
Radiation-Induced Oxidation Reactions of 2-Selenouracil in Aqueous Solutions: Comparison with Sulfur Analog of Uracil

 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Oxidation of 2-SeU by ●OH Radicals
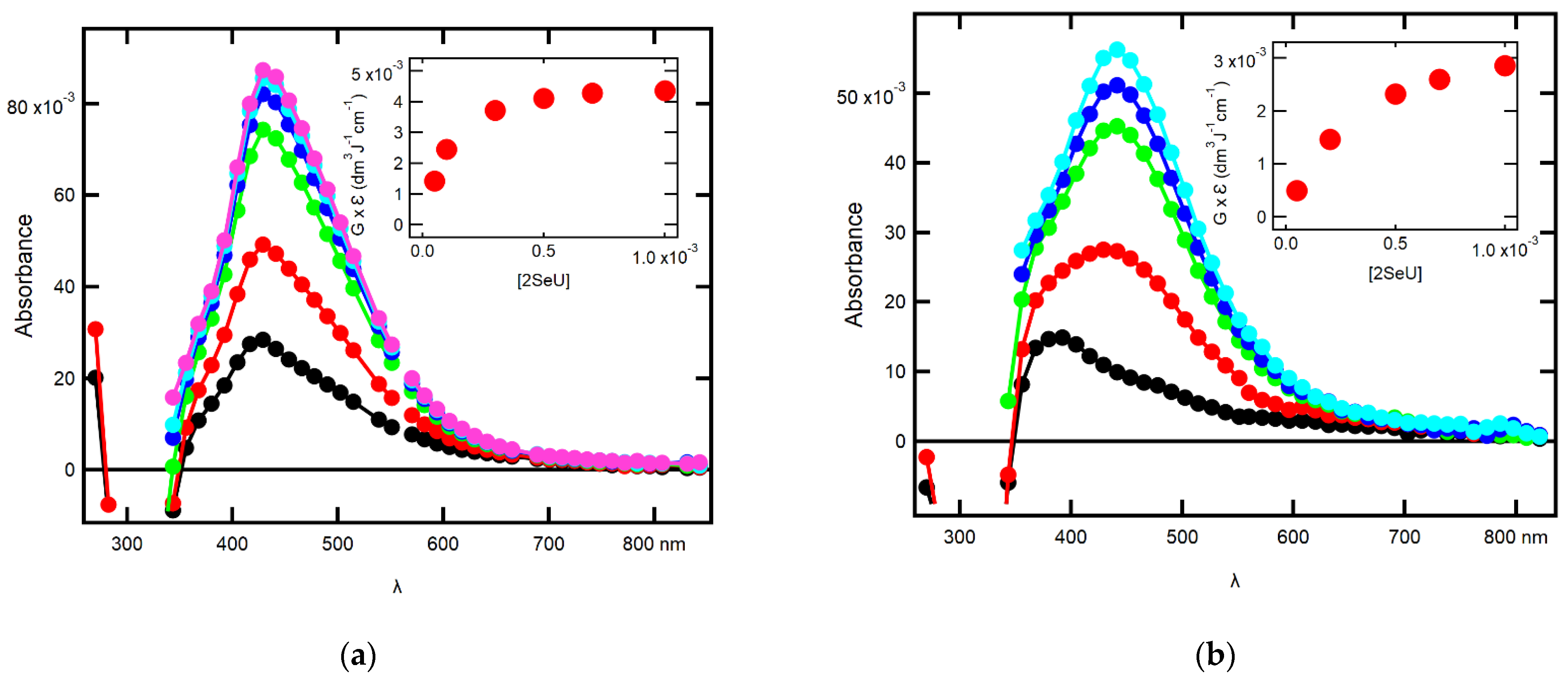
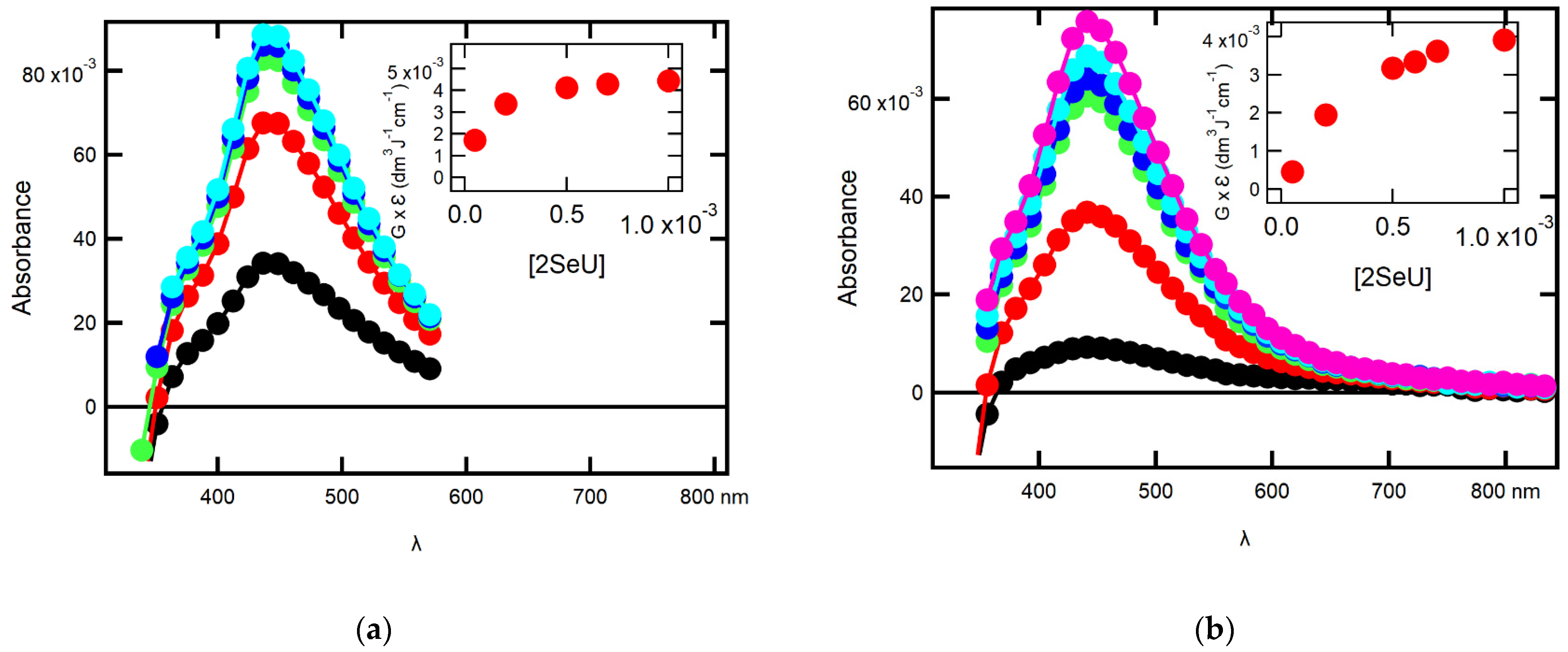
2.1.1. Influence of 2-SeU Concentration and pH on Absorption Spectra
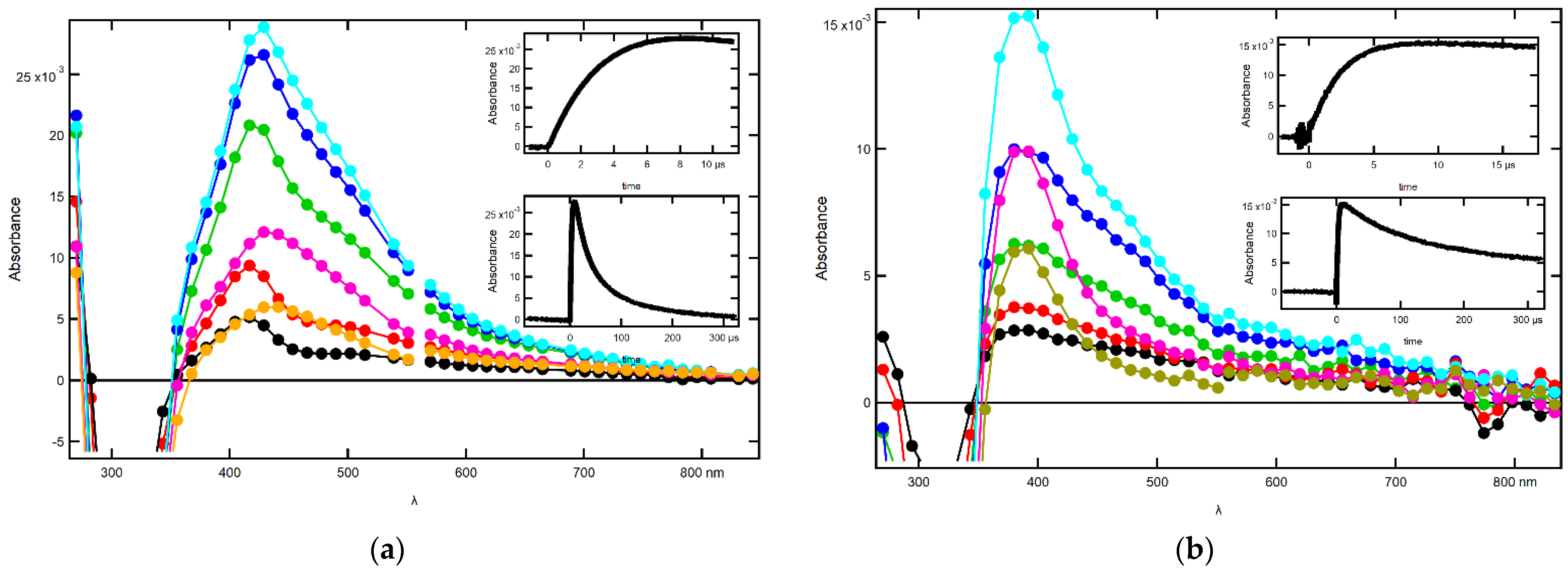
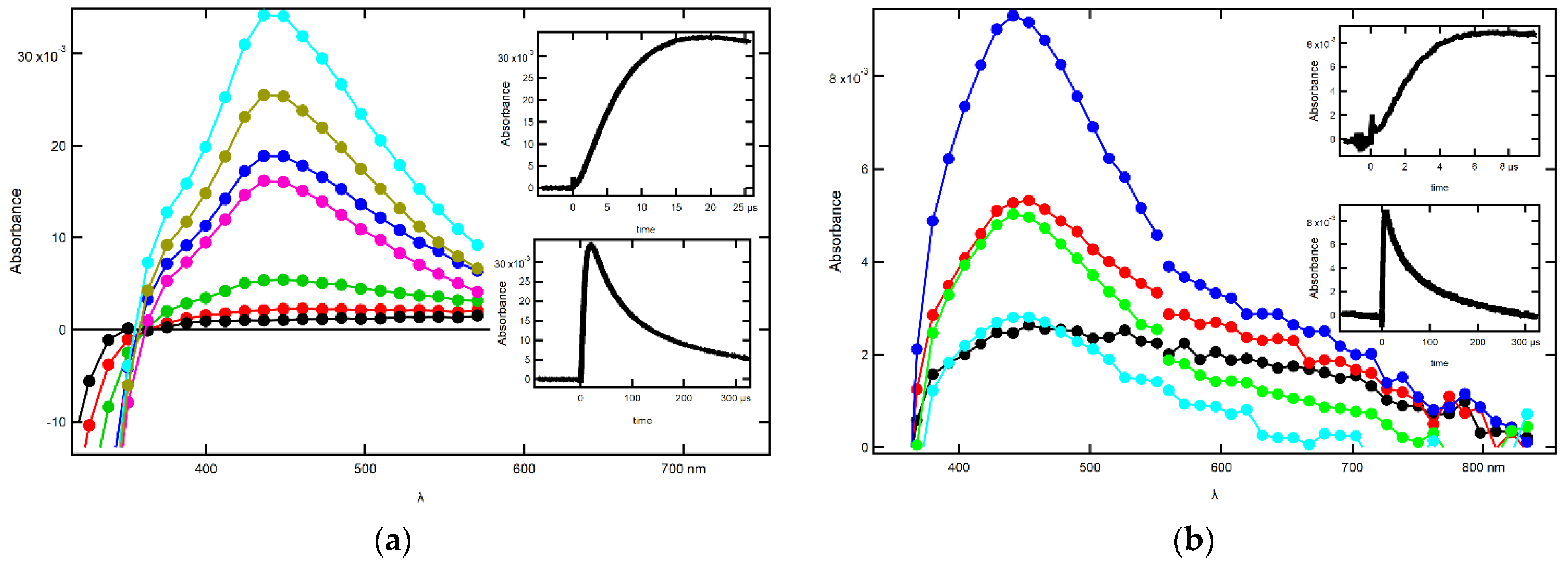
2.1.2. Influence of pH on the Time Evolution of the Absorption Spectra at Low Concentrations of 2-SeU
2.1.3. Influence of pH on the Time Evolution of Absorption Spectra at High Concentrations of 2-SeU
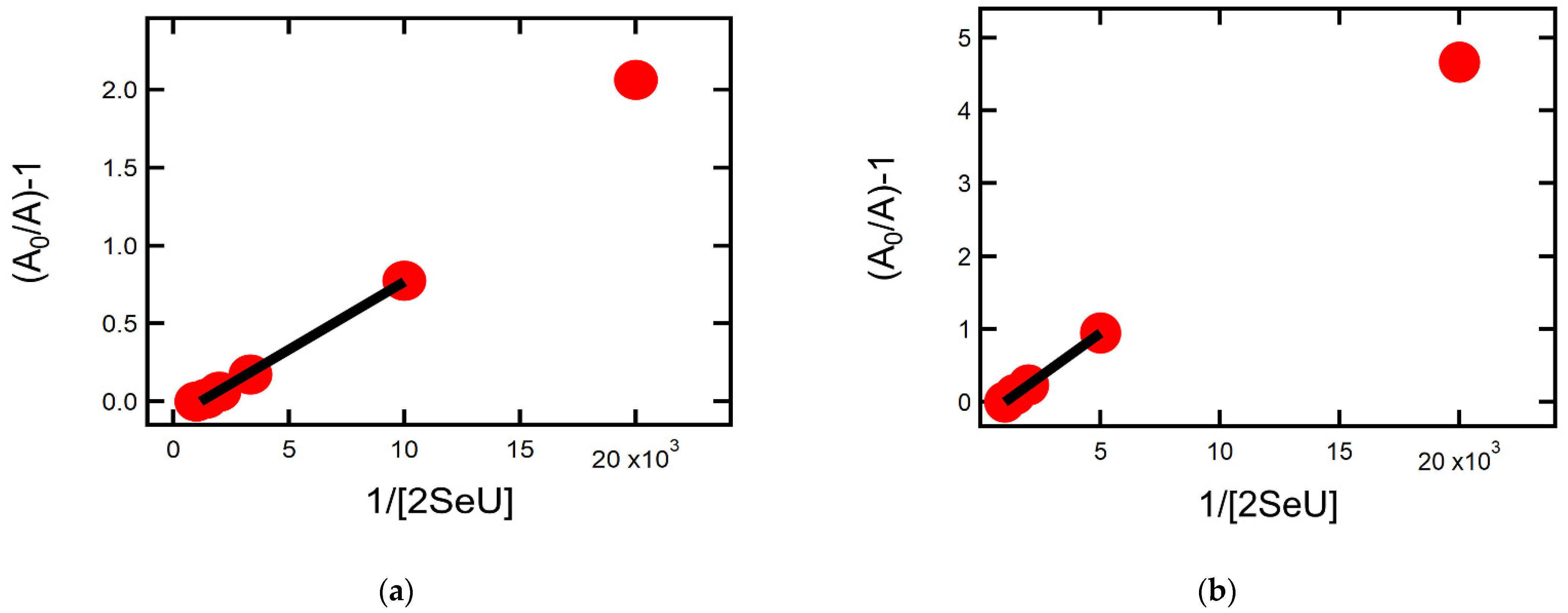
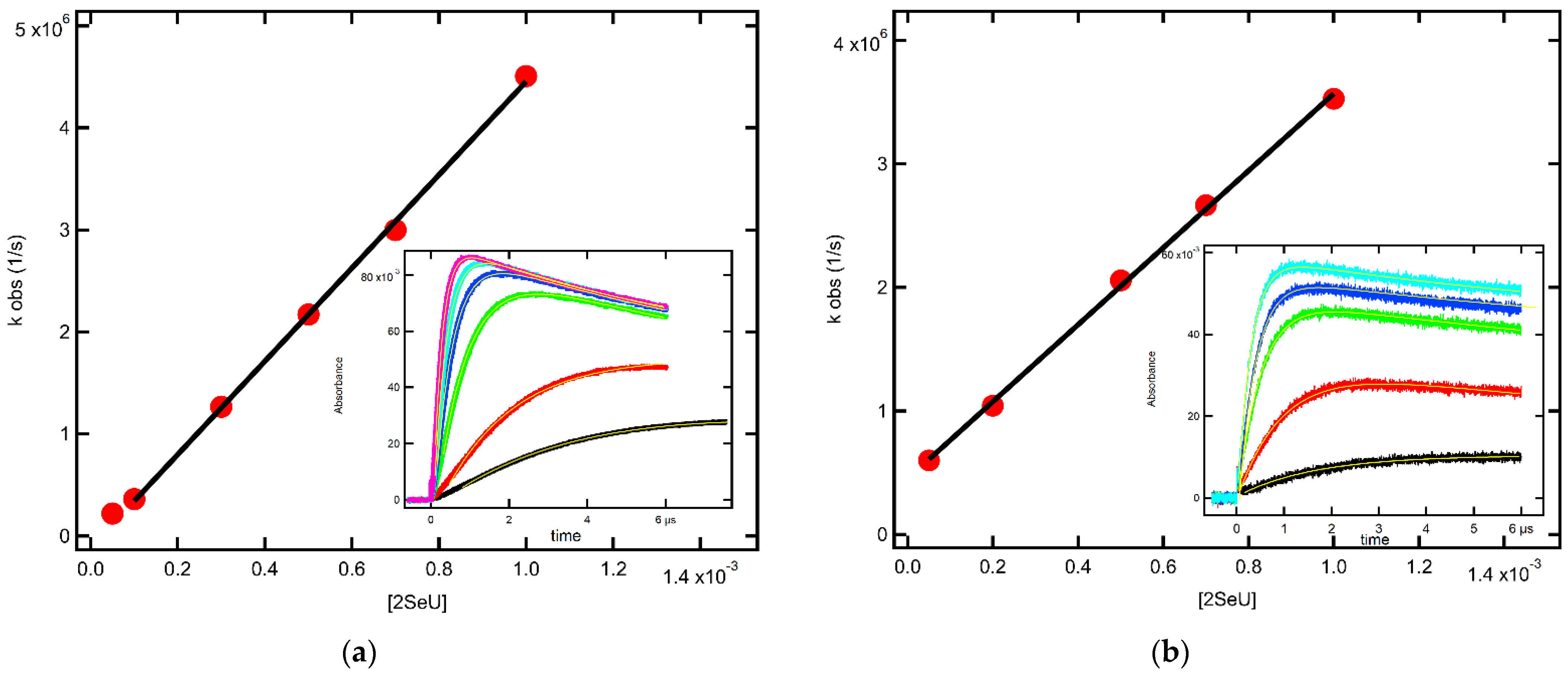
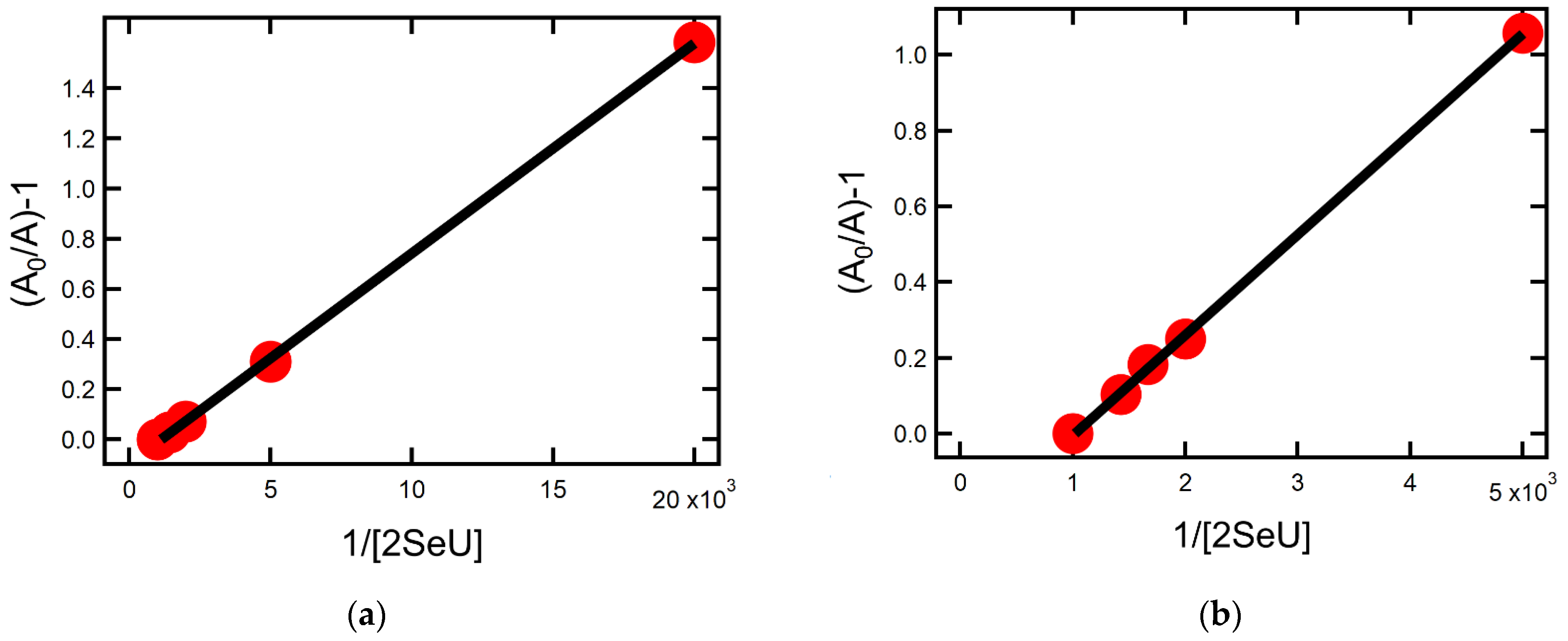
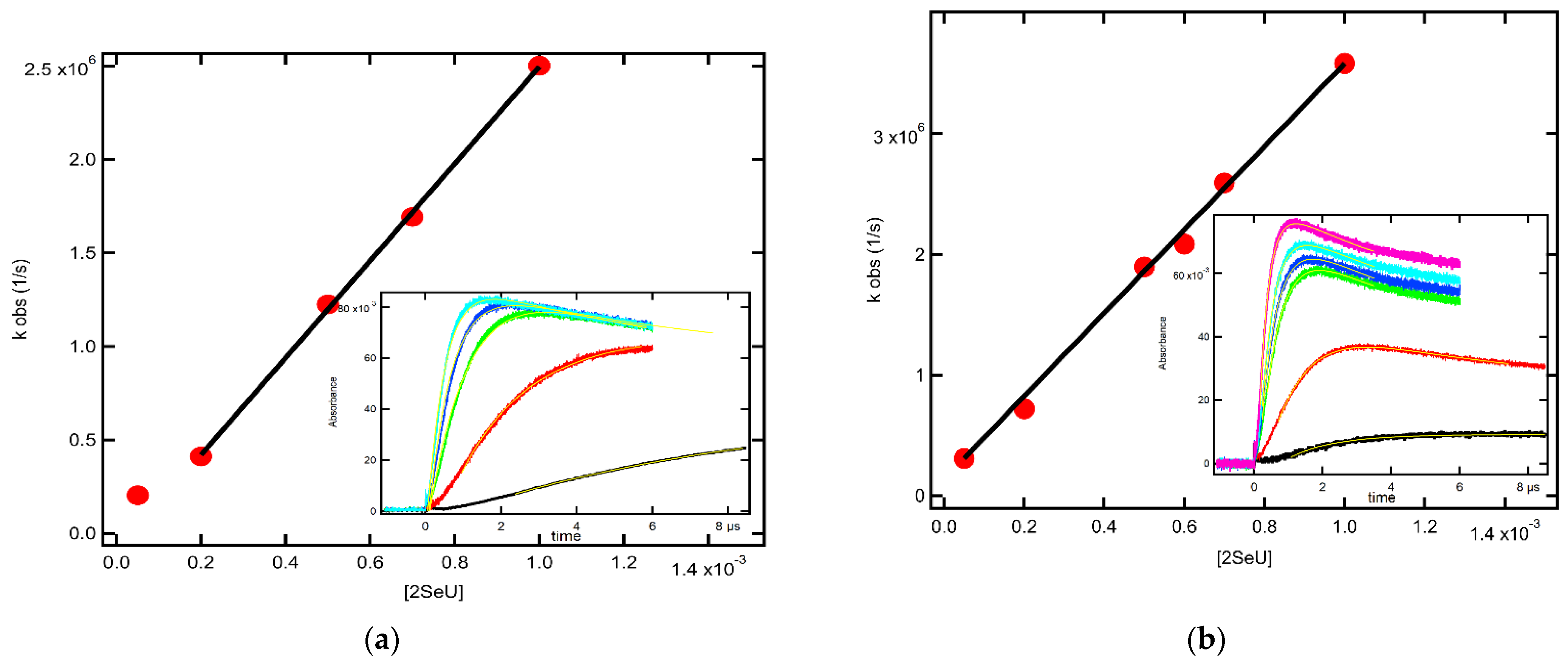
2.1.4. Equilibrium Constants and Rate Constants of Reactions Involved in Equilibrium
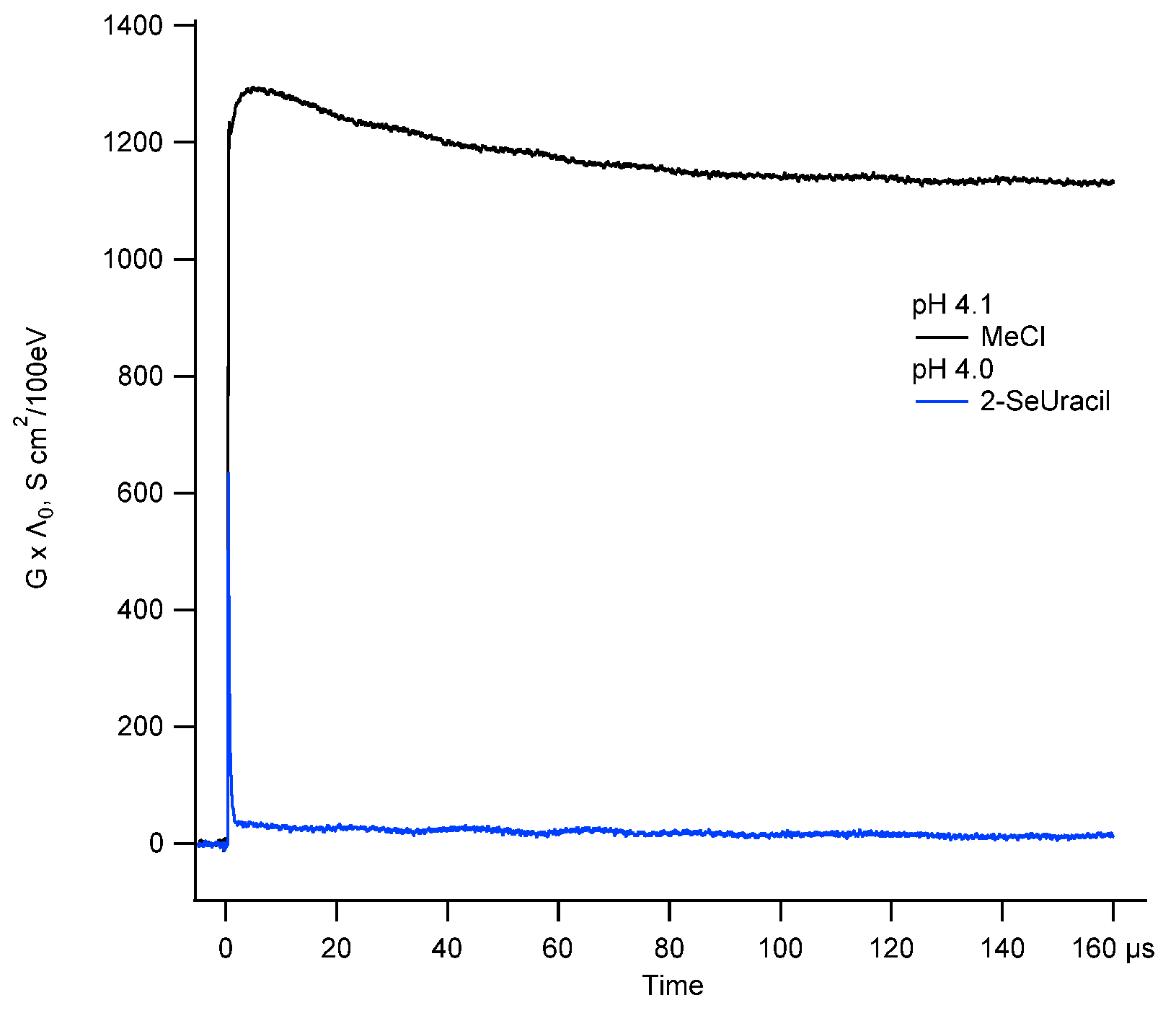
2.1.5. Oxidation of 2-SeU by ●OH Radicals at pH 4: Time-Resolved Conductivity
2.2. Oxidation of 2-SeU by ●N3 Radicals
2.2.1. Influence of 2-SeU Concentration and pH on Absorption Spectra
2.2.2. Influence of pH on the Time Evolution of Absorption Spectra at Low Concentrations of 2-SeU
2.2.3. Influence of pH on the Time Evolution of Absorption Spectra at High Concentrations of 2-SeU
2.2.4. Equilibrium Constants and Rate Constants of Reactions Involved in Equilibrium
2.3. Theoretical Calculations
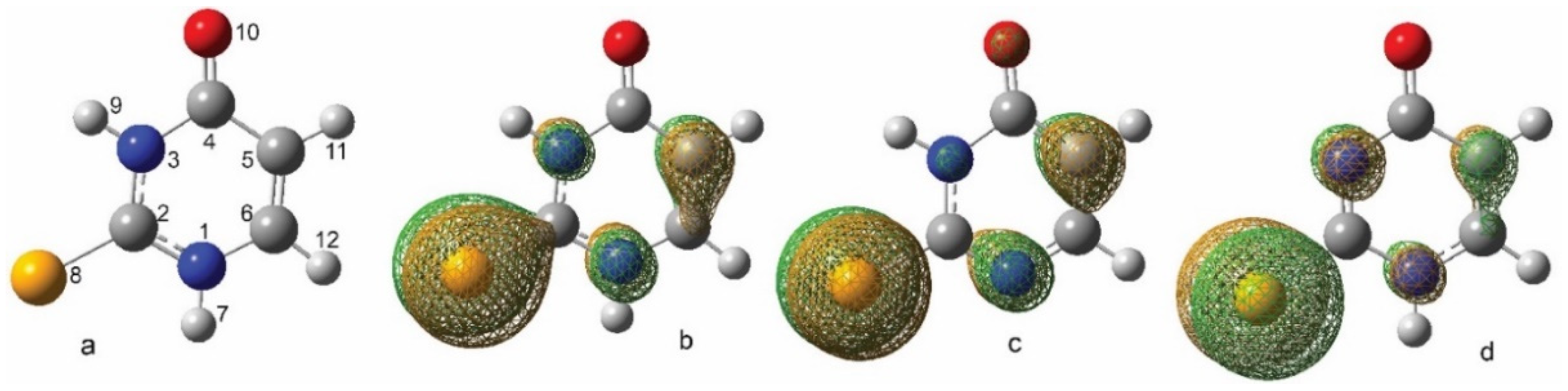
2.3.1. Neutral and Deprotonated Forms of 2-SeU
2.3.2. Monomeric Intermediates Generated in Neutral and Deprotonated Forms of 2-SeU
2.3.3. Dimeric Intermediates Generated in Neutral and Deprotonated Forms of 2-SeU
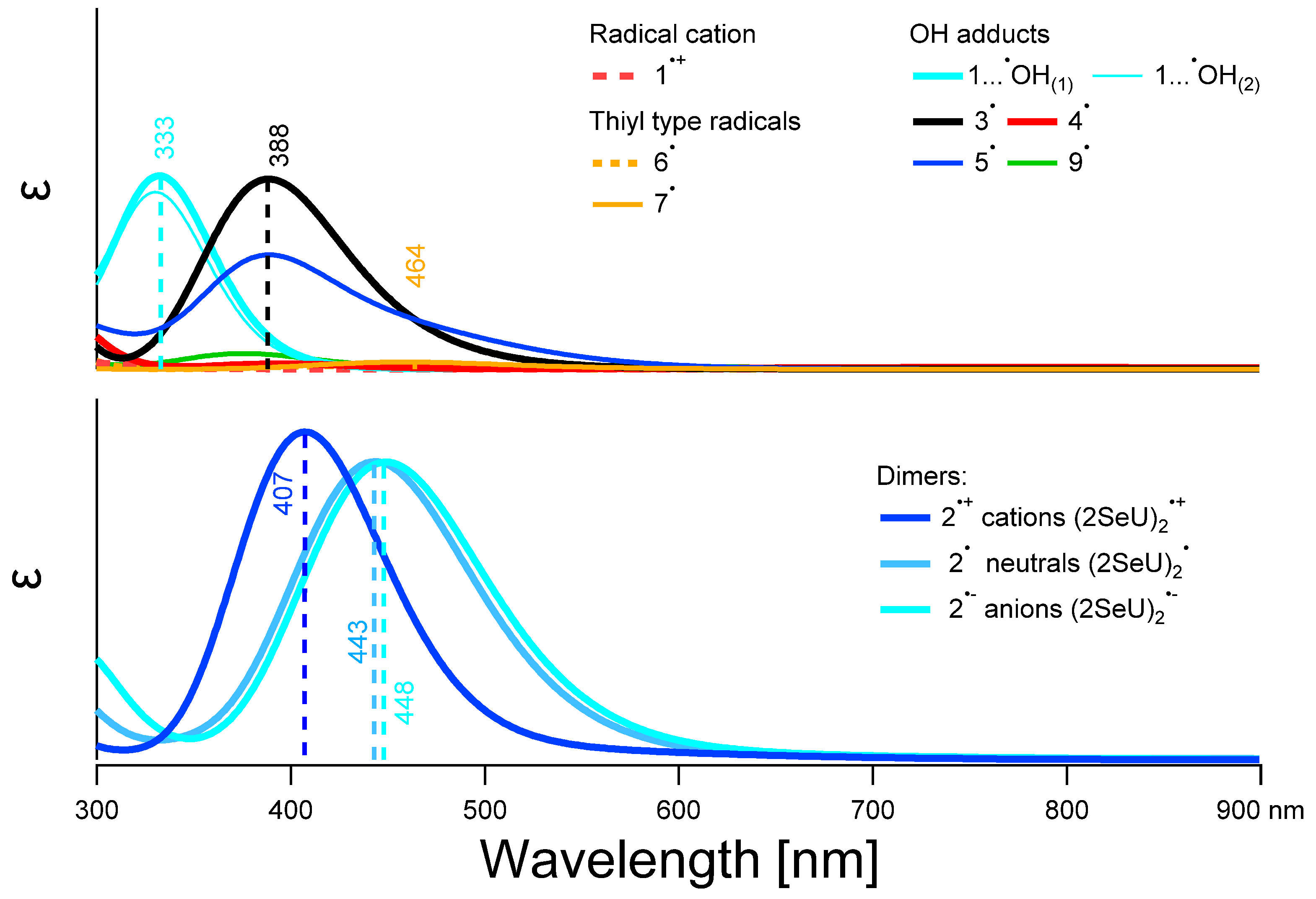
2.3.4. UV-Vis Spectra of Monomeric and Dimeric Intermediates Derived from 2-SeU
3. Discussion
3.1. Characterization of Radical Species: Spectral, Kinetic, and Energetic Parameters
3.2. Justification of the Reaction Pathway Involving Hemibonded ●OH Adducts to Selenium Atom: One-Electron Transfer vs. Proton-Coupled Electron Transfer
