
Structural Modeling of TRPA1 Ion Channel—Determination of the Binding Site for Antagonists


Abstract
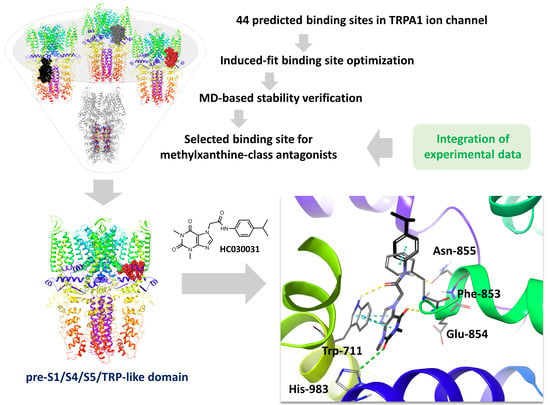
:
1. Introduction
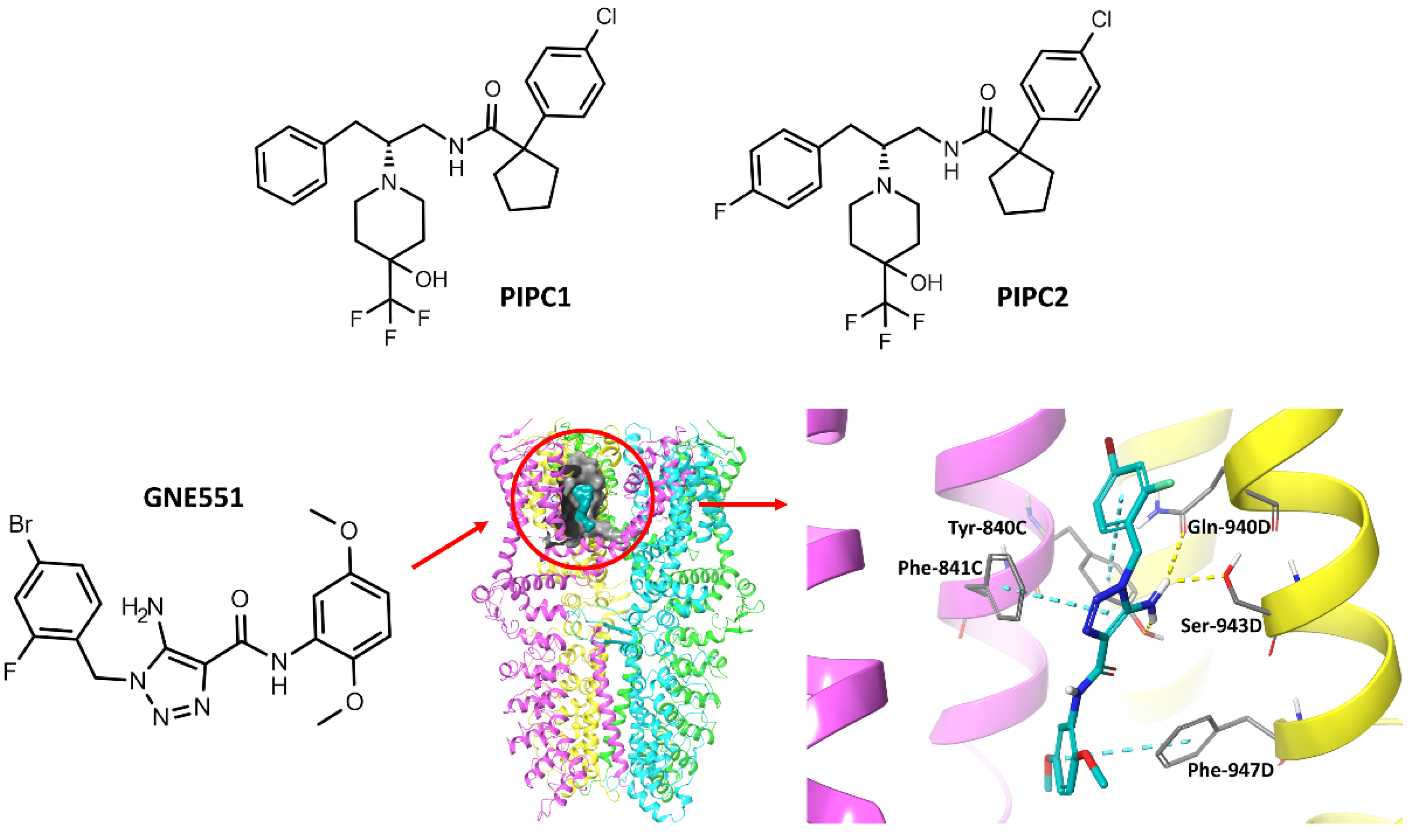
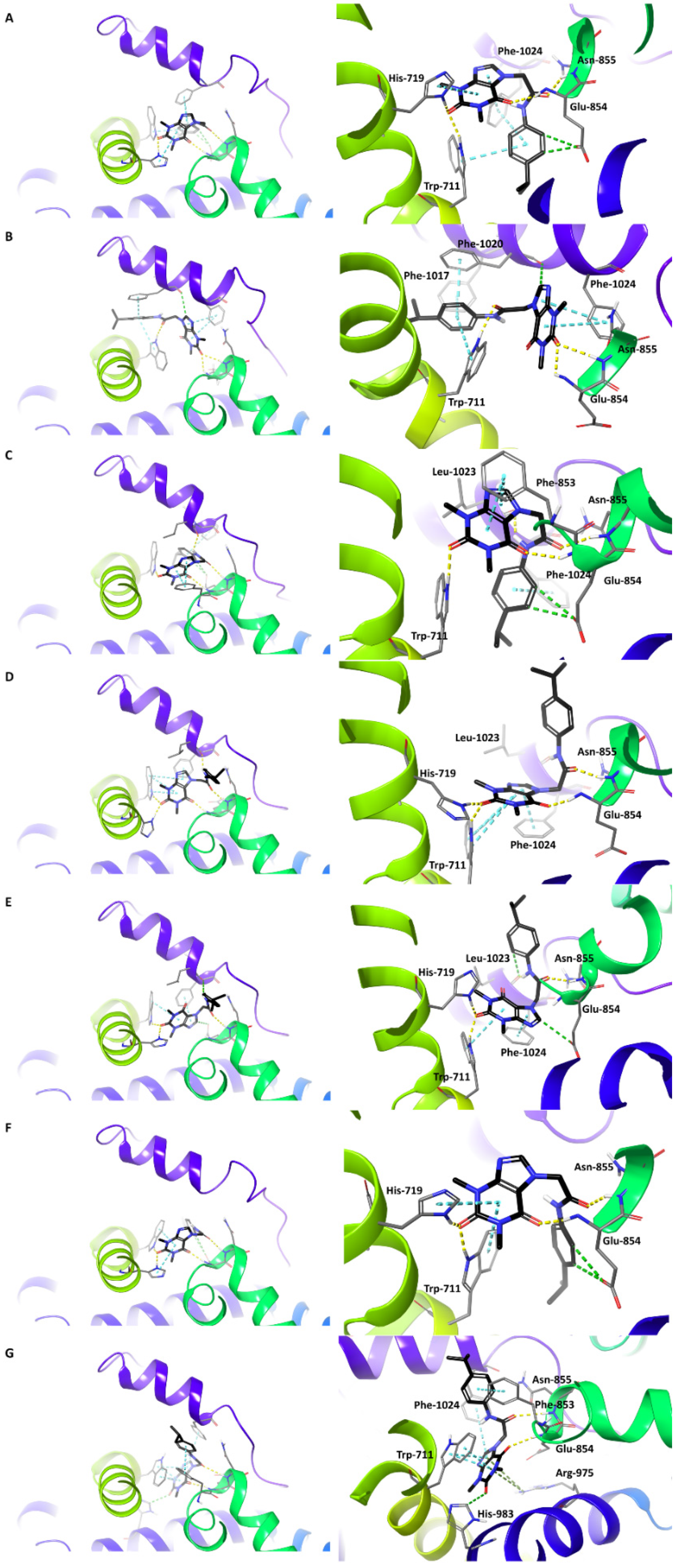
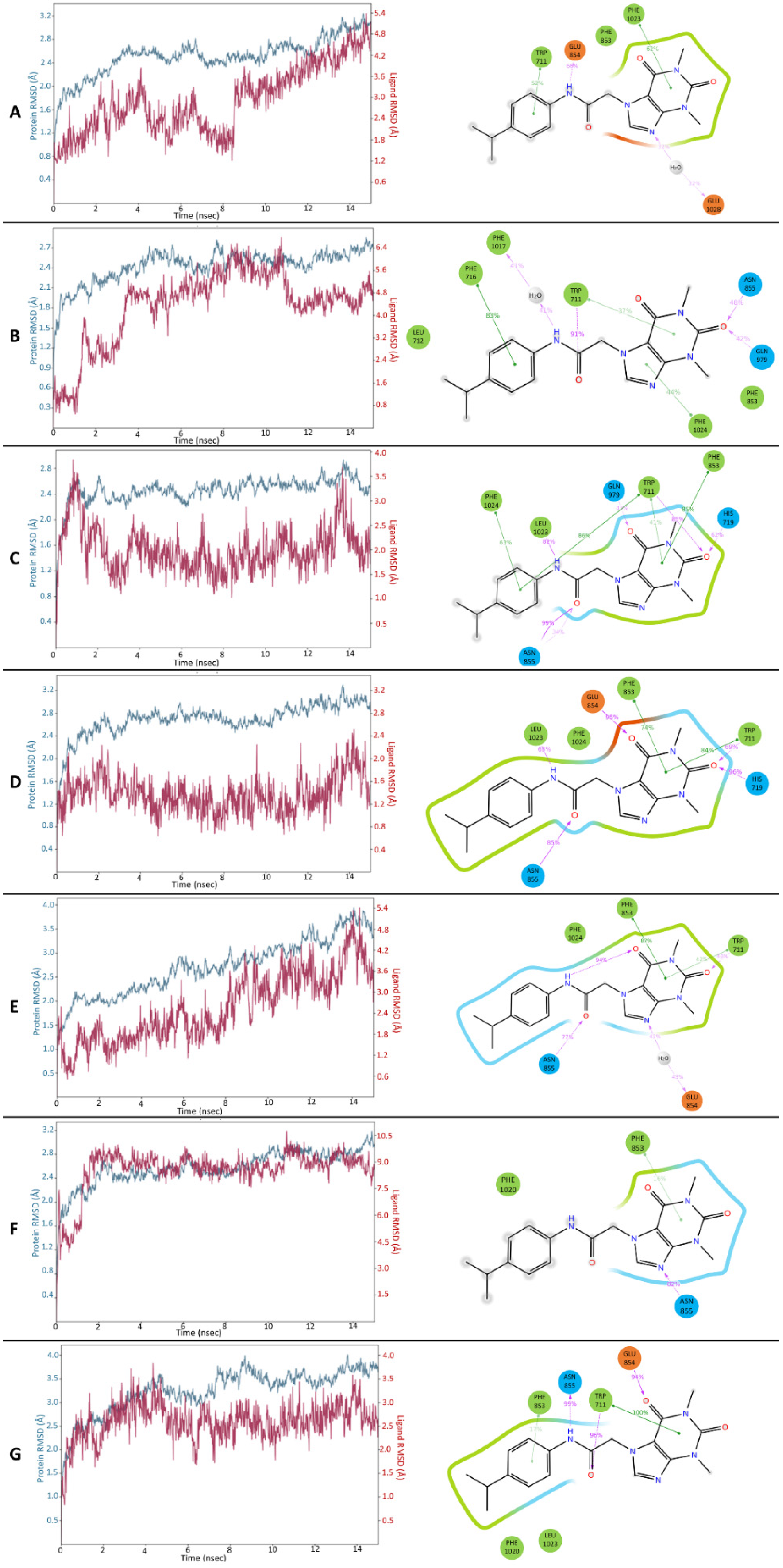
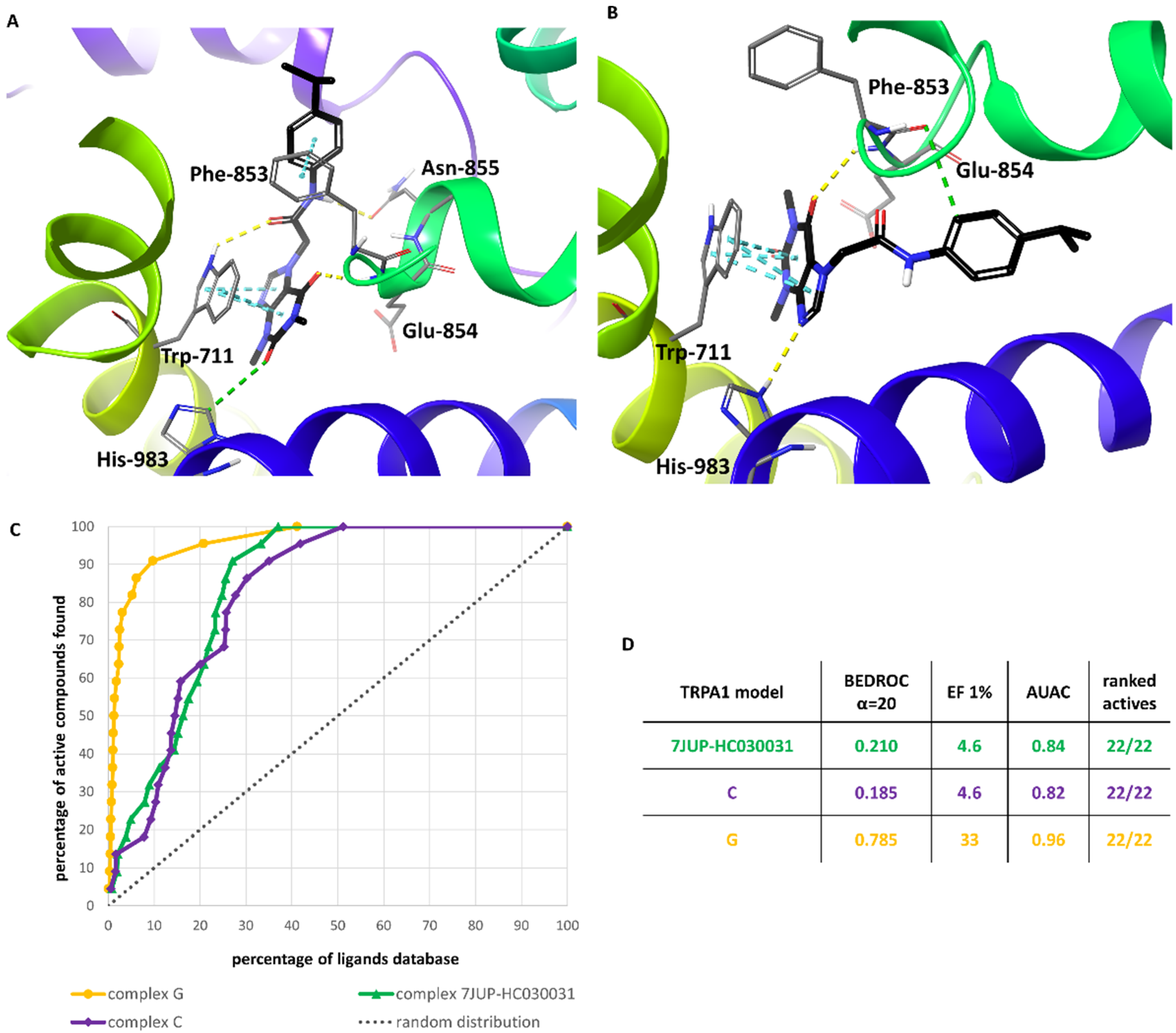
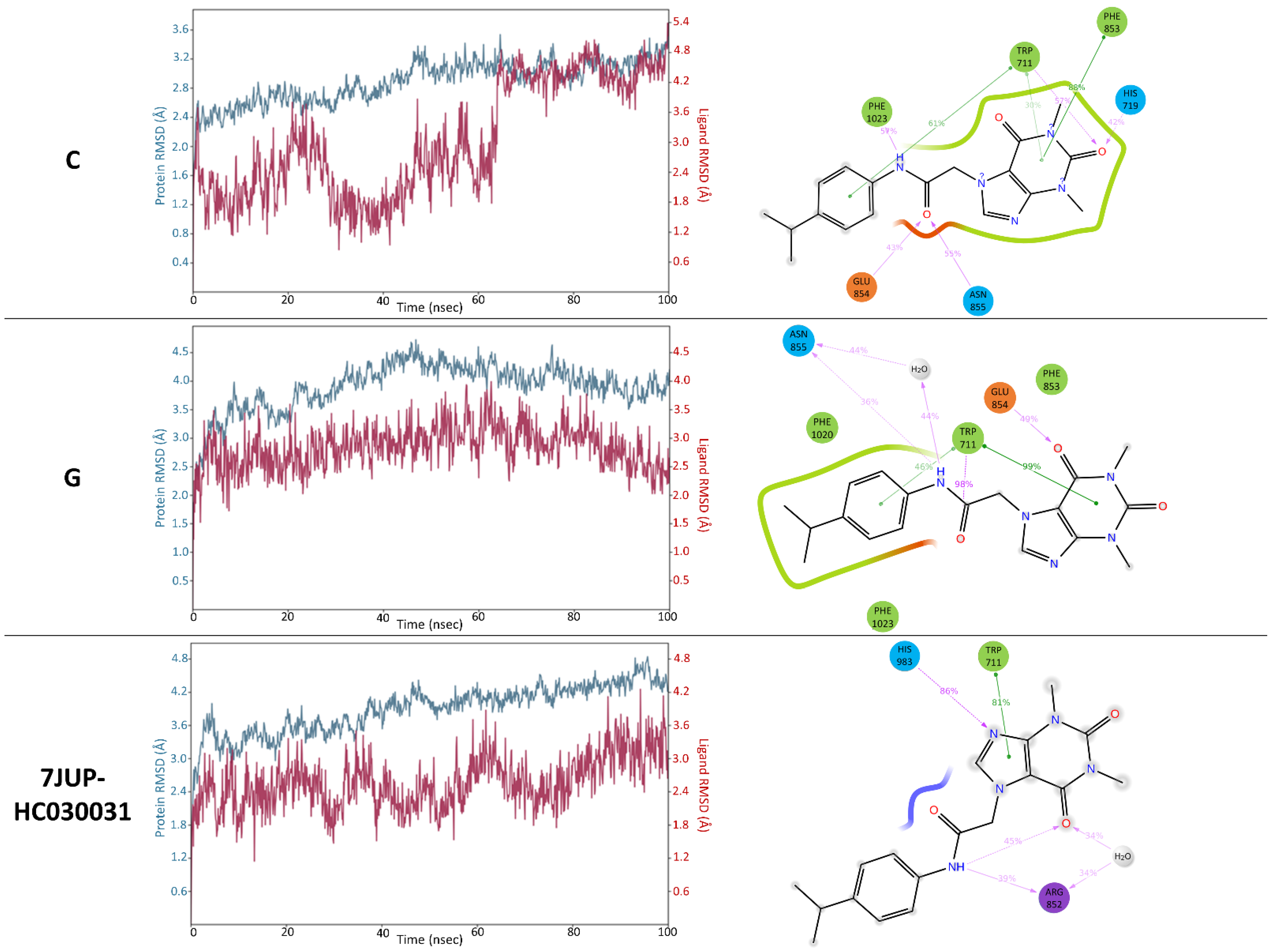
2. Results and Discussion
3. Materials and Methods
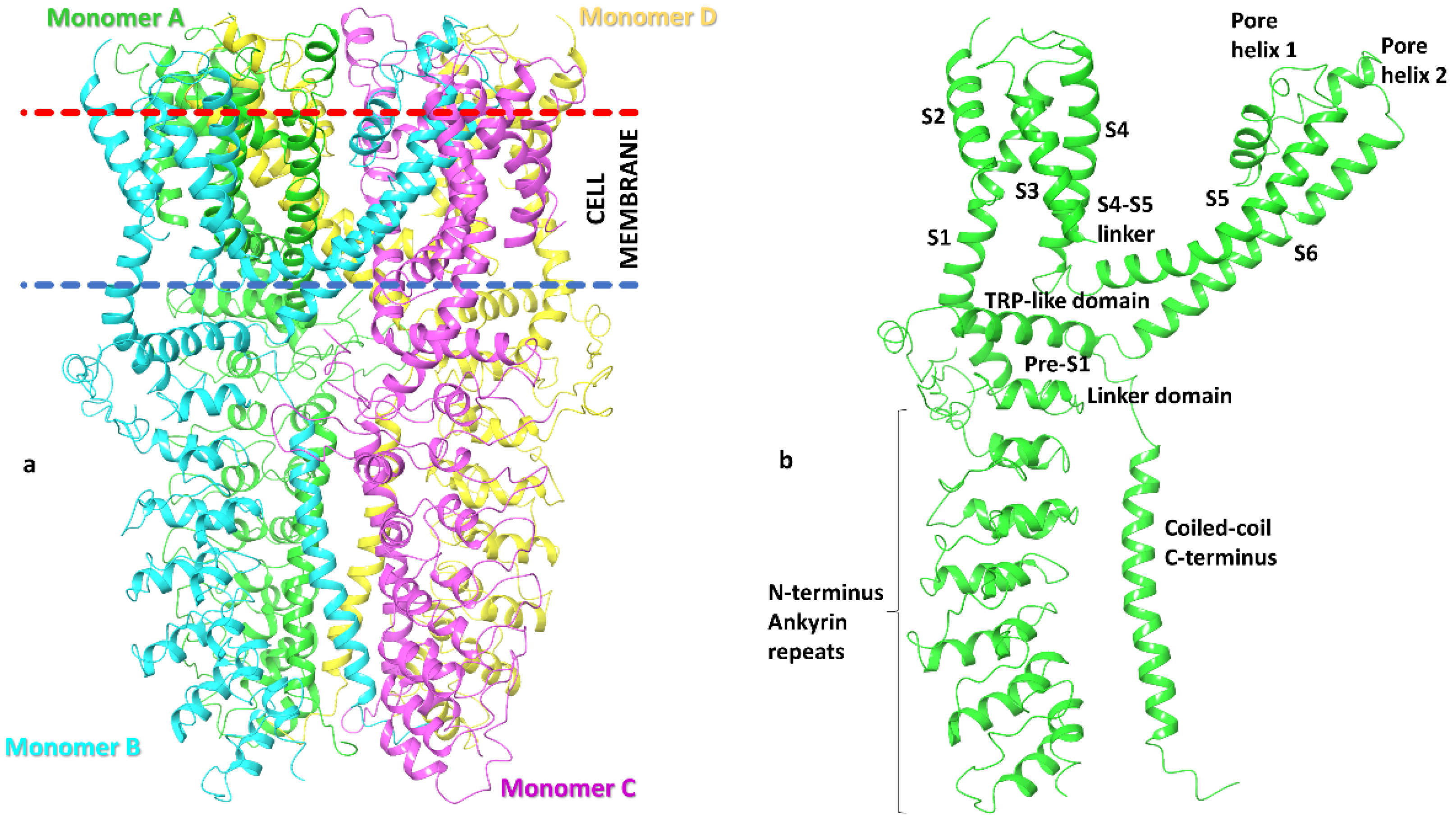
3.1. Preparation of TRPA1 Ion Channel Models
3.2. TRPA1 Binding Site Prediction
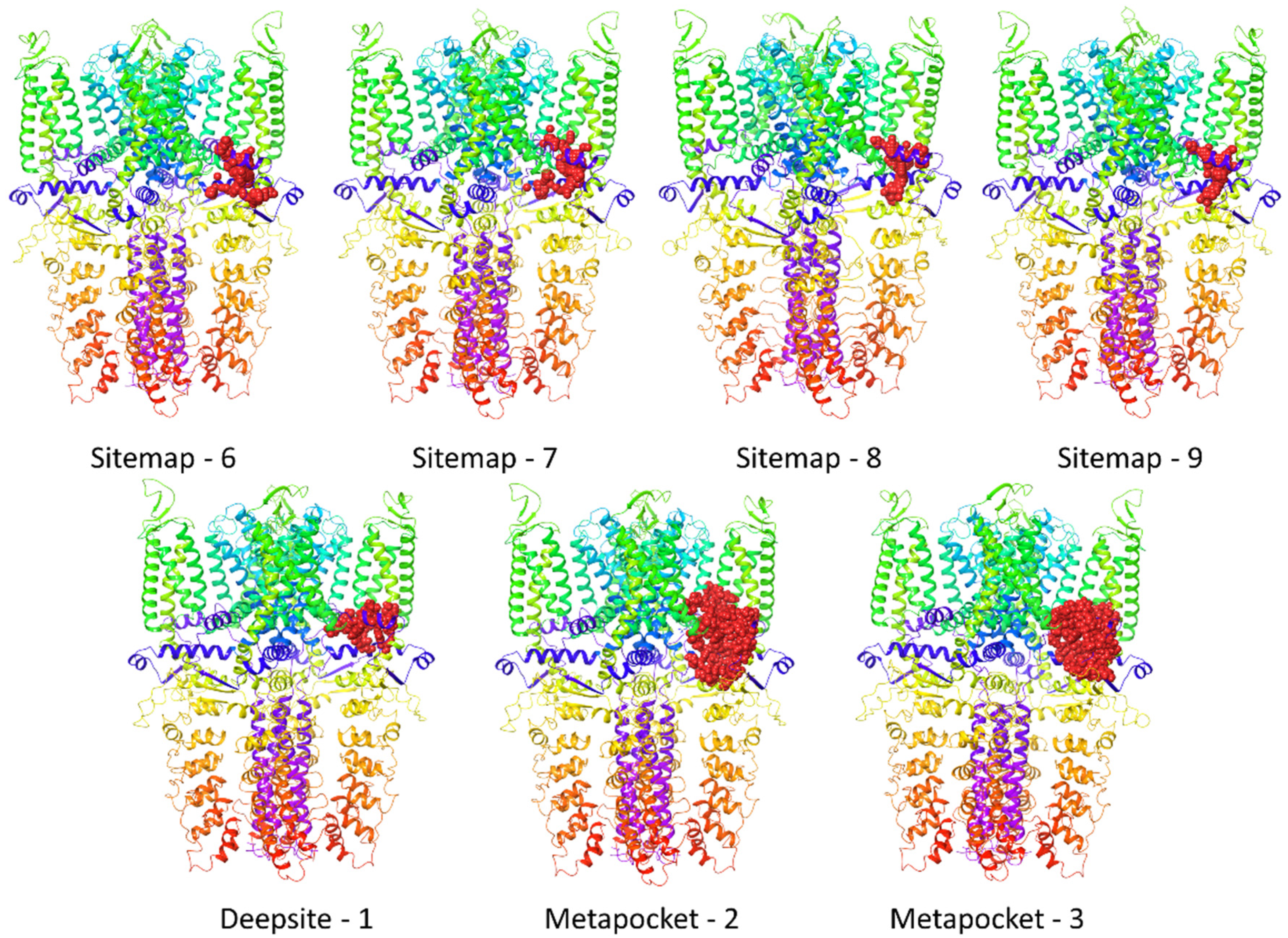
3.2.1. SiteMap (Schrödinger, LLC, New York, NY, 2021)
3.2.2. MetaPocket 2.0 Server
3.2.3. DeepSite
3.2.4. CASTp 3.0
3.3. The Procedure of HC-030031 Docking to TRPA1 Binding Sites
3.4. MD Simulations of HC-030031-TRPA1 Complex
3.5. Validation of the Selected HC-030031-TRPA1 Interaction Models—The Retrospective Virtual Screening Benchmark
3.6. Comparison of the Optimized Models with the PDB Structure 7JUP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Logashina, Y.A.; Korolkova, Y.V.; Kozlov, S.A.; Andreev, Y.A. TRPA1 Channel as a Regulator of Neurogenic Inflammation and Pain: Structure, Function, Role in Pathophysiology, and Therapeutic Potential of Ligands. Biochem. Mosc. 2019, 84, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.C.; Pryde, D.C.; Yoger, K.E.; Padilla, K.M.; Antonio, B.M.; Han, S.; Shanmugasundaram, V.; Gerlach, A.C. TRPA1 Ankyrin Repeat Six Interacts with a Small Molecule Inhibitor Chemotype. Proc. Natl. Acad. Sci. USA 