
Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi





Abstract
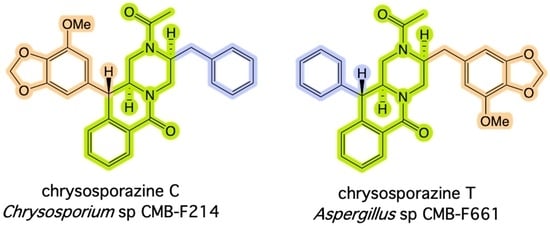
:
1. Introduction
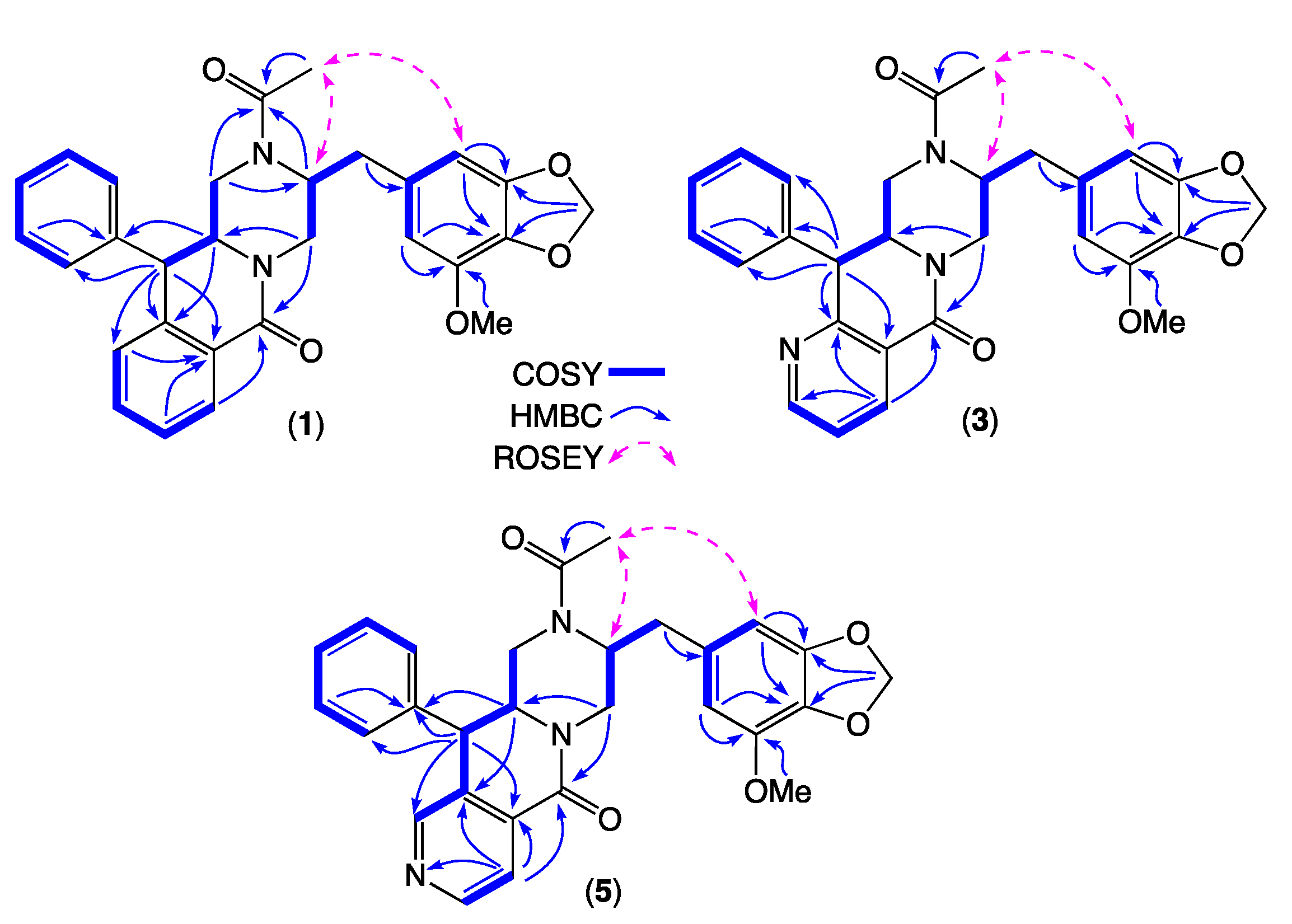
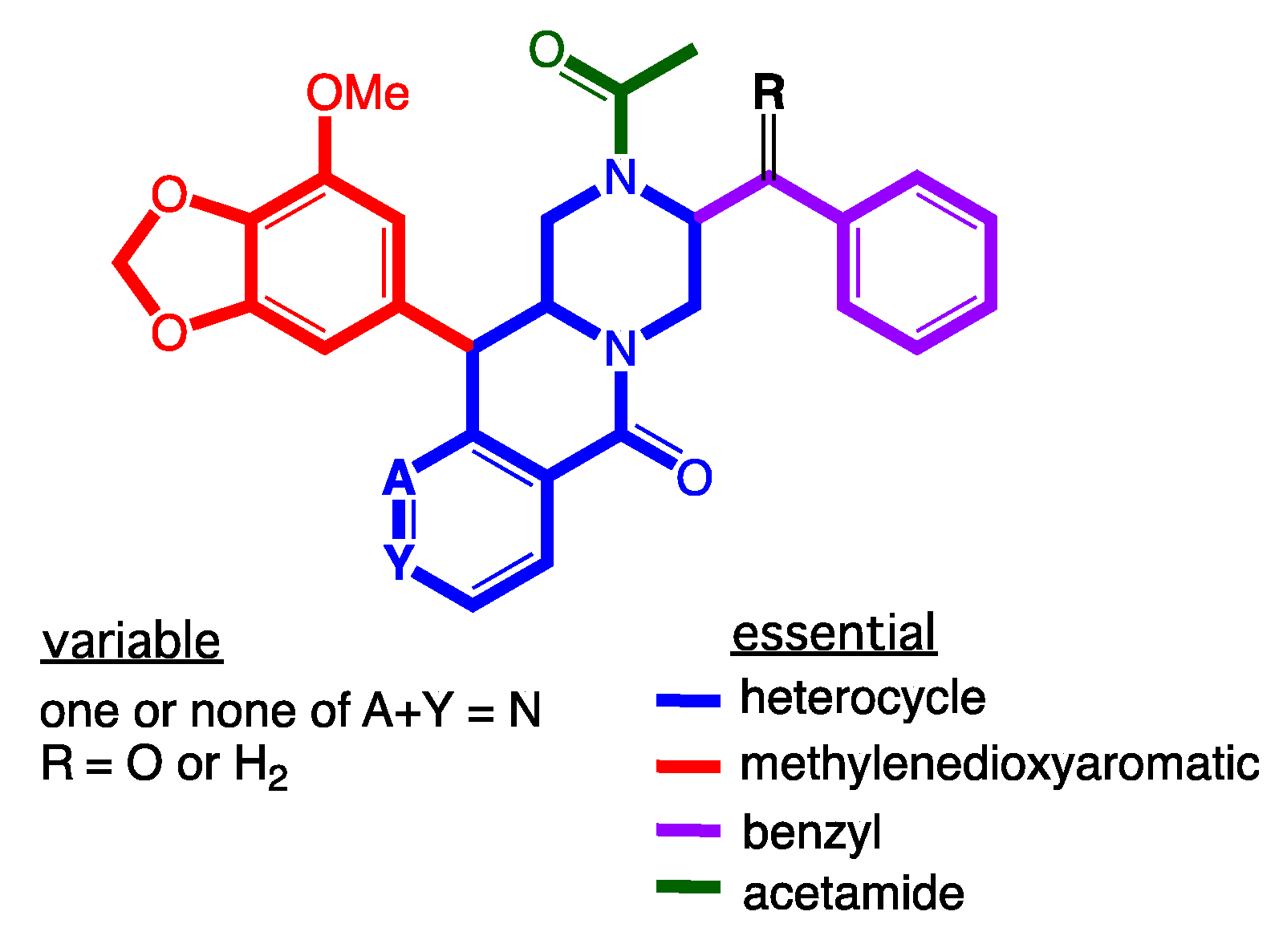
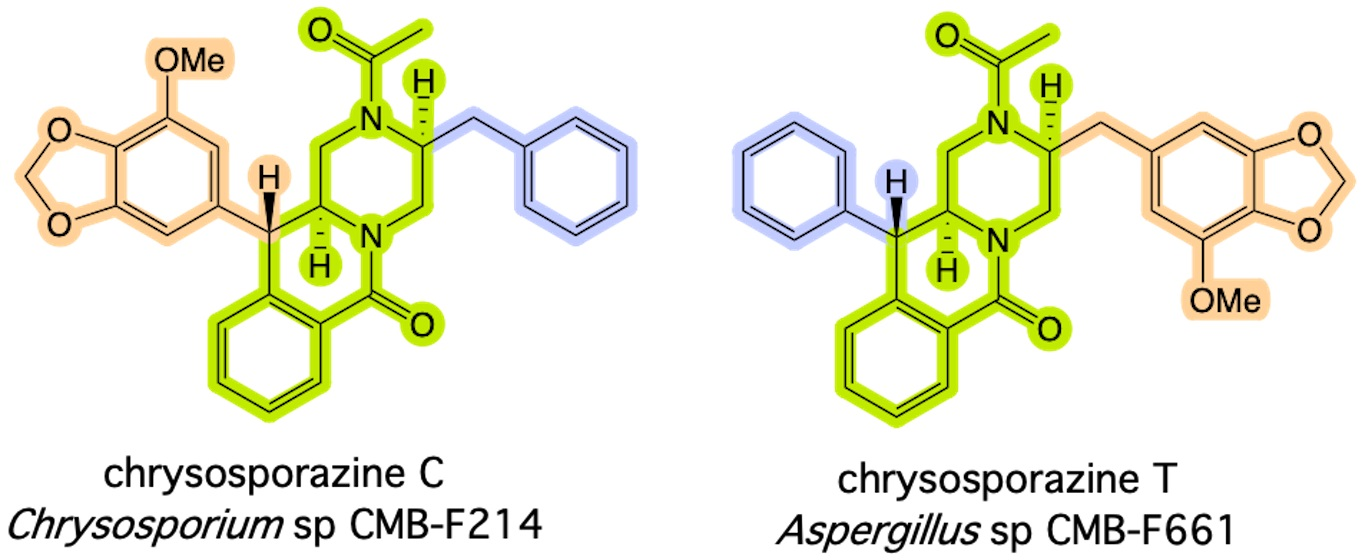
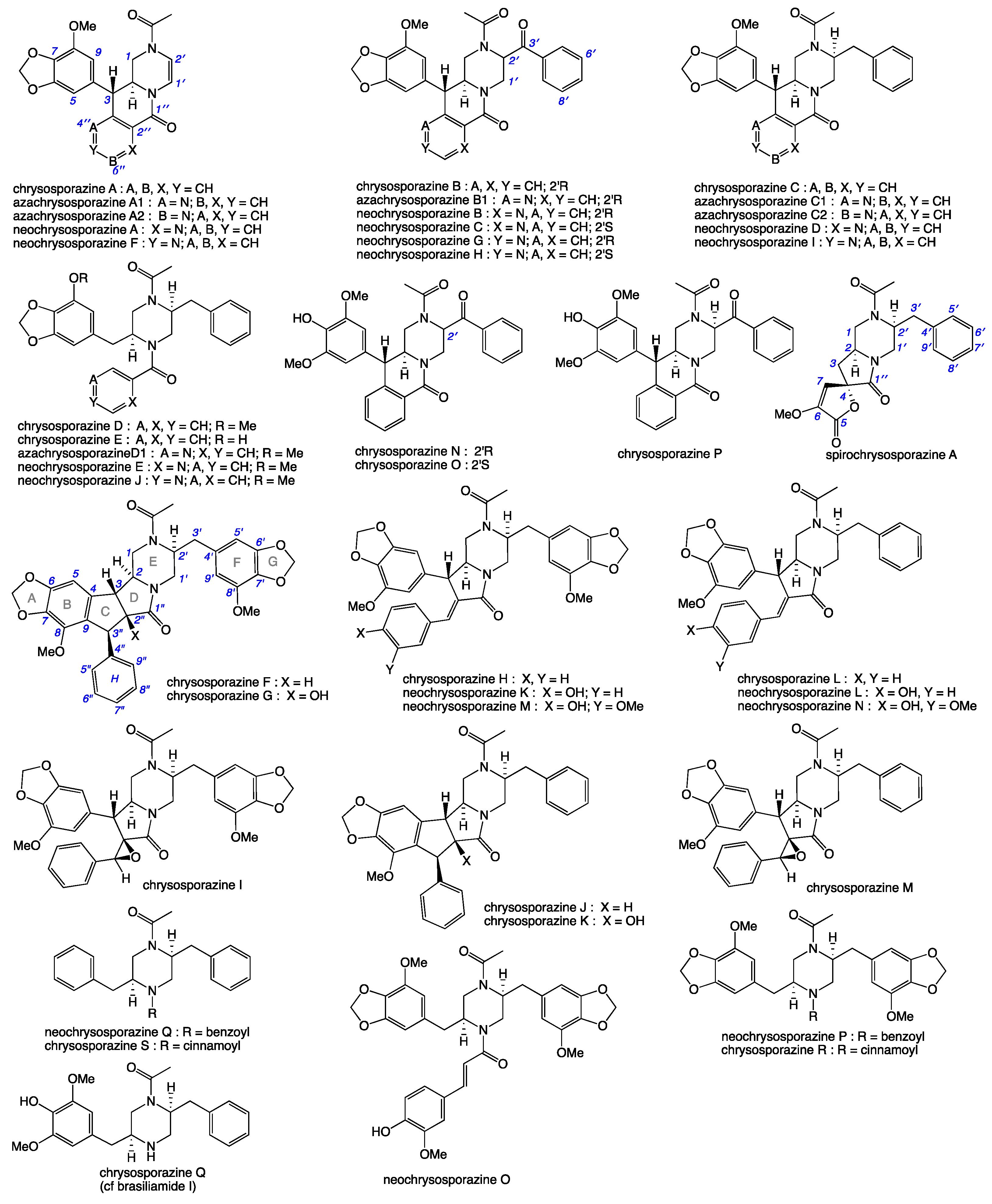
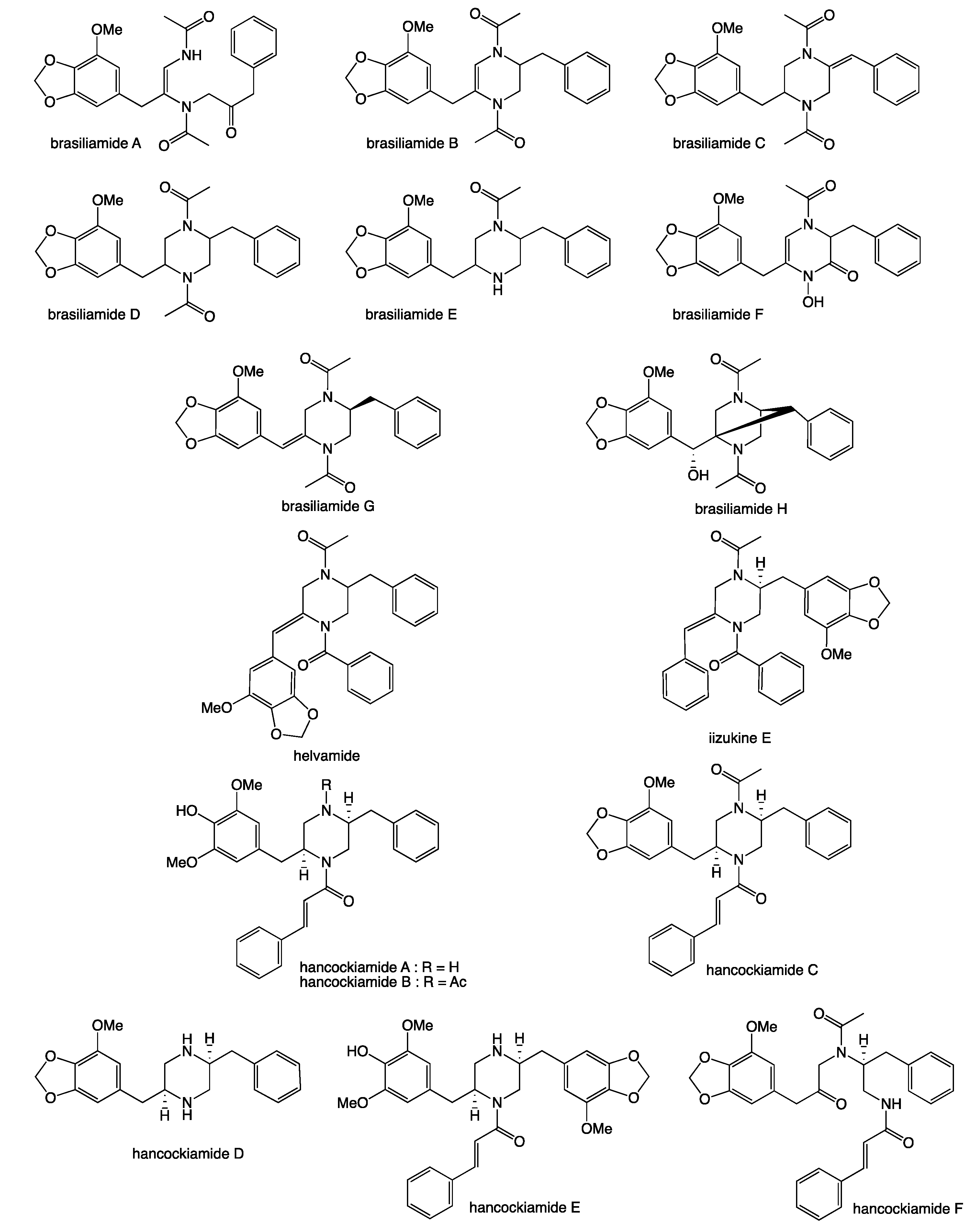
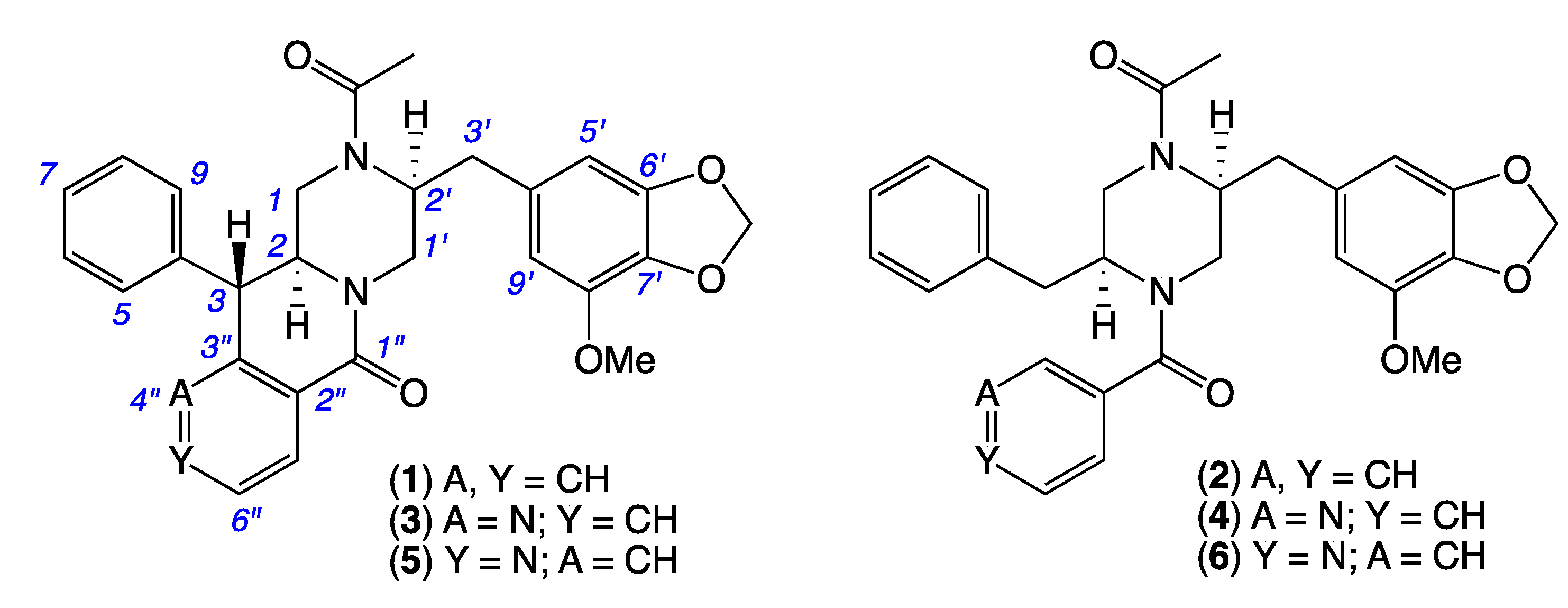
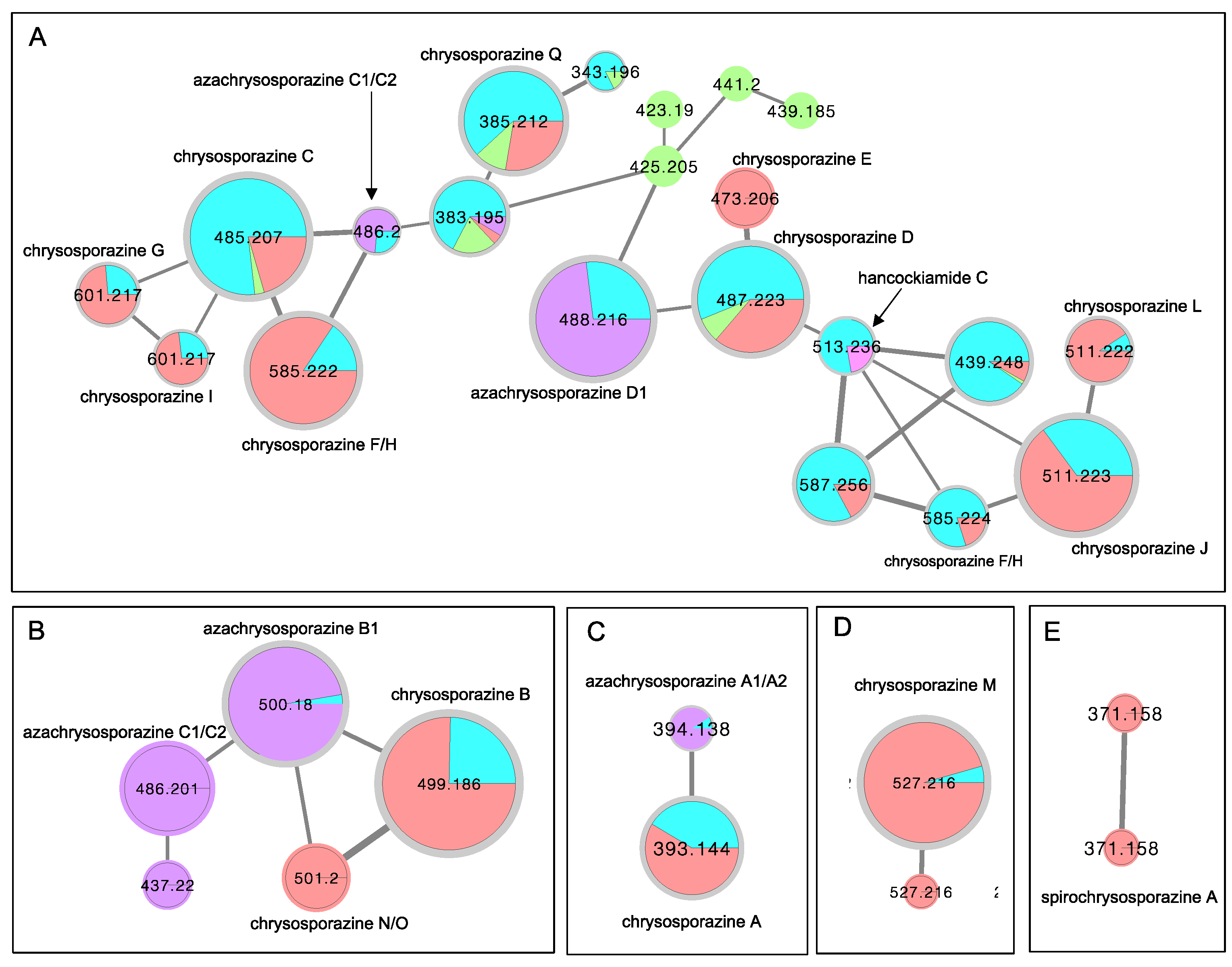
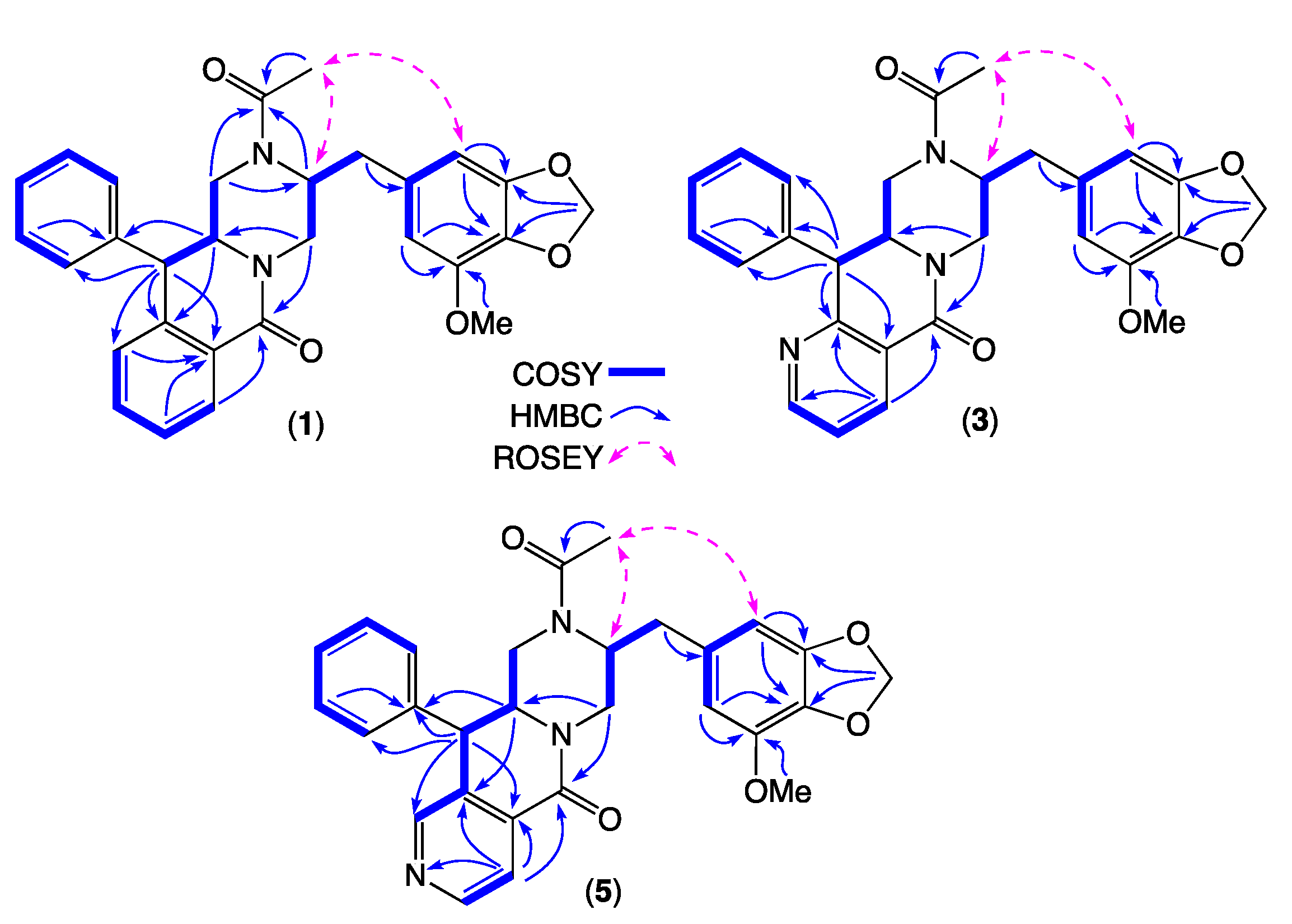
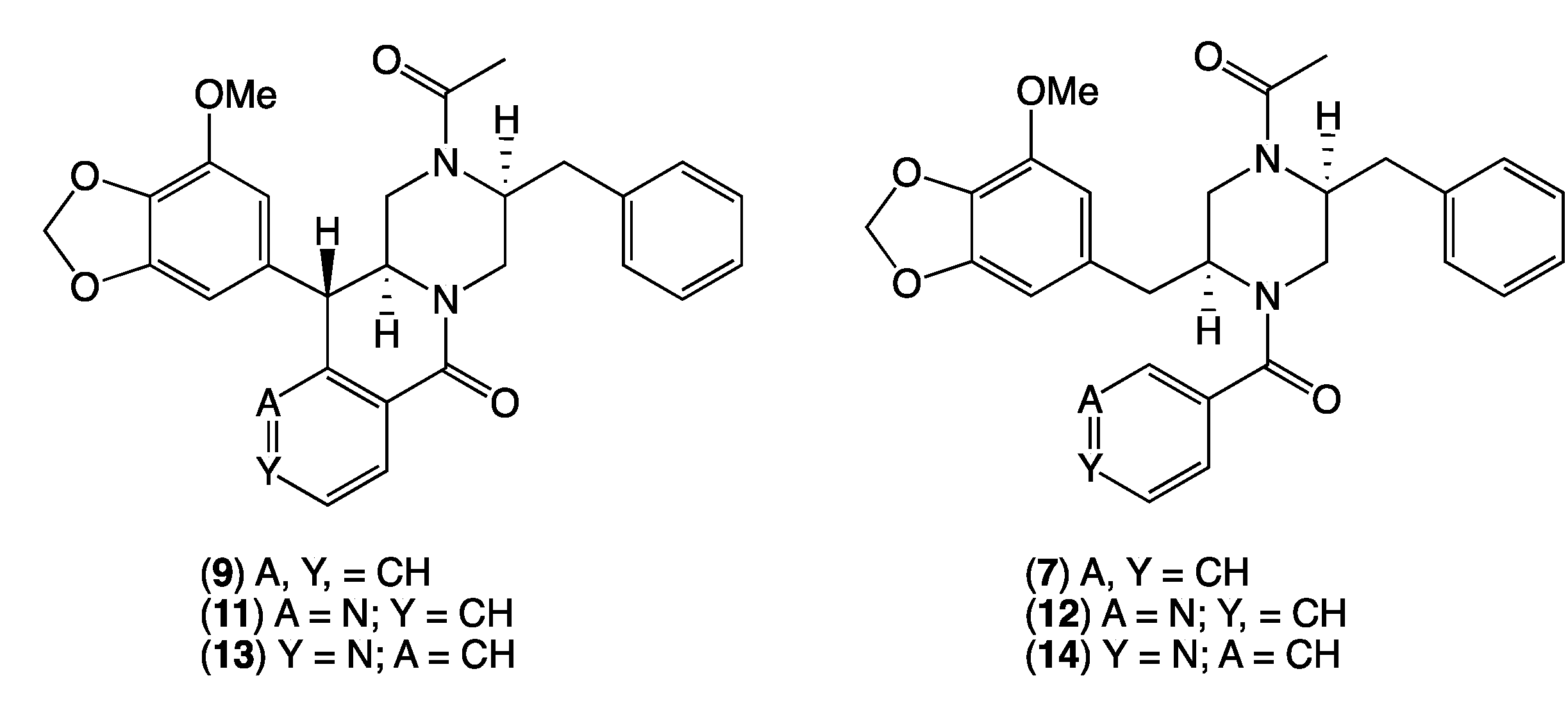
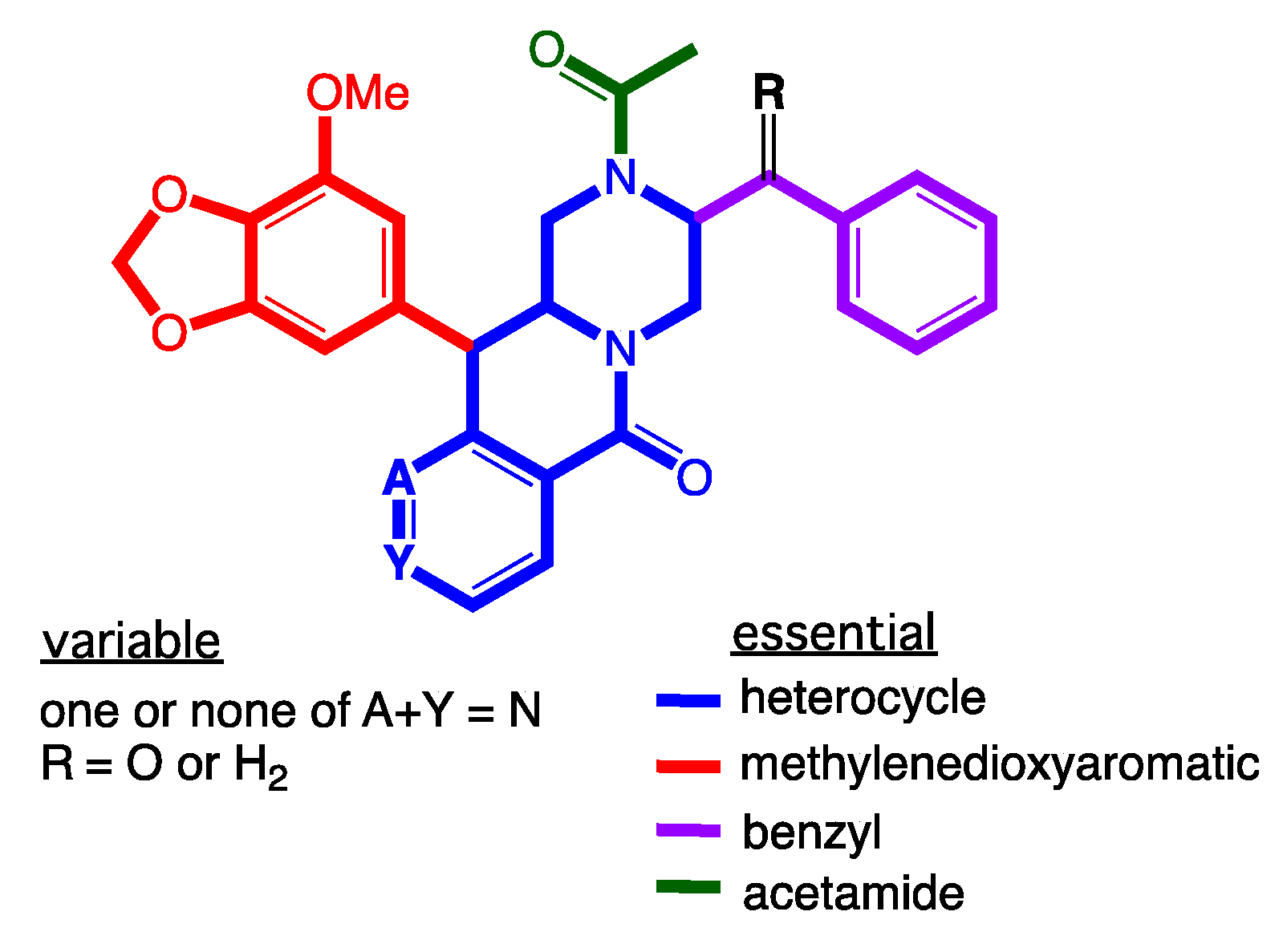
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Isolation
3.3. Fungal Taxonomy
3.4. Global Natural Product Social (GNPS) Molecular Networking