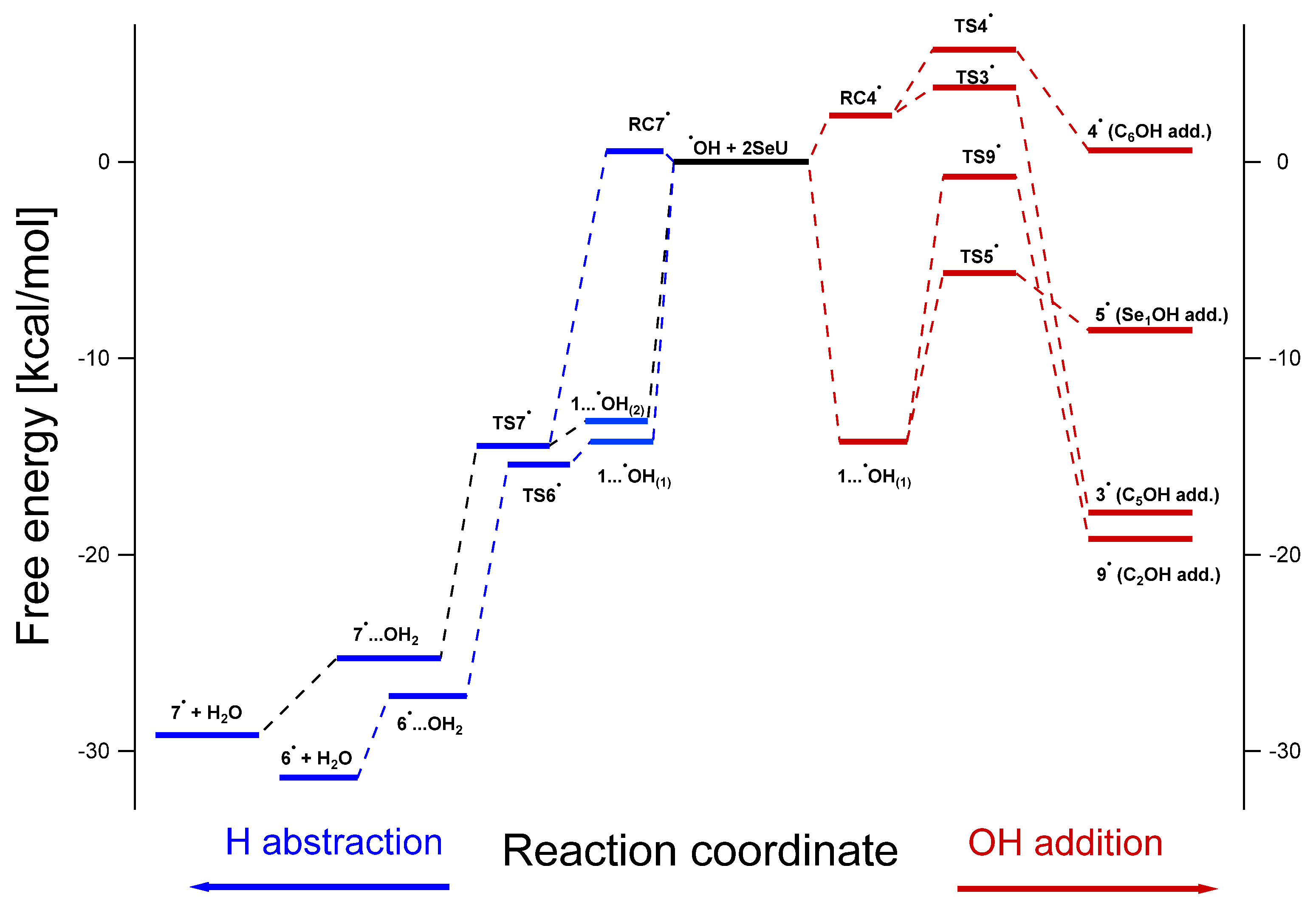
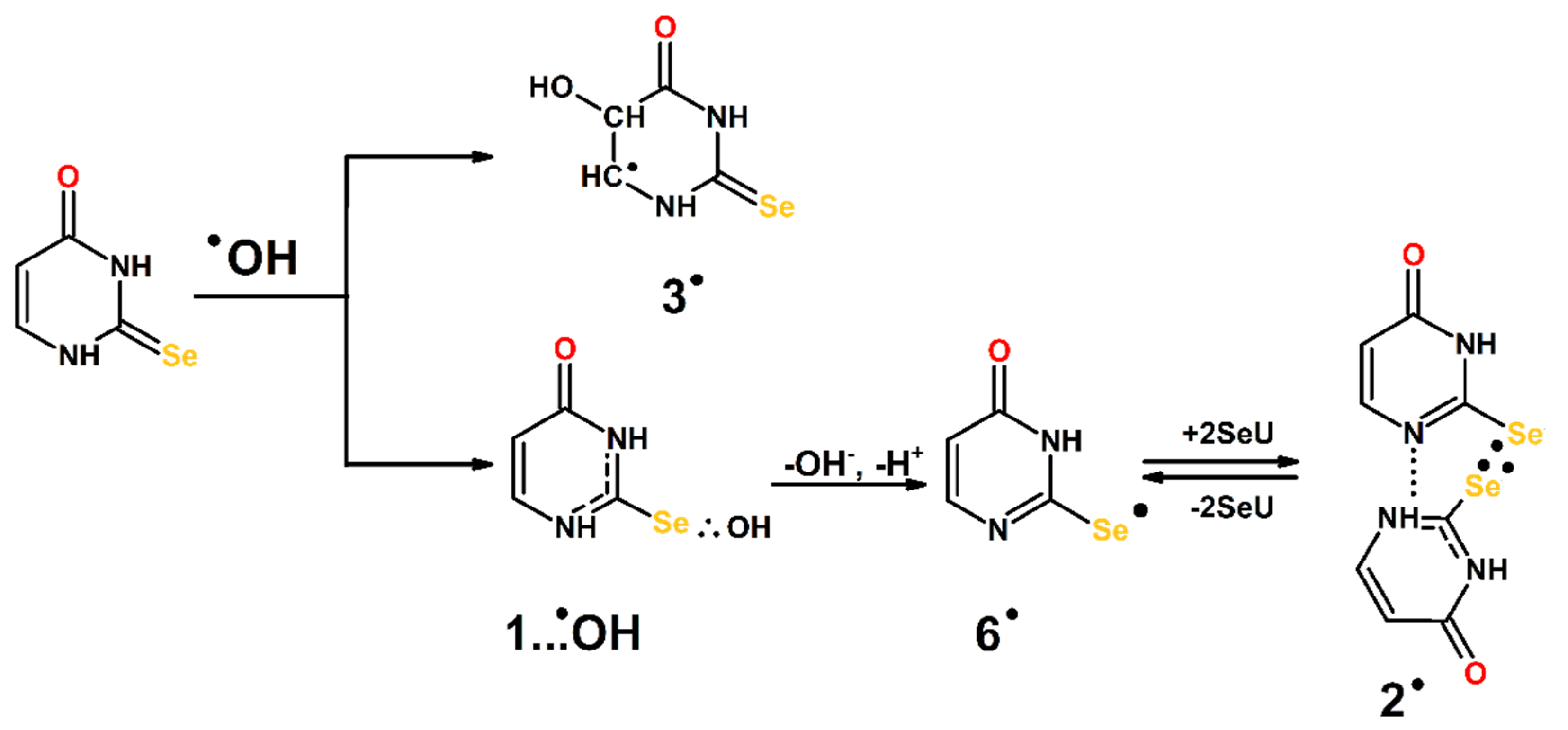
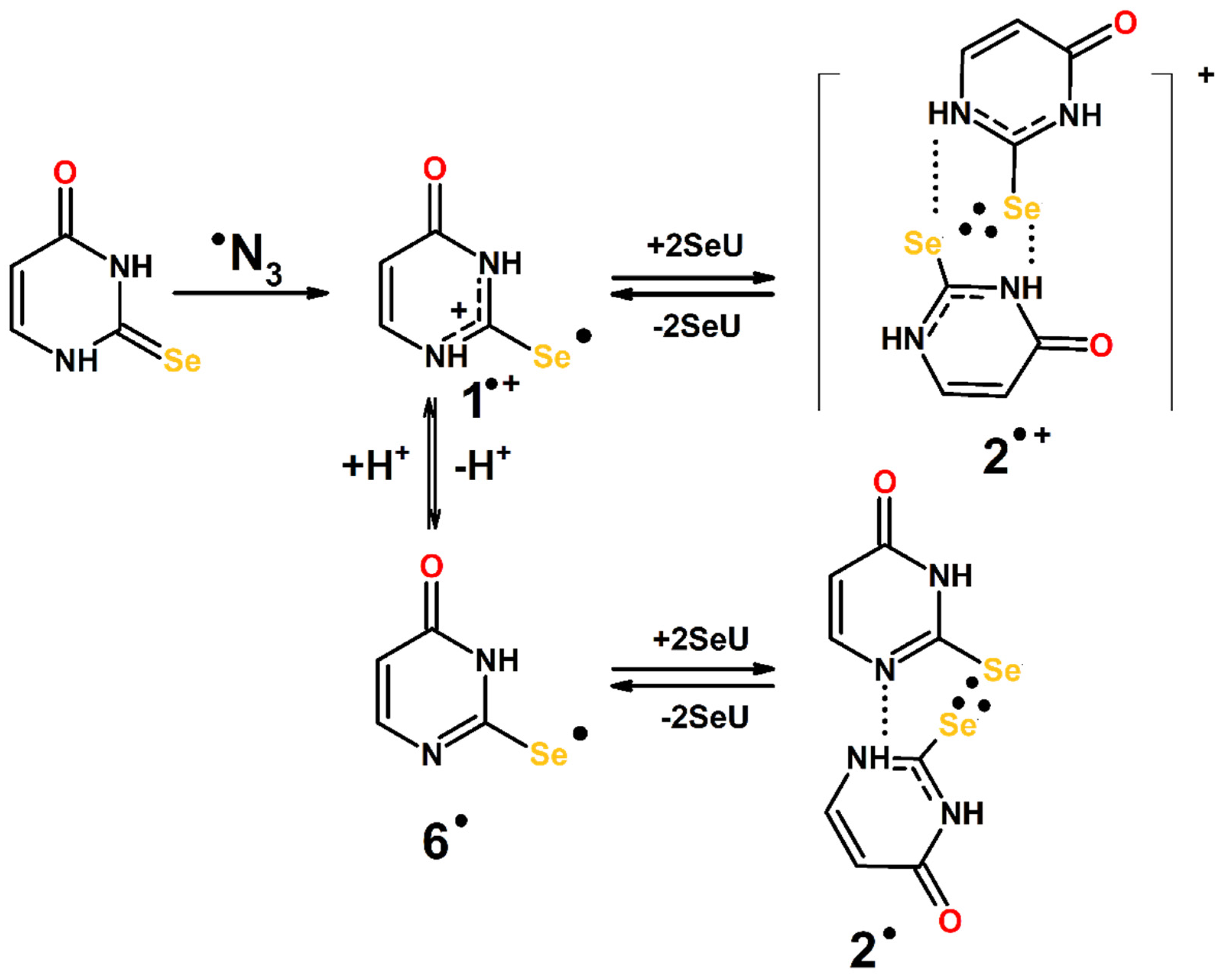
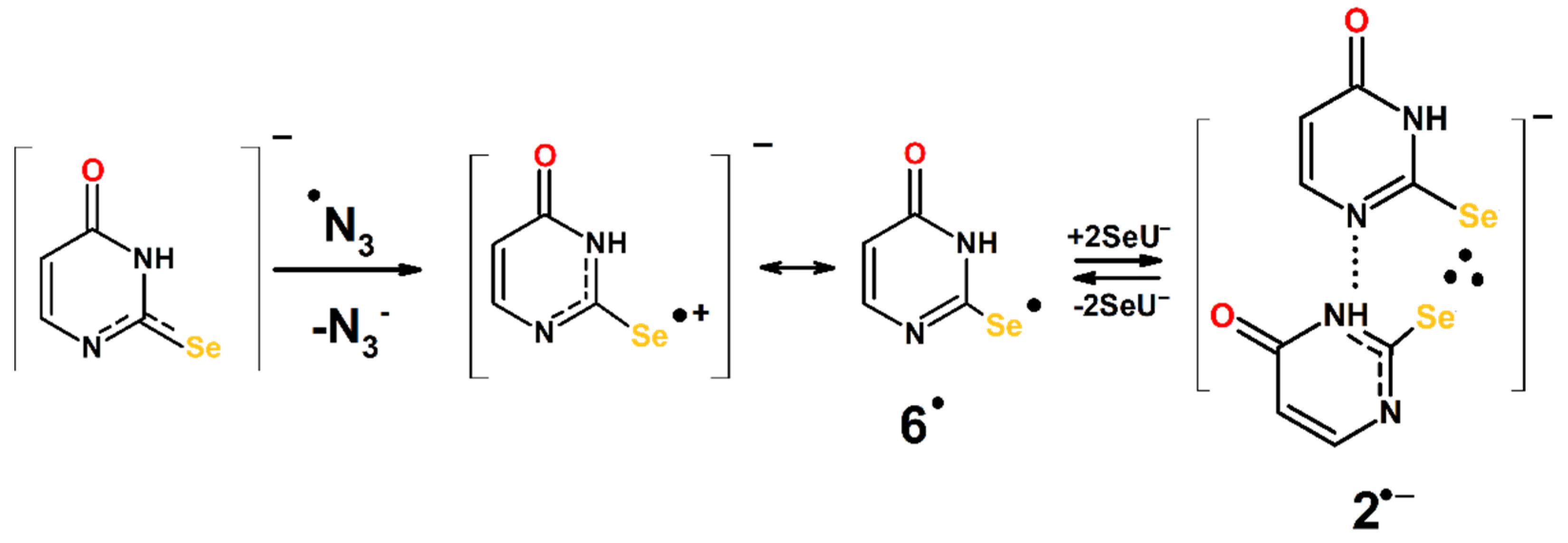
3.3. Mechanism of the ●OH- and ●N3-Induced Oxidation of 2-Selenouracil
3.3.1. Mechanism of the ●OH-Induced Oxidation of 2-Selenouracil
3.3.2. Mechanism of ●N3-Induced Oxidation of 2-Selenouracil
4. Materials and Methods
4.1. Chemicals
4.2. Preparation of Solutions
4.3. Pulse Radiolysis
4.4. Theoretical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Reich, H.J.; Hondal, R.J. Why Nature Choose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. The antioxidant role of selenium and seleno-compounds. Biomed. Pharmacother. 2003, 57, 133–144. [Google Scholar] [CrossRef]
- Mugesh, G.; Du Mont, W.W.; Sies, H. Chemistry of Biologically Important Synthetic Organoselenium Compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B.T. Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology. Chem. Rev. 2004, 104, 6255–6285. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Giles, G.I.; Giles, N.M.; Sies, H. Sulfur and Selenium: The Role of Oxidation State in Protein Structure and Function. Angew. Chem. Int. Ed. 2003, 42, 4742–4758. [Google Scholar] [CrossRef]
- Birringer, M.; Pilawa, S.; Flohé, L. Trends in selenium biochemistry. Nat. Prod. Rep. 2002, 19, 693–718. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Arthur, J.R. Selenium, selenoproteins and human health: A review. Pub. Health Nutr. 2001, 4, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Stolwijk, J.M.; Garje, R.; Sieren, J.C.; Buettner, G.R.; Zakharia, Y. Understanding the Redox Biology of Selenium in the Search of Targeted Cancer Therapies. Antioxidants 2020, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Foster, S.J.; Ganther, H.E. Identification of selenocysteine in glutathione peroxidase by mass spectroscopy. Biochemistry 1983, 22, 5853–5858. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Rad. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Wessjohann, L.A.; Schneider, A.; Abbas, M.; Brand, W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol. Chem. 2007, 388, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- CRC. Handbook of Chemistry and Physics, 87th ed.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Steinmann, D.; Nauser, T.; Koppenol, W.H. Selenium andsulfur in exchange reactions. A comparative study. J. Org. Chem. 2010, 75, 6696–6699. [Google Scholar] [CrossRef]
- Pearson, R.G.; Sobel, H.R.; Songstad, J. Nucleophilic reactivity constants toward methyl iodide and trans-dichlorodi(pyridine)platinum (II). J. Am. Chem. Soc. 1968, 90, 319–326. [Google Scholar] [CrossRef]
- Zielinski, Z.; Presseau, N.; Amorati, R.; Valgimigli, L.; Pratt, D.A. Redox Chemistry of Selenenic Acids and the Insight It Brings on Transition State Geometry in the Reactions of Peroxyl Radicals. J. Am. Chem. Soc. 2014, 136, 1570–1578. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merenyi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueois solutions: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Mishra, B.; Maity, D.K.; Priyadarsini, K.I.; Mohan, H.; Mittal, J.P. One-Electron Oxidation of Selenourea in Aqueous Solution. J. Phys. Chem. A 2004, 108, 1552–1559. [Google Scholar] [CrossRef]
- Mishra, B.; Sharma, A.; Naumov, S.; Priyadarsini, K.I. Novel Reactions of One-Electron Oxidized Radicals of Selenomethionine in Comparison with Methionine. J. Phys. Chem. B 2009, 113, 7709–7715. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Schuchmann, M.N.; Schuchmann, H.-P.; Knolle, W.; Von Sonntag, J.; Von Sonntag, C. Radical Cations in the OH-Radical-Induced Oxidation of Thiourea and Tetramethylthiourea in Aqueous Solution. J. Am. Chem. Soc. 1999, 121, 238–245. [Google Scholar] [CrossRef]
- Bobrowski, K.; Lubis, R. Intermolecular charge transfer involving tryptophan, tyrosine, and three-electron-bonded intermediated derived from methionine. Int. J. Radiat. Biol. 1986, 50, 1039–1050. [Google Scholar]
