsorption Properties and Effective Catalytic Performance. Molecules 2020, 25, 4063. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Li, X.; Song, Y. Sludge-based mesoporous activated carbon: The effect of hydrothermal pretreatment on material preparation and adsorption of bisphenol A. J. Chem. Technol. Biotechnol. 2020, 95, 1666–1674. [Google Scholar] [CrossRef]
- Hossain, M.Z.; Wu, W.; Xu, W.Z.; Chowdhury, M.B.I.; Jhawar, A.K.; Machin, D.; Charpentier, P.A. High-Surface-Area Mesoporous Activated Carbon from Hemp Bast Fiber Using Hydrothermal Processing. J. Carbon Res. 2018, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Le Van, K.; Luong, T. Preparation of Pore-Size Controllable Activated Carbon from Rice Husk Using Dual Activating Agent and Its Application in Supercapacitor. J. Chem. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Guo, H.; Srinivasan, M.; Yaming, N. A simple method for developing mesoporosity in activated carbon. Sep. Purif. Technol. 2003, 31, 47–52. [Google Scholar] [CrossRef]
- Hu, Z.; Srinivasan, M. Mesoporous high-surface-area activated carbon. Microporous Mesoporous Mater. 2001, 43, 267–275. [Google Scholar] [CrossRef]
- Hu, Z.; Srinivasan, M.; Ni, Y. Novel activation process for preparing highly microporous and mesoporous activated carbons. Carbon 2001, 39, 877–886. [Google Scholar] [CrossRef]
- Juárez-Galán, J.M.; Silvestre-Albero, A.; Silvestre-Albero, J.; Rodriguez-Reinoso, F. Synthesis of activated carbon with highly developed “mesoporosity”. Microporous Mesoporous Mater. 2009, 117, 519–521. [Google Scholar] [CrossRef]
- Ariyadejwanich, P.; Tanthapanichakoon, W.; Nakagawa, K.; Mukai, S.; Tamon, H. Preparation and characterization of mesoporous activated carbon from waste tires. Carbon 2003, 41, 157–164. [Google Scholar] [CrossRef]
- Molina-Sabio, M.; Gonzalez, M.; Rodriguez-Reinoso, F.; Sepúlveda-Escribano, A. Effect of steam and carbon dioxide activation in the micropore size distribution of activated carbon. Carbon 1996, 34, 505–509. [Google Scholar] [CrossRef]
- Guo, J.; Lua, A. Kinetic study on pyrolytic process of oil-palm solid waste using two-step consecutive reaction model. Biomass Bioenergy 2001, 20, 223–233. [Google Scholar] [CrossRef]
- Junpirom, S.; Do, D.; Tangsathitkulchai, C. A carbon activation model with application to longan seed char gasification. Carbon 2005, 43, 1936–1943. [Google Scholar] [CrossRef]
- García, R.; Pizarro, C.; Lavín, A.G.; Bueno, J.L. Characterization of Spanish biomass wastes for energy use. Bioresour. Technol. 2012, 103, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Rouquerol, J.; Rouquerol, F.; Sing, K.S.W.; Llewellyn, P.; Maurin, G. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Alvarez, J.; Lopez, G.; Amutio, M.; Bilbao, J.; Olazar, M. Preparation of adsorbents from sewage sludge pyrolytic char by carbon dioxide activation. Process. Saf. Environ. Prot. 2016, 103, 76–86. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Erkiaga, A.; Alvarez, J.; Barbarias, I.; Olazar, M. Preparation of adsorbents derived from waste tires. Chem. Eng. Trans. 2012, 29, 811–816. [Google Scholar]