2018, 115, 12301–12306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, M.M.; McAlexander, M.A.; Bíró, T.; Szallasi, A. Transient Receptor Potential Channels as Therapeutic Targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Armache, J.-P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 Ion Channel Suggests Regulatory Mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hackos, D.H. TRPA1 as a Drug Target--Promise and Challenges. Naunyn Schmiedebergs Arch. Pharm. 2015, 388, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.R.; Gerhold, K.A.; Bifolck-Fisher, A.; Liu, Q.; Patel, K.N.; Dong, X.; Bautista, D.M. TRPA1 Is Required for Histamine-Independent, Mas-Related G Protein-Coupled Receptor-Mediated Itch. Nat. Neurosci. 2011, 14, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Morales, J.F.; Gracheva, E.O.; Julius, D. Cytoplasmic Ankyrin Repeats of Transient Receptor Potential A1 (TRPA1) Dictate Sensitivity to Thermal and Chemical Stimuli. Proc. Natl. Acad. Sci. USA 2011, 108, E1184–E1191. [Google Scholar] [CrossRef] [Green Version]
- Moparthi, L.; Kjellström, S.; Kjellbom, P.; Filipovic, M.R.; Zygmunt, P.M.; Johanson, U. Electrophile-Induced Conformational Switch of the Human TRPA1 Ion Channel Detected by Mass Spectrometry. Int. J. Mol. Sci. 2020, 21, 6667. [Google Scholar] [CrossRef]
- Suo, Y.; Wang, Z.; Zubcevic, L.; Hsu, A.L.; He, Q.; Borgnia, M.J.; Ji, R.-R.; Lee, S.-Y. Structural Insights into Electrophile Irritant Sensing by the Human TRPA1 Channel. Neuron 2020, 105, 882–894.e5. [Google Scholar] [CrossRef]
