
Investigation of Metastable Low Dimensional Halometallates

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.1.1. [1,10-phenH][Pb3.5I8] and [1,10-phenH2][Pb5I12]·(H2O)
2.1.2. [1,10-phen][Pb2I4]
2.1.3. [1,10-phen]2[Pb5Br10]
2.1.4. [1,10-phenH][SbI4]·(H2O)
2.2. Crystal Structure Solutions
2.2.1. [1,10-phenH][Pb3.5I8]
2.2.2. [1,10-phenH2][Pb5I12]·(H2O)
2.2.3. [1,10-phen][Pb2I4]
2.2.4. [1,10-phen]2[Pb5Br10]
2.2.5. [1,10-phenH][SbI4]·(H2O)
2.3. Crystal Structure Description
2.3.1. [1,10-phenH][Pb3.5I8]
2.3.2. [1,10-phenH2][Pb5I12]·(H2O)
2.3.3. [1,10-phen][Pb2I4]
2.3.4. [1,10-phen]2[Pb5Br10]
2.3.5. [1,10-phenH][SbI4]·(H2O)
2.4. Infrared Spectroscopy
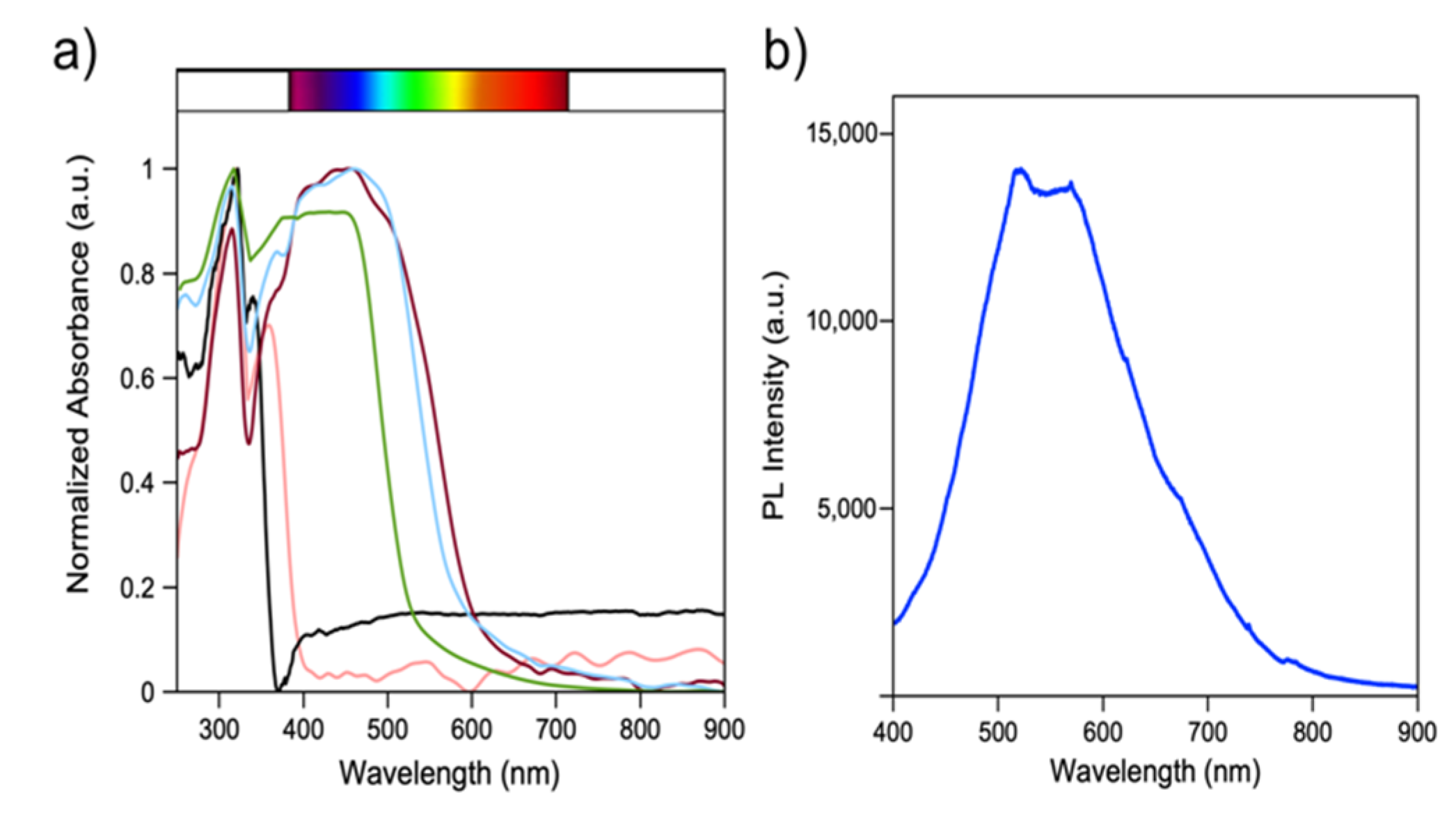
2.5. Optical Properties
2.6. Bandgap Calculations
3. Materials and Methods
3.1. Syntheses
3.1.1. [1,10-phenH][Pb3.5I8] and [1,10-phenH2][Pb5I12]·(H2O)
3.1.2. [1,10-phen][Pb2I4]
3.1.3. [1,10-phen]2[Pb5Br10]
3.1.4. [1,10-phenH][SbI4]·(H2O)
3.2. Single-Crystal X-ray Diffraction (SXRD)
3.3. Powder X-ray Diffraction (PXRD)
3.4. FTIR Spectroscopy
3.5. Optical Properties
3.6. Calculation Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jonderian, A.; Ting, M.; McCalla, E. Metastability in Li–La–Ti–O Perovskite Materials and Its Impact on Ionic Conductivity. Chem. Mater. 2021, 33, 4792–4804. [Google Scholar] [CrossRef]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef]
- Li, W.; Wang, Z.; Deschler, F.; Gao, S.; Friend, R.H.; Cheetham, A.K. Chemically Diverse and Multifunctional Hybrid Organic–Inorganic Perovskites. Nat. Rev. Mater. 2017, 2, 16099. [Google Scholar] [CrossRef]
- Lin, H.; Zhou, C.; Tian, Y.; Siegrist, T.; Ma, B. Low-Dimensional Organometal Halide Perovskites. ACS Energy Lett. 2018, 3, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Saparov, B.; Mitzi, D.B. Organic–Inorganic Perovskites: Structural Versatility for Functional Materials Design. Chem. Rev. 2016, 116, 4558–4596. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.; Nie, W.; Blancon, J.-C.; Stoumpos, C.C.; Asadpour, R.; Harutyunyan, B.; Neukirch, A.J.; Verduzco, R.; Crochet, J.J.; Tretiak, S.; et al. High-Efficiency Two-Dimensional Ruddlesden–Popper Perovskite Solar Cells. Nature 2016, 536, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Akkerman, Q.A.; Manna, L. What Defines a Halide Perovskite? ACS Energy Lett. 2020, 5, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Burns, M.C.; Tershansy, M.A.; Ellsworth, J.M.; Khaliq, Z.; Peterson, L.; Smith, M.D.; zur Loye, H.-C. Layered Heterometallic Iodoplumbate Containing a Novel Pb3Cu6I16 Net: Structure and Optical Properties. Inorg. Chem. 2006, 45, 10437–10439. [Google Scholar] [CrossRef]