- Sanaullah; Wilson, G.S.; Glass, R.S. The effect of pH and complexation of amino acid functionality on the redox chemistry of methionine and X-ray structure of [Co(en)2(L-Met)](ClO4)2.H2O. J. Inorg. Biochem. 1994, 55, 87–99. [Google Scholar] [CrossRef]
- Steinmann, D.; Nauser, T.; Beld, J.; Tanner, M.; Gunther, D.; Bounds, P.L.; Koppenol, W.H. Kinetics of Tyrosyl Radical Reduction by Selenocysteine. Biochemistry 2008, 47, 9602–9607. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Priyadarsini, K.I.; Mohan, H. Effect of pH on One-Electron Oxidation Chemistry of Organoselenium Compounds in Aqueous Solutions. J. Phys. Chem. A 2006, 110, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Badiello, R.; Fielden, E.M. Pulse radiolysis of selenium-containing radioprotectors. I. Selenourea. Int. J. Radiat. Biol. 1970, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, K.I.; Mishra, B. Radical cations of some water-soluble organoselenium compounds: Insights from pulse radiolysis studies. Radiat. Phys. Chem. 2008, 77, 1294–1299. [Google Scholar] [CrossRef]
- Tamba, M.; Bonora, S.; Badiello, R. Pulse Radiolysis of Selenium Containing Compounds: Selenomethionine. Z. Naturforsch. 1974, 29, 571–572. [Google Scholar] [CrossRef]
- Nauser, T.; Dockheer, S.; Kissner, R.; Koppenol, W.H. Catalysis of Electron Transfer by Selenocysteine. Biochemistry 2006, 45, 6038–6043. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Kumbhare, L.B.; Jain, V.K.; Priyadarsini, K.I. Pulse radiolysis Studies on Reactions of Hydroxyl Radicals with Selenocystine Derivatives. J. Phys. Chem. B 2008, 112, 4441–4446. [Google Scholar] [CrossRef]
- Tamba, M.; Badiello, R. Pulse- and gamma-radiolysis of selenium-containing compounds: Selenocystine. Int. J. Radiat. Biol. 1973, 23, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, K.I.; Singh, B.G. Selenium Centered Radicals: How Do They Diiffer from Sulfur Centered Radicals? Proc. Natl. Acad. Sci. India Sect. A Phys. Sci. 2016, 86, 459–464. [Google Scholar] [CrossRef]