- Zhao, J.; Lin King, J.V.; Paulsen, C.E.; Cheng, Y.; Julius, D. Irritant-Evoked Activation and Calcium Modulation of the TRPA1 Receptor. Nature 2020, 585, 141–145. [Google Scholar] [CrossRef]
- Chernov-Rogan, T.; Gianti, E.; Liu, C.; Villemure, E.; Cridland, A.P.; Hu, X.; Ballini, E.; Lange, W.; Deisemann, H.; Li, T.; et al. TRPA1 Modulation by Piperidine Carboxamides Suggests an Evolutionarily Conserved Binding Site and Gating Mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 26008–26019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton, H.T.; Phan, T.X.; Abramyan, A.M.; Shi, L.; Ahern, G.P. Identification of a Putative Binding Site Critical for General Anesthetic Activation of TRPA1. Proc. Natl. Acad. Sci. USA 2017, 114, 3762–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Reese, R.; Vu, S.; Rougé, L.; Shields, S.D.; Kakiuchi-Kiyota, S.; Chen, H.; Johnson, K.; Shi, Y.P.; Chernov-Rogan, T.; et al. A Non-Covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 2020, 109, 273–284.e4. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, M.J.; Filipovic, M.R.; Leffler, A.; de la Roche, J.; Kistner, K.; Fischer, M.J.; Fleming, T.; Zimmermann, K.; Ivanovic-Burmazovic, I.; Nawroth, P.P.; et al. Methylglyoxal Activates Nociceptors through Transient Receptor Potential Channel A1 (TRPA1): A Possible Mechanism of Metabolic Neuropathies. J. Biol. Chem. 2012, 287, 28291–28306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrini, A.; Joseph, V.; Dourado, M.; Reese, R.M.; Shields, S.D.; Rougé, L.; Bravo, D.D.; Chernov-Rogan, T.; Austin, C.D.; Chen, H.; et al. A TRPA1 Inhibitor Suppresses Neurogenic Inflammation and Airway Contraction for Asthma Treatment. J. Exp. Med. 2021, 218, e20201637. [Google Scholar] [CrossRef]