3.5. Fractionation of a Scaled Up PDA Culture of Aspergillus sp. CMB-F661
3.6. Media MATRIX Cultivation Profiling of Aspergillus sp. CMB-F661
3.7. Precursor-Directed Biosynthesis Cultivation Profiling of Aspergillus sp. CMB-F661
3.8. Scaled-Up Cultivation of Aspergillus sp. CMB-F661 on PDA with Sodium Nicotinate
3.9. Scaled-Up Cultivation of Aspergillus sp. CMB-F661 on PDA with Sodium Isonicotinate
3.10. Fractionation of a Scaled Up M1 Culture of Spiromastix sp. CMB-F455
3.11. Media MATRIX Cultivation Profiling of Spiromastix sp. CMB-F455
3.12. Precursor-Directed Biosynthesis Cultivation Profiling of Spiromastix sp. CMB-F455
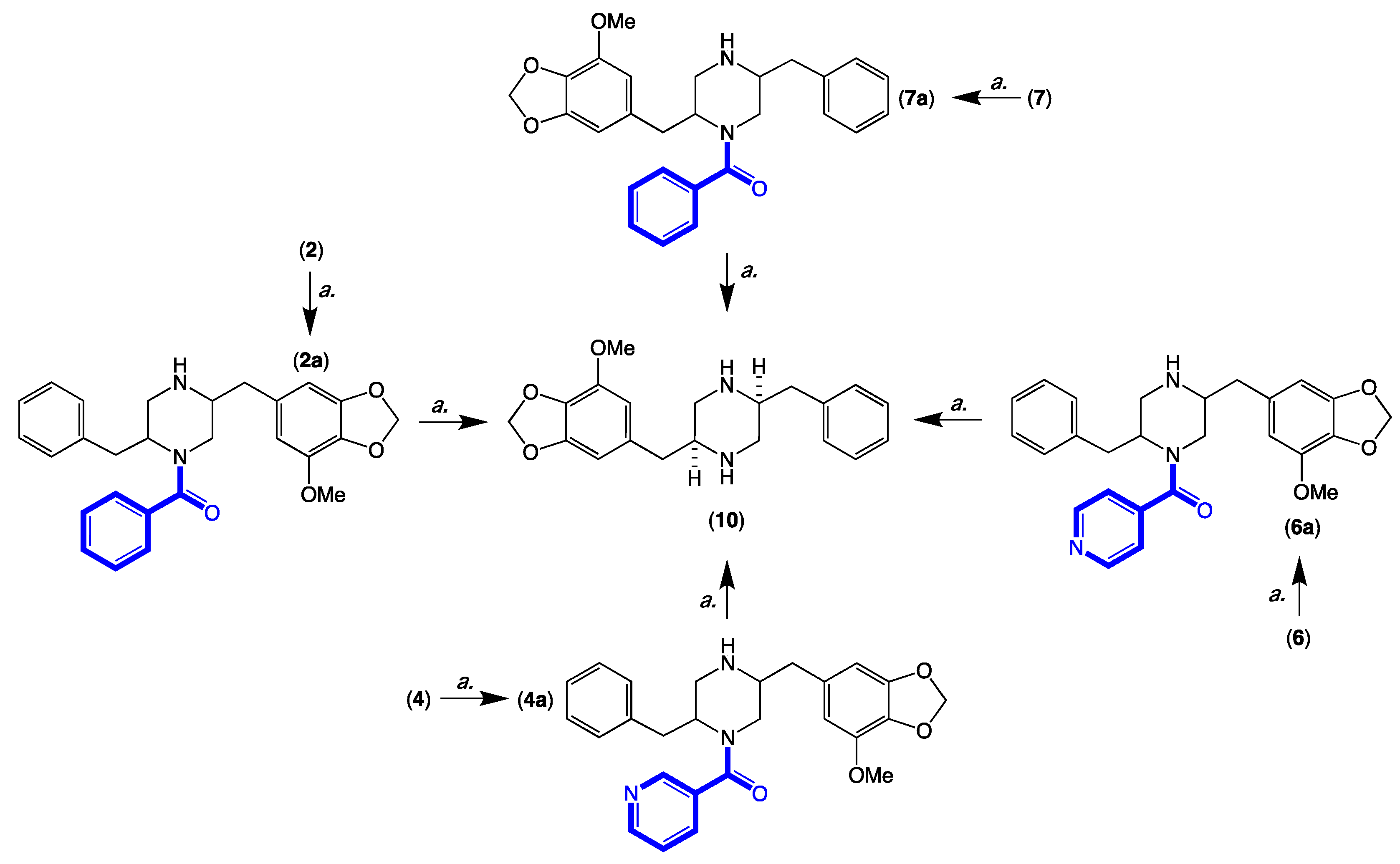