- Gonçalves, G.D.C.; Pereira, N.C.; Veit, M.T. Production of bio-oil and activated carbon from sugarcane bagasse and molasses. Biomass Bioenergy 2016, 85, 178–186. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to Read and Interpret FTIR Spectroscope of Organic Material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R.; Sukmafitri, A.; Zaen, R. Amorphous content on the photocatalytic performance of micrometer-sized tungsten trioxide particles. Arab. J. Chem. 2020, 13, 2912–2924. [Google Scholar] [CrossRef]
- Pradhan, B.K.; Sandle, N. Effect of different oxidizing agent treatments on the surface properties of activated carbons. Carbon 1999, 37, 1323–1332. [Google Scholar] [CrossRef]
- Bouwman, R.; Freriks, I.L. Low-temperature oxidation of a bituminous coal. Infrared spectroscopic study of samples from a coal pile. Fuel 1980, 59, 315–322. [Google Scholar] [CrossRef]
- Burg, P.; Fydrych, P.; Cagniant, D.; Nanse, G.; Bimer, J.; Jankowska, A. The characterization of nitrogen-enriched activated carbons by IR, XPS and LSER methods. Carbon 2002, 40, 1521–1531. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Fanning, P.E.; Vannice, M. A DRIFTS study of the formation of surface groups on carbon by oxidation. Carbon 1993, 31, 721–730. [Google Scholar] [CrossRef]
- El-Hendawy, A.-N.A. Variation in the FTIR spectra of a biomass under impregnation, carbonization and oxidation conditions. J. Anal. Appl. Pyrolysis 2006, 75, 159–166. [Google Scholar] [CrossRef]
- He, X.; Geng, Y.; Qiu, J.; Zheng, M.; Long, S.; Zhang, X. Effect of activation time on the properties of activated carbons prepared by microwave-assisted activation for electric double layer capacitors. Carbon 2010, 48, 1662–1669. [Google Scholar] [CrossRef]
- Kwiatkowski, J.F. Activated Carbon: Classifications, Properties and Applications; Nova Science Publishers, Incorporated: Hauppauge, NY, USA, 2011. [Google Scholar]
- Shafeeyan, M.S.; Daud, W.M.A.W.; Houshmand, A.; Shamiri, A. A review on surface modification of activated carbon for carbon dioxide adsorption. J. Anal. Appl. Pyrolysis 2010, 89, 143–151. [Google Scholar] [CrossRef]
- Bandosz, T.; Ania, C. Chapter 4 Surface chemistry of activated carbons and its characterization. Heavy Met. Environ. Orig. Interact. Remediat. 2006, 159–229. [Google Scholar]
- Tangsathitkulchai, C.; Naksusuk, S.; Wongkoblap, A.; Phadungbut, P.; Borisut, P. Equilibrium and Kinetics of CO2 Adsorption by Coconut Shell Activated Carbon Impregnated with Sodium Hydroxide. Processes 2021, 9, 201. [Google Scholar] [CrossRef]
- Guo, J.; Lua, A.C. Characterization of chars pyrolyzed from oil palm stones for the preparation of activated carbons. J. Anal. Appl. Pyrolysis 1998, 46, 113–125. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W.; Salzberg, H.W. Adsorption Surface Area and Porosity. J. Electrochem. Soc. 1967, 114, 279C. [Google Scholar] [CrossRef]
- Boehm, H.P. Chemical Identification of Surface Groups. In Advances in Catalysis; Eley, D.D., Pines, H., Weisz, P.B., Eds.; Academic Press: Cambridge, MA, USA, 1966; pp. 179–274. [Google Scholar]
- Boehm, H. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Seung Kim, Y.; Rae Park, C. Chapter 13—Titration Method for the Identification of Surface Functional Groups. In Materials Science and Engineering of Carbon; Inagaki, M., Kang, F., Eds.; Butterworth-Heinemann: Oxford, UK, 2016; pp. 273–286. [Google Scholar]
- Goertzen, S.L.; Thériault, K.D.; Oickle, A.M.; Tarasuk, A.C.; Andreas, H.A. Standardization of the Boehm titration. Part I. CO2 expulsion and endpoint determination. Carbon 2010, 48, 1252–1261. [Google Scholar] [CrossRef]