- Klement, G.; Eisele, L.; Malinowsky, D.; Nolting, A.; Svensson, M.; Terp, G.; Weigelt, D.; Dabrowski, M. Characterization of a Ligand Binding Site in the Human Transient Receptor Potential Ankyrin 1 Pore. Biophys. J. 2013, 104, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Saito, S.; Mori, Y.; Itoh, S.G.; Okumura, H.; Tominaga, M. Structural Basis of TRPA1 Inhibition by HC-030031 Utilizing Species-Specific Differences. Sci. Rep. 2016, 6, 37460. [Google Scholar] [CrossRef] [Green Version]
- Ye, W.; Tu, Y.-H.; Cooper, A.J.; Zhang, Z.; Katritch, V.; Liman, E.R. Activation Stoichiometry and Pore Architecture of TRPA1 Probed with Channel Concatemers. Sci. Rep. 2018, 8, 17104. [Google Scholar] [CrossRef] [Green Version]
- Kravchenko, A.D.; Pyatigorskaya, N.V.; Brkich, G.E.; Yevsieieva, L.V.; Kyrychenko, A.V.; Kovalenko, S.M. Synthesis, Molecular Docking, ADMET Study and In Vitro Pharmacological Research of 7-(2-Chlorophenyl)-4-(4-Methylthiazol-5-Yl)-4,6,7,8-Tetrahydroquinoline-2,5(1H,3H)-Dione as a Promising Non-Opioid Analgesic Drug. Res. Results Pharmacol. 2022, 8, 1–11. [Google Scholar] [CrossRef]
- Christensen, A.P.; Akyuz, N.; Corey, D.P. The Outer Pore and Selectivity Filter of TRPA1. PLoS ONE 2016, 11, e0166167. [Google Scholar] [CrossRef]