- Hiller, K.-O.; Masloch, B.; Göbl, M.; Asmus, K.-D. Mechanism of the hydroxyl radical induced oxidation of methionine in aqueous solution. J. Am. Chem. Soc. 1981, 103, 2734–2743. [Google Scholar] [CrossRef]
- Nauser, T.; Steinmann, D.; Grassi, G.; Koppenol, W.H. Why Selenocysteine Replaces Cysteine in Thiredoxin Reductase: A Radical Hypothesis. Biochemistry 2014, 53, 5017–5022. [Google Scholar] [CrossRef] [PubMed]
- Nauser, T.; Steinmann, D.; Koppenol, W.H. Why do proteins use selenocysteine instead of cysteine? Amino Acids 2012, 42, 39–44. [Google Scholar] [CrossRef]
- Schöneich, C. Thiyl radicals and induction of protein degradation. Free Radic. Res. 2016, 50, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Stadtman, T.C. Specific occurrence of selenium in enzymes and amino acid tRNAs. FASEB J. 1987, 1, 375–379. [Google Scholar] [CrossRef]
- Ching, W.M.; Tsai, L.; Wittwer, J. Selenium-Containing Transfer RNAs. Curr. Top. Cell Regul. 1985, 27, 495–507. [Google Scholar]
- Ching, W.M. Characterization of selenium-containing tRNAGlu from Clostridium sticklandii. Arch. Biochem. Biophys. 1986, 244, 137–146. [Google Scholar] [CrossRef]
- Sun, H.; Sheng, J.; Hassan, A.E.A.; Jiang, S.; Gan, J.; Huang, Z. Novel RNA base pair with higher specificity using single selenium atom. Nucleic Acid Res. 2012, 40, 5171–5179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, V.I. Analysis of action of wooble nucleoside modifications on codon-anticodon pairing within the ribosome. J. Mol. Biol. 1994, 240, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Payne, N.R.; Geissler, A.; Button, A.; Sasuclark, A.R.; Schroll, A.L.; Ruggles, E.L.; Gladyshev, V.N.; Hondal, R.J. Comparison of the redox chemistry of sulfur- and selenium-containing analogs of uracil. Free Radic. Biol. Med. 2017, 104, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Kulik, K.; Sadowska, K.; Wielgus, E.; Pacholczyk-Sienicka, B.; Sochacka, E.; Nawrot, B. Different Oxidation Pathways of 2-Selenouracil and 2-Thiouracil, Natural Components of Transfer RNA. Int. J. Mol. Sci. 2020, 21, 5956. [Google Scholar] [CrossRef] [PubMed]