- Ravikovitch, P.I.; Vishnyakov, A.; Russo, R.; Neimark, A.V. Unified Approach to Pore Size Characterization of Microporous Carbonaceous Materials from N2, Ar, and CO2 Adsorption Isotherms†. Langmuir 2000, 16, 2311–2320. [Google Scholar] [CrossRef]
- Do, D.; Do, H. Pore Characterization of Carbonaceous Materials by DFT and GCMC Simulations: A Review. Adsorpt. Sci. Technol. 2003, 21, 389–423. [Google Scholar] [CrossRef]
- Johnson, J.K.; Zollweg, J.A.; Gubbins, K.E. The Lennard-Jones equation of state revisited. Mol. Phys. 1993, 78, 591–618. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Proximate Analysis (Dry Basis) [wt.%] | ||
|---|---|---|---|
| Fixed Carbon | Volatile Matters | Ash | |
| Longan seed (this work) | 22.34 | 76.51 | 1.15 |
| Longan seed 1 | 19.60 | 78.70 | 1.70 |
| Oil palm shell 2 | 19.80 | 77.60 | 2.60 |
| Coconut shell 3 | 19.40 | 79.20 | 0.60 |
| Peanut shell 3 | 16.50 | 81.00 | 2.50 |
| Eucalyptus sawdust 3 | 15.38 | 83.70 | 0.90 |
| corn cob 3 | 14.60 | 83.00 | 2.40 |
| Samples | Proximate Analysis (Dry Basis) [wt.%] | Ultimate Analysis (Dry Basis) [wt.%] | ||||||
|---|---|---|---|---|---|---|---|---|
| Fixed Carbon | Volatile | Ash | C | H | N | S | Odiff. | |
| Longan seed | 23.34 | 76.51 | 1.15 | 47.56 | 6.55 | 1.09 | 0.03 | 44.76 |
| C500-90 | 83.58 | 14.81 | 1.61 | 85.92 | 3.55 | 2.32 | 0.00 | 8.20 |
| A850-60 | 86.25 | 11.57 | 2.18 | 91.91 | 0.77 | 2.38 | 0.00 | 4.93 |
| A850-120 | 83.21 | 11.49 | 5.3 | 85.58 | 0.85 | 2.47 | 0.00 | 11.10 |
| A850-180 | 83.02 | 11.45 | 5.53 | 81.16 | 0.98 | 2.48 | 0.00 | 15.38 |
| A850-240 | 82.58 | 11.32 | 6.1 | 76.75 | 0.88 | 2.50 | 0.00 | 19.87 |
| A850-60-1 | 83.40 | 10.93 | 5.67 | 84.00 | 0.63 | 2.16 | 0.00 | 13.21 |
| A850-60-2 | 82.57 | 10.89 | 6.54 | 81.71 | 0.61 | 1.99 | 0.00 | 15.70 |
| A850-60-3 | 80.08 | 10.38 | 9.54 | 76.60 | 0.44 | 1.90 | 0.00 | 21.06 |
| Sample | Sample | Avg.pore | VMic. | VMes. | VT | VMes/Vmic | SBET | % Carbon | Yield |
|---|---|---|---|---|---|---|---|---|---|
| no. | code | dia.(nm) | (cm3/g) (%) | (cm3/g) (%) | (cm3/g) | (%) | (m2/g) | Burn-off | (%) |
| 1 | A850-60 | 1.67 | 0.248 (91.1) | 0.024 (8.9) | 0.272 | 9.5 | 652 | 27.7 | 72.3 |
| 2 | A850-120 | 1.88 | 0.343 (78.5) | 0.094 (21.5) | 0.437 | 27.6 | 932 | 42.1 | 57.9 |
| 3 | A850-180 | 1.91 | 0.340 (76.9) | 0.102 (23.1) | 0.442 | 30.0 | 926 | 52.2 | 47.8 |
| 4 | A850-240 | 2.06 | 0.409 (69.7) | 0.178 (30.3) | 0.587 | 43.5 | 1140 | 68.8 | 31.2 |
| 5 | A850-60-1 | 1.89 | 0.375 (78.1) | 0.105 (21.9) | 0.480 | 27.8 | 980 | 46.1 | 53.9 |
| 6 | A850-60-2 | 2.32 | 0.466 (63.3) | 0.270 (36.7) | 0.736 | 58.1 | 1271 | 62.1 | 37.9 |
| 7 | A850-60-3 | 2.42 | 0.600 (55.9) | 0.474 (44.1) | 1.074 | 78.9 | 1773 | 78.4 | 21.6 |
| 8 | A900-60 | 1.73 | 0.253 (86.0) | 0.041 (14.0) | 0.294 | 16.3 | 684 | 34.8 | 65.2 |
| 9 | A900-120 | 1.96 | 0.376 (77.5) | 0.109 (22.5) | 0.485 | 29.1 | 1046 | 58.8 | 41.2 |
| 10 | A900-180 | 2.16 | 0.406 (70.6) | 0.169 (29.4) | 0.575 | 41.6 | 1115 | 63.9 | 36.1 |
| 11 | A900-240 | 2.30 | 0.547 (67.0) | 0.270 (33.0) | 0.817 | 49.3 | 1499 | 92.7 | 7.3 |
| 12 | A900-60-1 | 2.06 | 0.434 (77.0) | 0.130 (23.0) | 0.564 | 29.9 | 1093 | 56.2 | 43.8 |
| 13 | A900-60-2 | 2.49 | 0.583 (62.0) | 0.357 (38.0) | 0.940 | 61.3 | 1585 | 76.5 | 23.5 |
| 14 | A900-60-3 | 2.54 | 0.576 (55.0) | 0.471 (45.0) | 1.047 | 81.8 | 1730 | 93.7 | 6.3 |