- Terrett, J.A.; Chen, H.; Shore, D.G.; Villemure, E.; Larouche-Gauthier, R.; Déry, M.; Beaumier, F.; Constantineau-Forget, L.; Grand-Maître, C.; Lépissier, L.; et al. Tetrahydrofuran-Based Transient Receptor Potential Ankyrin 1 (TRPA1) Antagonists: Ligand-Based Discovery, Activity in a Rodent Asthma Model, and Mechanism-of-Action via Cryogenic Electron Microscopy. J. Med. Chem. 2021, 64, 3843–3869. [Google Scholar] [CrossRef] [PubMed]
- Validation Protocol—3J9P. Available online: http://files.rcsb.org/pub/pdb/validation_reports/j9/3j9p/3j9p_full_validation.pdf (accessed on 26 February 2021).
- Validation Protocol—6PQQ. Available online: https://files.rcsb.org/pub/pdb/validation_reports/pq/6pqq/6pqq_full_validation.pdf (accessed on 26 February 2021).
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Souaille, C.; Estienne, F.; Baurin, N.; Kroemer, R.T. Large-Scale Comparison of Four Binding Site Detection Algorithms. J. Chem. Inf. Model. 2010, 50, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Huang, B. MetaPocket: A Meta Approach to Improve Protein Ligand Binding Site Prediction. OMICS J. Integr. Biol. 2009, 13, 325–330. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, Y.; Lin, B.; Schroeder, M.; Huang, B. Identification of Cavities on Protein Surface Using Multiple Computational Approaches for Drug Binding Site Prediction. Bioinformatics 2011, 27, 2083–2088. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Preti, D.; Saponaro, G.; Szallasi, A. Transient Receptor Potential Ankyrin 1 (TRPA1) Antagonists. Pharm. Pat. Anal. 2015, 4, 75–94. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Truchon, J.-F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Charifson, P.S. Improved Scoring of Ligand-Protein Interactions Using OWFEG Free Energy Grids. J. Med. Chem. 2001, 44, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.C.; Sonar, P.K.; Rathore, R.; Saraf, S.K. Structural Insights into the Molecular Design of HER2 Inhibitors. Open Pharm. Sci. J. 2016, 3, 164–181. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ankyrin Repeats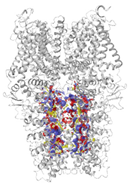 | Channel Pore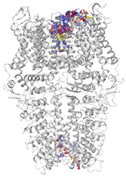 | Transmembrane Region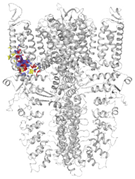 | Pre-S1/S4/S5/ TRP-Like Domain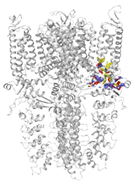 | Below TRP-Like Domain Area | ||
|---|---|---|---|---|---|---|
| 3J9P-based model | number of sites | 6 | 3 | 1 | 5 | 5 |
| average Dscore ± SD | 1.029 ± 0.080 | 1.064 ± 0.110 | 1.213 | 1.019 ± 0.027 | 1.045 ± 0.038 | |
| 6PQQ-based model | number of sites | 4 | 4 | 1 | 4 | 6 |
| average Dscore ± SD | 1.011 ± 0.014 | 1.010 ± 0.052 | 1.041 | 1.137 ± 0.047 | 0.971 ± 0.056 | |
| Total average Dscore ± SD | 1.020 ± 0.013 | 1.037 ± 0.038 | 1.127 ± 0.122 | 1.078 ± 0.074 | 1.008 ± 0.052 | |
| Binding Site 1 | Binding Site 2 | Binding Site 3 | ||
|---|---|---|---|---|
| 3J9P-based model | score | 0.9989 | 0.9968 | 0.9991 |
| site | linker domain | transmembrane domain (S1/S2/S3) | ankyrin repeats | |
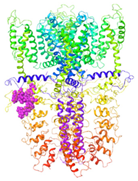 | 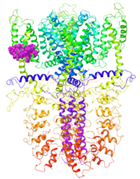 |  | ||