- Sochacka, E.; Bartos, P.; Kraszewska, B.; Nawrot, B. Desulfuration of 2-thiouridine with hydrogen peroxide in the physiological pH range 6.6–7.6 is pH dependent and results in two distinct products. Bioorg. Med. Chem. Lett. 2013, 23, 5803–5805. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, B.; Sochacka, E.; Duchler, M. tRNA structural and functional changes induced by oxidative stress. Cell Mol. Life Sci. 2011, 68, 4023–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, C.; Mo, O.; Yanez, M. A theoretical study of hydration effects on the protropic tautomerism of selenouracils. Org. Biomol. Chem. 2007, 5, 3092–3099. [Google Scholar] [CrossRef] [PubMed]
- Skotnicki, K.; Taras-Goslinska, K.; Janik, I.; Bobrowski, K. Radiation-Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions. Molecules 2019, 24, 4402. [Google Scholar] [CrossRef] [Green Version]
- Schuler, R.H.; Hartzell, A.L.; Behar, B. Track effects in radiation chemistry. Concentration dependence for the scavenging of OH by ferrocyanide in N2O-saturated aqueous solutions. J. Phys. Chem. 1981, 85, 192–199. [Google Scholar] [CrossRef]
- Eyring, E.M. Fast Reactions in Solutions. Surv. Prog. Chem. 1964, 2, 57–89. [Google Scholar]
- Wardman, P. The reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1753. [Google Scholar] [CrossRef] [Green Version]
- Prasanthkumar, K.P.; Suresh, C.H.; Aravindakumar, C.T. Oxidation Reactions of 2-Thiouracil: A Theoretical and Pulse Radiolysis Study. J. Phys. Chem. A 2012, 116, 10712–10720. [Google Scholar] [CrossRef]
- Mautner, H.G. The synthesis and properties of some selenopurines and selenopyrimidines. J. Am. Chem. Soc. 1956, 78, 5292–5294. [Google Scholar] [CrossRef]