- Li, B.; Dai, Q.; Yun, S.; Tian, J. Insights into iodoplumbate Complex Evolution of Precursor Solutions for Perovskite Solar Cells: From Aging to Degradation. J. Mater. Chem. A 2021, 9, 6732–6748. [Google Scholar] [CrossRef]
- Sharenko, A.; Mackeen, C.; Jewell, L.; Bridges, F.; Toney, M.F. Evolution of Iodoplumbate Complexes in Methylammonium Lead Iodide Perovskite Precursor Solutions. Chem. Mater. 2017, 29, 1315–1320. [Google Scholar] [CrossRef]
- Wu, L.-M.; Wu, X.-T.; Chen, L. Structural Overview and Structure–Property Relationships of Iodoplumbate and Iodobismuthate. Coord Chem. Rev. 2009, 253, 2787–2804. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, H.-Y.; Li, J.; Xu, Y.-R.; Xu, Y.-P.; Yang, X.; Zou, G.-D. Hybrid Iodoplumbates with Metal Complexes: Syntheses, Crystal Structures, Bandgaps and Photoelectric Properties. Dalton Trans. 2020, 49, 1803–1810. [Google Scholar] [CrossRef]
- Zhang, B.; Li, J.; Li, L.-Z.; Ren, X.-C.; Pang, M.; Shao, Y.-N. Two New Hybrid Iodoplumbates with Chain-like Cations. Cryst. Growth Des. 2021, 21, 5317–5324. [Google Scholar] [CrossRef]
- Zhu, H.-G.; Xu, Y.; Yu, Z.; Wu, Q.-J.; Fun, H.-K.; You, X.-Z. The Synthesis, Structure of 1-D Lead Halide Adducts: PbI2(L)(L= 2 2′-bipyridine, 1,10-phenanthroline). Polyhedron 1999, 18, 3491–3495. [Google Scholar] [CrossRef]
- Goforth, A.M.; Smith, M.D.; Peterson, L.; zur Loye, H.-C. Preparation and Characterization of Novel Inorganic-Organic Hybrid Materials Containing Rare, Mixed-Halide Anions of Bismuth(III). Inorg. Chem. 2004, 43, 7042–7049. [Google Scholar] [CrossRef]
- Goforth, A.M.; Peterson, L.; Smith, M.D.; zur Loye, H.-C. Syntheses and Crystal Structures of Several Novel Alkylammonium Iodobismuthate Materials Containing the 1,3-bis-(4-piperidinium)propane Cation. J. Solid State Chem. 2005, 178, 3529–3540. [Google Scholar] [CrossRef]
- Tershansy, M.A.; Goforth, A.M.; Gardinier, J.R.; Smith, M.D.; Peterson, L.; zur Loye, H.-C. Solvothermal Syntheses, High- and Low-Temperature Crystal Structures, and Thermochromic Behavior of [1,2-diethyl-3,4,5-trimethyl-pyrazolium]4[Bi4I16] and [1,10-phenanthrolinium][BiI4]·(H2O). Solid State Sci. 2007, 9, 410–420. [Google Scholar] [CrossRef]
- Mitzi, D.B. Synthesis, Structure, and Properties of Organic-Inorganic Perovskites and Related Materials. In Progress in Inorganic Chemistry; Karlin, K.D., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1999; pp. 1–121. [Google Scholar]