| 6PQQ-based model | score | 0.9997 | 0.9987 | |
| site | pre-S1/S4/S5/TRP-like domain | ankyrin repeats | ||
 | 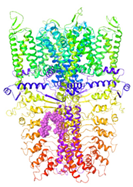 |
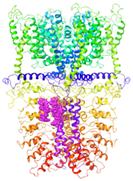
| Binding Site 1 | Binding Site 2 | Binding Site 3 | ||
| 3J9P-based model | score | 4.39 | 3.76 | 3.70 |
| Site | pre-S1/S4/S5/TRP-like domain | Ankyrin repeats | pre-S1/S4/S5/TRP-like domain | |
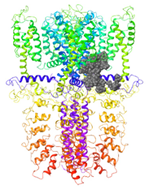 | 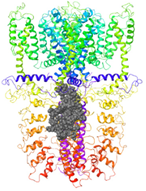 | 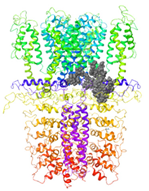 | ||
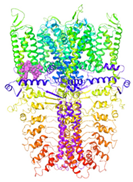
| 6PQQ-based model | score | 8.34 | 5.21 | 4.52 |
| Site | Channel pore | pre-S1/S4/S5/TRP-like domain | pre-S1/S4/S5/TRP-like domain | |
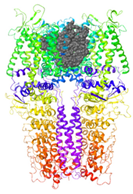 | 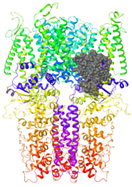 | 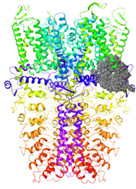 |
| Ankyrin Repeats | Channel Pore | Transmembrane Region | Below TRP-Like Domain Area | ||
|---|---|---|---|---|---|
| 3J9P-based model | number of sites | 8 | 3 | 3 | 4 |
| sites | 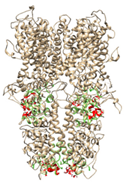 |  | 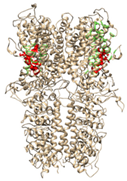 |  | |
| 6PQQ-based model | number of sites | 5 | 2 | 11 | |
| sites |  |  |  |
| 3J9P-Based Model | 6PQQ-Based Model | Total | |
|---|---|---|---|
| Ankyrin repeats | 11 | 6 | 17 |
| Channel pore | 4 | 6 | 10 |
| Transmembrane region | 3 | 3 | 6 |
| pre-S1/S4/S5/ TRP-like domain18 | 7 | 7 | 14 |
| TRP-like domain | 5 | 6 | 11 |
| HC-030031-TRPA1 Complex | Binding Site | IFDScore (kcal/mol) | Docking Score (kcal/mol) |
|---|---|---|---|