| Precursor | Activation Method | Mesopore Volume, cm3/g | BET Surface Area | References |
|---|---|---|---|---|
| (% Total Pore Volume) | (m2/g) | |||
| Coconut shell | ZnCl2 + CO2 | 1.364 (71%) | 2191 | [21,22] |
| Waste tires | HCl + steam | 1.620 (74%) | 1119 | [24] |
| Date stems | H3PO4 | 0.993 (95%) | 1455 | [6] |
| Rice husk | KOH | 0.691 (46%) | 2696 | [15] |
| Chitosan flakes | NaOH | 0.157 (62%) | 318 | [8] |
| Coconut leaves | H3PO4 | 1.276 (93%) | 982 | [9] |
| Longan seed | OTA method | 0.474 (44%) | 1773 | This study |
| Porous Properties | Activation Method | Empirical Equations | R2 |
|---|---|---|---|
| (a) BET surface area (m2/g) | 2-step | y = 668.72ln(x) − 1638.7 | 0.9312 |
| OTA | y = 1157.5ln(x) − 3462.4 | 0.9582 | |
| (b) Total pore volume (cm3/g) | 2-step | y = 0.4289ln(x) − 1.2032 | 0.9194 |
| OTA | y = 0.8556ln(x) − 2.7839 | 0.9432 | |
| (c) Micropore volume (cm3/g) | 2-step | y = 0.2336ln(x) − 0.5562 | 0.9111 |
| OTA | y = 0.3642ln(x) − 1.0278 | 0.9457 | |
| (d) Mesopore volume (cm3/g) | 2-step | y = 0.1951ln(x) − 0.6463 | 0.9151 |
| OTA | y = 0.4914ln(x) − 1.7561 | 0.8943 |
| Sample | Sample | Basic Groups | Acidic Groups | Total Groups |
|---|---|---|---|---|
| Number | code | (mmol/g carbon) | (mmol/g carbon) | (mmol/g carbon) |
| 1 | A850-60 | 1.090 | 0.069 | 1.159 |
| 2 | A850-60-OX | 0.894 | 0.393 | 1.286 |
| 3 | A850-60-HT | 1.331 | 0.002 | 1.333 |
| 4 | A850-60-1 | 1.295 | 0.152 | 1.447 |
| 5 | A850-60-1-OX | 1.144 | 0.467 | 1.611 |
| Sample No. | Sample Code | Pore Volume for Pore width (cm3/g) | Total Pore Volume (cm3/g) | ||||
|---|---|---|---|---|---|---|---|
| 0–0.6 nm | 0.6–1.4 nm | 1.4–2 nm | 2–3 nm | 3–4 nm | |||
| 1 | A850-60 | 0.000 | 0.242 | 0.006 | 0.002 | 0.022 | 0.272 |
| 2 | A850-120 | 0.000 | 0.306 | 0.037 | 0.006 | 0.088 | 0.437 |
| 3 | A850-180 | 0.000 | 0.291 | 0.049 | 0.002 | 0.100 | 0.442 |
| 4 | A850-240 | 0.000 | 0.307 | 0.102 | 0.012 | 0.166 | 0.587 |
| 5 | A850-60-1 | 0.000 | 0.309 | 0.066 | 0.046 | 0.059 | 0.480 |
| 6 | A850-60-2 | 0.000 | 0.339 | 0.127 | 0.176 | 0.094 | 0.736 |
| 7 | A850-60-3 | 0.000 | 0.207 | 0.393 | 0.330 | 0.144 | 1.074 |
| Type | Interacting Site | Energy Well Depth (ε/kB), K | Collision Diameter (σ), nm |
|---|---|---|---|
| N2 (Spherical model) | N2 | 101.5 | 0.3615 |
| Activated carbon | C | 28.0 | 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawtae, P.; Tangsathitkulchai, C. A New Approach for Controlling Mesoporosity in Activated Carbon by the Consecutive Process of Air Oxidation, Thermal Destruction of Surface Functional Groups, and Carbon Activation (the OTA Method). Molecules 2021, 26, 2758. https://doi.org/10.3390/molecules26092758
Lawtae P, Tangsathitkulchai C. A New Approach for Controlling Mesoporosity in Activated Carbon by the Consecutive Process of Air Oxidation, Thermal Destruction of Surface Functional Groups, and Carbon Activation (the OTA Method). Molecules. 2021; 26(9):2758. https://doi.org/10.3390/molecules26092758
Chicago/Turabian StyleLawtae, Panuwat, and Chaiyot Tangsathitkulchai. 2021. "A New Approach for Controlling Mesoporosity in Activated Carbon by the Consecutive Process of Air Oxidation, Thermal Destruction of Surface Functional Groups, and Carbon Activation (the OTA Method)" Molecules 26, no. 9: 2758. https://doi.org/10.3390/molecules26092758