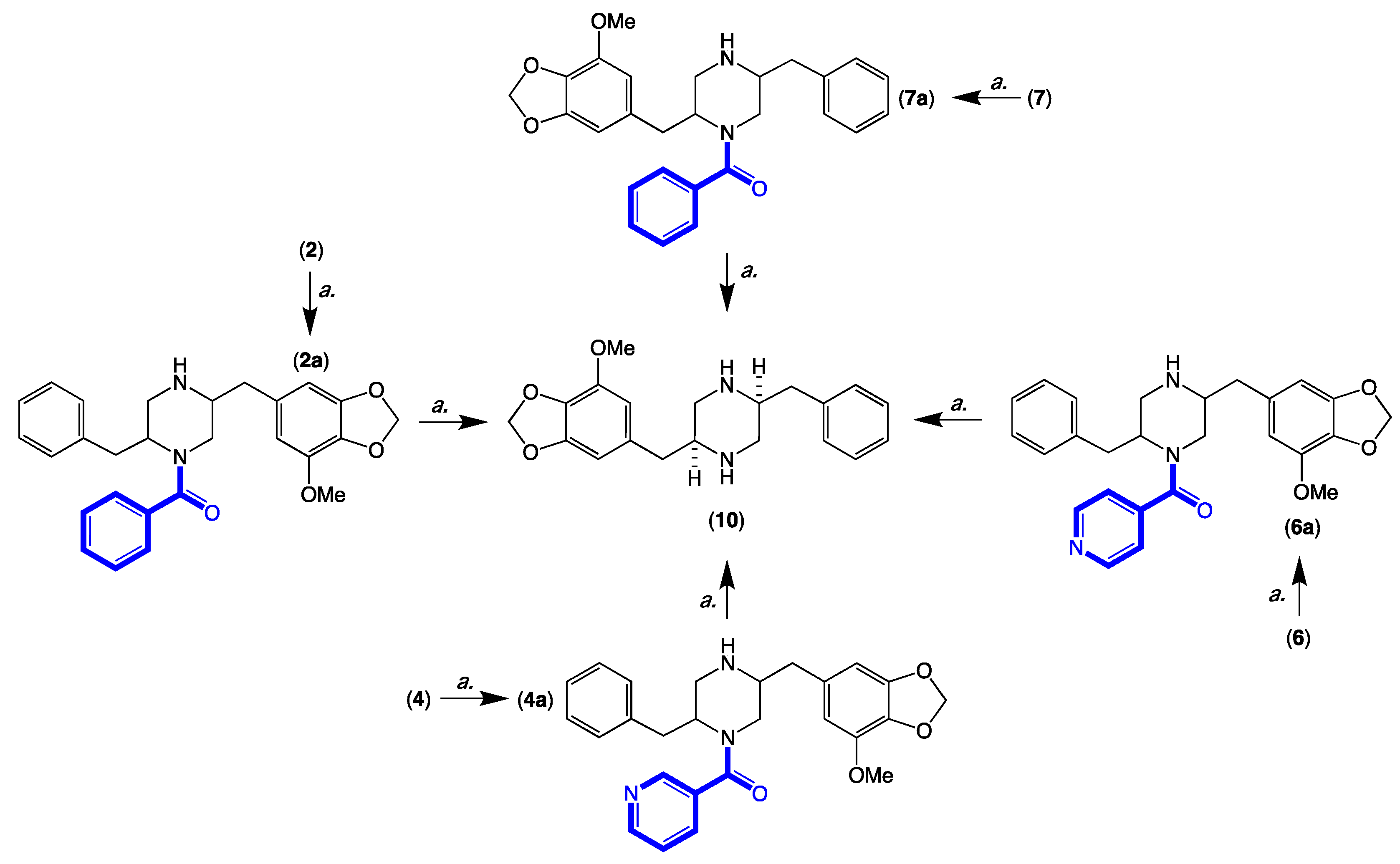
3.13. Acid Hydrolysis and Chemical Correlation of 2, 4, 6 and 7 to the Common Product 10
3.14. Antibacterial Assay
3.15. Antifungal Assay
3.16. Cytotoxicity (MTT) Assay
3.17. P-Glycoprotein Mediated MDR Reversal Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Scopularides revisited: Molecular networking guided exploration of lipodepsipeptides in Australian marine fish gastrointestinal tract-derived fungi. Mar. Drugs 2019, 17, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, O.G.; Khalil, Z.G.; Capon, R.J. Prolinimines: N-Amino-l-Pro-methyl ester (hydrazine) Schiff bases from a fish gastrointestinal tract-derived fungus, Trichoderma