| A | Sitemap-6 | −4983.80 | −9908 |
| B | Sitemap-7 | −4982.47 | −9681 |
| C | Sitemap-8 | −4985.76 | −9874 |
| D | Sitemap-9 | −4990.55 | −12,833 |
| E | Metapocket-2 | −4986.84 | −11,574 |
| F | Metapocket-3 | −4981.45 | −9303 |
| G | Deepsite-1 | −4984.21 | −10,698 |
| 3J9P-Based Model | 6PQQ-Based Model | |
|---|---|---|
| PDB original structure | 3J9P | 6PQQ |
| Membrane amino acids | 720–739 | 719–738 |
| 767–785 | 768–793 | |
| 807–823 | 799–820 | |
| 830–850 | 833–853 | |
| 870–892 | 863–891 | |
| 934–957 | 935–961 | |
| Added loops in each homomonomer | 664–669 | 669–676 |
| 748–763 | 754–760 | |
| 786–802 | 1026–1038 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawalska, A.; Kołaczkowski, M.; Bucki, A. Structural Modeling of TRPA1 Ion Channel—Determination of the Binding Site for Antagonists. Molecules 2022, 27, 3077. https://doi.org/10.3390/molecules27103077
Gawalska A, Kołaczkowski M, Bucki A. Structural Modeling of TRPA1 Ion Channel—Determination of the Binding Site for Antagonists. Molecules. 2022; 27(10):3077. https://doi.org/10.3390/molecules27103077
Chicago/Turabian StyleGawalska, Alicja, Marcin Kołaczkowski, and Adam Bucki. 2022. "Structural Modeling of TRPA1 Ion Channel—Determination of the Binding Site for Antagonists" Molecules 27, no. 10: 3077. https://doi.org/10.3390/molecules27103077
APA StyleGawalska, A., Kołaczkowski, M., & Bucki, A. (2022). Structural Modeling of TRPA1 Ion Channel—Determination of the Binding Site for Antagonists. Molecules, 27(10), 3077. https://doi.org/10.3390/molecules27103077