- Hug, G.L.; Wang, Y.; Schöneich, C.; Jiang, P.-Y.; Fessenden, R.W. Multiple time scales in pulse radiolysis. Application to bromide solutions and dipeptides. Radiat. Phys. Chem. 1999, 54, 559–566. [Google Scholar] [CrossRef]
- Schuler, R.H.; Patterson, L.K.; Janata, E. Yield for the scavenging of hydroxyl radicals in the radiolysis of nitrous oxide-saturated aqueous solutions. J. Phys. Chem. 1980, 84, 2088–2090. [Google Scholar] [CrossRef]
- Janata, E. Pulse radiolysis conductivity measurements in aqueous solutions with nanosecond time resolution. Radiat. Phys. Chem. 1982, 19, 17–21. [Google Scholar] [CrossRef]
- Veltwisch, D.; Janata, E.; Amus, K.-D. Primary processes in the reaction of OH-radicals with sulphoxides. J. Chem. Soc. Perkin Trans. 1980, 1, 146–153. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Vydrov, A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, J.T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Doig, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113. [Google Scholar] [CrossRef] [Green Version]
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.S.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennuci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Janik, I.; Carmichael, I.; Tripathi, G.N.R. Transient Raman spectra, structure, and thermochemistry of the thiocyanate dimer radical anion in water. J. Chem. Phys. 2017, 146, 214305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janik, I.; Tripathi, G.N.R. The selenocyanate dimer radical anion in water: Transient Raman spectra, structure, and reaction dynamics. J. Chem. Phys. 2019, 150, 094304. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Dupont, C.; Dumont, E.; Jacquemin, D. Superior Performance of Range-Separated Hybrid Functionals for Describing σ* ← σ UV–Vis Signatures of Three-Electron Two-Center Anions. J. Phys. Chem. A 2012, 116, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Milhoj, B.O.; Sauer, S.P.A. Kinetics and Thermodynamics of the Reaction between the •OH Radical and Adenine: A Theoretical Investigation. J. Phys. Chem. A 2015, 119, 6516–6527. [Google Scholar] [CrossRef] [PubMed]