sp. CMB-F563. Org. Lett. 2018, 20, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.G.; Khalil, Z.G.; Capon, R.J. N-Amino-l-Pro-methyl ester from an Australian fish gut-derived fungus: Challenging the distinction between natural product and artifact. Mar. Drugs 2021, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.G.; Khalil, Z.G.; Santiago, V.; Capon, R.J. Metarhizides A–C and metarhizosides A–B: PKS-NRPS macrolides and aromatic glycosides from an Australian fish gut-derived fungus, Metarhizium sp. CMB-F624. Tetrahedron 2022, 113, 132759. [Google Scholar] [CrossRef]
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Chrysosporazines A-E: P-Glycoprotein inhibitory piperazines from an Australian marine fish gastrointestinal tract-derived fungus, Chrysosporium sp. CMB-F214. Org. Lett. 2019, 21, 8097–8100. [Google Scholar] [CrossRef]
- Mohamed, O.G.; Salim, A.A.; Khalil, Z.G.; Elbanna, A.H.; Bernhardt, P.V.; Capon, R.J. Chrysosporazines F-M: P-Glycoprotein inhibitory phenylpropanoid piperazines from an Australian marine fish derived fungus, Chrysosporium sp. CMB-F294. J. Nat. Prod. 2020, 83, 497–504. [Google Scholar] [CrossRef]
- Elbanna, A.H.; Agampodi Dewa, A.; Khalil, Z.G.; Capon, R.J. Precursor-directed biosynthesis mediated amplification of minor aza phenylpropanoid piperazines in an Australian marine fish-gut-derived fungus, Chrysosporium sp. CMB-F214. Mar. Drugs 2021, 19, 478. [Google Scholar] [CrossRef]