- Tershansy, M.A.; Goforth, A.M.; Peterson, L., Jr.; Burns, M.C.; Smith, M.D.; zur Loye, H.-C. Syntheses and Crystal Structures of New Chain-Containing Iodometallate Compounds: [H1,10-phen](H2O)1.41[AgI2], [H1,10-phen](H2O)1.42[CuI2]; [Co (tpy)2][Bi2I8],[Fe(tpy)2][Bi2I8]; [Co(1,10-phen)3][Pb3I8]·H2O, and [Fe(1,10-phen)3][Pb3I8]·0.5(H2O). Solid State Sci. 2007, 9, 895–906. [Google Scholar] [CrossRef]
- Krautscheid, H.; Vielsack, F. Discrete and polymeric iodoplumbates with Pb3I10 Building Blocks: [Pb3I10]4−, [Pb7I22]8−, [Pb10I28]8−, 1∞[Pb3I10]4− and 2∞[Pb7I18]4−. J. Chem. Soc. Dalton Trans. 1999, 2731–2735. [Google Scholar] [CrossRef]
- Yang, H.; Yin, J.; Xu, X.; Fei, H. Enhanced Intrinsic White-Light Emission Upon Near-UV Excitation by Crystal Engineering of Cationic Lead Bromide Layered Materials. J. Mater. Chem. C 2019, 7, 7090–7095. [Google Scholar] [CrossRef]
- Li, H.-H.; Wu, Y.-L.; Dong, H.-J.; Wang, M.; Huang, S.-W.; Chen, Z.-R. Structure and Property Variations of Lead Iodide–Organic Coordination Polymers Tuned by Substituted Groups on Phenanthroline. CrystEngComm 2011, 13, 6766–6773. [Google Scholar] [CrossRef]
- Chamorro, J.R.; McQueen, T.M. Progress toward Solid State Synthesis by Design. Acc. Chem. Res. 2018, 51, 2918–2925. [Google Scholar] [CrossRef] [PubMed]
- Martinolich, A.J.; Neilson, J.R. Toward Reaction-by-Design: Achieving Kinetic Control of Solid State Chemistry with Metathesis. Chem. Mater. 2017, 29, 479–489. [Google Scholar] [CrossRef]
- Kanatzidis, M.G. Discovery-Synthesis, Design, and Prediction of Chalcogenide Phases. Inorg. Chem. 2017, 56, 3158–3173. [Google Scholar] [CrossRef] [PubMed]
- Kanatzidis, M.G.; Poeppelmeier, K.R. Report from the third workshop on future directions of solid state chemistry: The status of solid state chemistry and its impact in the physical sciences. Prog. Solid State Chem. 2007, 36, 1–133. [Google Scholar] [CrossRef]
- Jansen, M.; Schön, J.C. “Design” in Chemical Synthesis—An Illusion? Angew. Chem. Int. Ed. 2006, 45, 3406–3412. [Google Scholar] [CrossRef]
- Felder, J.B.; Yeon, J.; Smith, M.D.; zur Loye, H.-C. Application of a Mild Hydrothermal Method to the Synthesis of Mixed Transition-Metal(II)/Uranium(IV) Fluorides. Inorg. Chem. Front. 2017, 4, 368–377. [Google Scholar] [CrossRef]
- Yeon, J.; Sefat, A.S.; Tran, T.T.; Halasyamani, P.S.; zur Loye, H.-C. Crystal Growth, Structure, Polarization and Magnetic Properties of Cesium Vanadate, Cs2V3O8: A Structure-Property Study. Inorg. Chem. 2013, 52, 6179–6186. [Google Scholar] [CrossRef]