- Frances-Monerris, A.; Merchan, M.; Roca-Sanjuan, D. Mechanism of the OH Radical Addition to Adenine from Quantum-Chemistry Determinations of Reaction Paths and Spectroscopic Tracking of the Intermediates. J. Org. Chem. 2017, 82, 276–288. [Google Scholar] [CrossRef]
- Fourré, I.; Bergés, J. Structural and topological characterization of three-electron bond: The SO radicals. J. Phys. Chem. A 2004, 108, 898–906. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH 4 (HO●) | pH 10 (HO●) | pH 6 (N3●) | pH 10 (N3●) | |
|---|---|---|---|---|
| K | 13,100 M−1 | 4400 M−1 | 11,900 M−1 | 3900 M−1 |
| kforward | 4.6 × 109 M−1s−1 | 3.1 × 109 M−1s−1 | 2.6 × 109 M−1s−1 | 3.7 × 109 M−1s−1 |
| kbackward | 3.6 × 105 s−1 1 | 7.0 × 105 s−1 1 | 2.2 × 105 s−1 1 | 9.5 × 105 s−1 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skotnicki, K.; Janik, I.; Sadowska, K.; Leszczynska, G.; Bobrowski, K. Radiation-Induced Oxidation Reactions of 2-Selenouracil in Aqueous Solutions: Comparison with Sulfur Analog of Uracil. Molecules 2022, 27, 133. https://doi.org/10.3390/molecules27010133
Skotnicki K, Janik I, Sadowska K, Leszczynska G, Bobrowski K. Radiation-Induced Oxidation Reactions of 2-Selenouracil in Aqueous Solutions: Comparison with Sulfur Analog of Uracil. Molecules. 2022; 27(1):133. https://doi.org/10.3390/molecules27010133
Chicago/Turabian StyleSkotnicki, Konrad, Ireneusz Janik, Klaudia Sadowska, Grazyna Leszczynska, and Krzysztof Bobrowski. 2022. "Radiation-Induced Oxidation Reactions of 2-Selenouracil in Aqueous Solutions: Comparison with Sulfur Analog of Uracil" Molecules 27, no. 1: 133. https://doi.org/10.3390/molecules27010133
APA StyleSkotnicki, K., Janik, I., Sadowska, K., Leszczynska, G., & Bobrowski, K. (2022). Radiation-Induced Oxidation Reactions of 2-Selenouracil in Aqueous Solutions: Comparison with Sulfur Analog of Uracil. Molecules, 27(1), 133. https://doi.org/10.3390/molecules27010133