- Bugaris, D.E.; zur Loye, H.-C. Materials Discovery by Flux Crystal Growth: Quaternary and Higher Oxides. Angew. Chem. Int. Ed. 2012, 51, 3780–3811. [Google Scholar] [CrossRef]
- Abeysinghe, D.; Huq, A.; Yeon, J.; Smith, M.D.; zur Loye, H.-C. In Situ Neutron Diffraction Studies of the Flux Crystal Growth of the Reduced Molybdates La4Mo2O11 and Ce4Mo2O11: Revealing Unexpected Mixed-Valent Transient Intermediates and Determining the Sequence of Events during Crystal Growth. Chem. Mater. 2018, 30, 1187–1197. [Google Scholar] [CrossRef]
- Campos-Vallette, M.M.; Clavijo, R.E.; Mendizabal, F.; Zamudio, W.; Baraona, R.; Diaz, G. Infrared Spectrum of the bis-(1,10-phenanthroline) Cu(I) and Cu(II) perchlorate Complexes. Vib Spectrosc. 1996, 12, 37–44. [Google Scholar] [CrossRef]
- Cook, D. Vibrational Spectra of Pyridinium Salts. Can. J. Chem. 1961, 39, 2009–2024. [Google Scholar] [CrossRef]
- Zhu, L.-H.; Zeng, M.-H.; Ye, B.-H.; Chen, X.-M. Hydrothermal Synthesis and Crystal Structure of a New Oxalato-bridged Lead(II) Polymer: {[Pb(phen)2(ox)]·5H20}n (phen = 1,10-phenanthroline, ox = oxalate). Z Anorg. Allg. Chem. 2004, 630, 952–955. [Google Scholar] [CrossRef]
- Wang, H.; Han, S.; Dun, L.; Zhang, B.; Chen, X.; Wang, J.; Li, C.; Liu, C. Crystal Structure, Thermal Behavior, Luminescence and Theoretical Calculation of a New Pb(II) Coordination Complex. J. Mol. Struct. 2020, 1222, 128950. [Google Scholar] [CrossRef]
- Glatfelter, A.; Dybowski, C.; Bai, S.; Kragten, D.; Blake, M.J.; Segarra, S.; Perry, D.L. Infrared Studies of Lead(II) Halide-1,10-phenanthroline Photosensitive Materials. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009, 71, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, N.; Inoue, H. Iron(II) and Nickel(II) Mixed-ligand Complexes Containing 1,10-phenanthroline and 4,7-diphenyl-1,10-phenanthroline. Transit. Met. Chem. 1999, 24, 210–217. [Google Scholar]
- Yue, C.-Y.; Sun, H.-X.; Liu, Q.-X.; Wang, X.-M.; Yuan, Z.-S.; Wang, J.; Wu, J.-H.; Hu, B.; Lei, X.-W. Organic Cation Directed Hybrid Lead Halides of Zero-Dimensional to Two-Dimensional Structures with Tunable Photoluminescence Properties. Inorg. Chem. Front. 2019, 6, 2709–2717. [Google Scholar] [CrossRef]
- Zhong, M.; Zhang, S.; Huang, L.; You, J.; Wei, Z.; Liu, X.; Li, J. Large-scale 2D PbI2 Monolayers: Experimental Realization and Their Indirect Bandgap Related Properties. Nanoscale 2017, 9, 3736–3741. [Google Scholar] [CrossRef] [PubMed]
- APEX3 Version 2019.1-0 and SAINT+ Version 8.40A.; Bruker Nano, Inc.: Madison, WI, USA, 2019.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-ray Sources for Single-Crystal Structure Determination. J. Appl Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. J. Chem. Phys. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Kubelka, P. New contributions to the Optics of Intensely Light-Scattering Materials. J. Opt. Soc. Am. 1948, 38, 448–457. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A Modular and Open-Source Software Project for Quantum Simulations of Materials. Can. J. Chem. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef] [Green Version]
- Otero-de-la-Roza, A.; Johnson, E.R. Van der Waals Interactions in Solids Using the Exchange-Hole Dipole Moment Model. J. Chem. Phys. 2012, 136, 174109. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Golec, B.; Das, P.; Bahou, M.; Lee, Y.P. Infrared Spectra of the 1-pyridinium (C5H5NH+) Cation and Pyridinyl (C5H5NH and 4-C5H6N) Radicals Isolated in Solid Para-hydrogen. J. Phys. Chem. A 2013, 117, 13680–13690. [Google Scholar] [CrossRef]
- Ghosh, B.; Chakraborty, S.; Wei, H.; Guet, C.; Li, S.; Mhaisalkar, S.; Mathews, N. Poor Photovoltaic Performance of Cs3Bi2I9: An Insight through First-Principles Calculations. J. Phys. Chem. C 2017, 121, 17062–17067. [Google Scholar] [CrossRef]
- Giorgi, G.; Fujisawa, J.I.; Segawa, H.; Yamashita, K. Small Photocarrier Effective Masses Featuring Ambipolar Transport in Methylammonium Lead Iodide Perovskite: A Density Functional Analysis. J. Phys. Chem. Lett. 2013, 4, 4213–4216. [Google Scholar] [CrossRef]
- Gou, G.Y.; Bennett, J.W.; Takenaka, H.; Rappe, A.M. Post Density Functional Theoretical Studies of Highly Polar Semiconductive Pb(Ti1-XNix)O3-x Solid Solutions: Effects of Cation Arrangement on Band Gap. Phys. Rev. B Condens. Matter 2011, 83, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lehner, A.J.; Fabini, D.H.; Evans, H.A.; Hébert, C.A.; Smock, S.R.; Hu, J.; Wang, H.; Zwanziger, J.W.; Chabinyc, M.L.; Seshadri, R. Crystal and Electronic Structures of Complex Bismuth Iodides A3Bi2I9 (A = K, Rb, Cs) Related to Perovskite: Aiding the Rational Design of Photovoltaics. Chem. Mater. 2015, 27, 7137–7148. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.J.; Ko, U.H.; Hwang, S.G.; Kim, Y.S.; Jong, U.G.; Kye, Y.H.; Ri, C.H. First-Principles Study on Material Properties and Stability of Inorganic Halide Perovskite Solid Solutions CsPb(I1-XBrx)3. Phys. Rev. Mater. 2020, 4, 1–9. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Formula | [C12H9N2][Pb3.5I8] (1) | [C12H10N2][Pb5I12]·(H2O) (2) | [C12H8N2][Pb2I4] (3) | [C12H8N2]2[Pb5Br10] (4) | [C12H9N2][SbI4]·(H2O) (5) |
|---|---|---|---|---|---|
| Formula weight | 2024.16 | 2756.97 | 1102.18 | 2195.46 | 828.58 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Triclinic |
| Space group | C2/m | Cmme | I2/a | P21/c | |
| a, Å | 26.2810(3) | 17.9551(3) | 15.6100(5) | 13.2524(14) | 7.7387(9) |
| b, Å | 13.0845(18) | 21.0222(3) | 16.0694(5) | 17.8894(15) | 11.1522(13) |
| c, Å | 4.4842(6) | 10.7977(2) | 7.9333(3) | 8.4643(9) | 12.0271(13) |
| α, deg. | 90 | 90 | 90 | 90 | 77.477(4) |
| β, deg. | 97.137(5) | 90 | 95.425(1) | 107.942(4) | 79.148(4) |
| γ, deg. | 90 | 90 | 90 | 90 | 70.754(4) |
| V, Å3 | 1530.1(4) | 4075.65(12) | 1981.10(12) | 1909.1(3) | 948.94(19) |
| ρcalcd, g/cm3 | 4.394 | 4.493 | 3.695 | 3.819 | 2.900 |
| Radiation (λ, Å) | MoKα, 0.71073 | MoKα, 0.71073 | MoKα, 0.71073 | MoKα, 0.71073 | MoKα, 0.71073 |
| µ, mm–1 | 30.014 | 29.686 | 23.200 | 32.462 | 7.958 |
| T, K | 301(2) | 299(2) | 299(2) | 302(2) | 301(2) |
| Crystal dim., mm3 | 0.05 × 0.04 × 0.02 | 0.07 × 0.06 × 0.05 | 0.05 × 0.02 × 0.01 | 0.07 × 0.03 × 0.01 | 0.06 × 0.03 × 0.02 |
| 2θ range, deg. | 2.81–31.77 | 4.81–56.58 | 5.07–59.19 | 5.55–52.74 | 4.80–59.95 |
| Reflections collected | 27,193 | 131,166 | 30,501 | 58,421 | 62,776 |
| Data/restraints/parameters | 1414/0/67 | 2684/0/69 | 2782/1/93 | 3906/0/196 | 5519/7/200 |
| Rint | 0.0524 | 0.0491 | 0.0601 | 0.0800 | 0.0340 |
| Goodness of fit | 1.120 | 1.181 | 1.057 | 1.175 | 1.065 |
| R1(I > 2σ(I)) | 0.0427 | 0.0164 | 0.0443 | 0.0422 | 0.0204 |
| wR2 (all data) | 0.1160 | 0.0303 | 0.1275 | 0.0851 | 0.0388 |
| Largest diff. peak/hole, e∙Å−3 | 3.80/−1.43 | 0.84/−0.66 | 1.39/−1.90 | 1.34/−1.24 | 0.56/−0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keerthisinghe, N.; Christian, M.S.; Berseneva, A.A.; Morrison, G.; Klepov, V.V.; Smith, M.D.; zur Loye, H.-C. Investigation of Metastable Low Dimensional Halometallates. Molecules 2022, 27, 280. https://doi.org/10.3390/molecules27010280
Keerthisinghe N, Christian MS, Berseneva AA, Morrison G, Klepov VV, Smith MD, zur Loye H-C. Investigation of Metastable Low Dimensional Halometallates. Molecules. 2022; 27(1):280. https://doi.org/10.3390/molecules27010280
Chicago/Turabian StyleKeerthisinghe, Navindra, Matthew S. Christian, Anna A. Berseneva, Gregory Morrison, Vladislav V. Klepov, Mark D. Smith, and Hans-Conrad zur Loye. 2022. "Investigation of Metastable Low Dimensional Halometallates" Molecules 27, no. 1: 280. https://doi.org/10.3390/molecules27010280