- Dewa, A.A.; Elbanna, A.H.; Khalil, Z.G.; Capon, R.J. Neochrysosporazines: Precursor-directed biosynthesis defines a marine-derived fungal natural product P-Glycoprotein inhibitory pharmacophore. J. Med. Chem. 2022, 65, 2610–2622. [Google Scholar] [CrossRef]
- Li, H.; Lacey, A.E.; Shu, S.; Kalaitzis, J.A.; Vuong, D.; Crombie, A.; Hu, J.; Gilchrist, C.L.M.; Lacey, E.; Piggott, A.M.; et al. Hancockiamides: Phenylpropanoid piperazines from Aspergillus hancockii are biosynthesised by a versatile dual single-module NRPS pathway. Org. Biomol. Chem. 2021, 19, 587–595. [Google Scholar] [CrossRef]
- Fujita, T.; Makishima, D.; Akiyama, K.; Hayashi, H. New convulsive compounds, brasiliamides A and B, from Penicillium brasilianum batista JV-379. Biosci. Biotechnol. Biochem. 2002, 66, 1697–1705. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T.; Hayashi, H. New brasiliamide congeners, brasiliamides C, D and E, from Penicillium brasilianum Batista JV-379. Biosci. Biotechnol. Biochem. 2004, 68, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fill, T.P.; Geris dos Santos, R.M.; Barisson, A.; Rodrigues-Filho, E.; Souza, A.Q. Co-production of bisphenylpropanoid amides and meroterpenes by an endophytic Penicillium brasilianum found in the root bark of Melia azedarach. Z Nat. C J. Biosci. 2009, 64, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fill, T.P.; Silva, B.F.D.; Rodrigues-Fo, E. Biosynthesis of phenylpropanoid amides by an endophytic Penicillium brasilianum found in root bark of Melia azedarach. J. Microbiol. Biotechnol. 2010, 20, 622–629. [Google Scholar] [PubMed]
- Liu, Y.; Mandi, A.; Li, X.M.; Meng, L.H.; Kurtan, T.; Wang, B.G. Peniciadametizine A, a dithiodiketopiperazine with a unique spiro[furan-2,7’-pyrazino[1,2-b][1,2]oxazine] skeleton, and a related analogue, peniciadametizine b, from the marine sponge-derived fungus Penicillium adametzioides. Mar. Drugs 2015, 13, 3640–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluka, J.; Kanokmedhakul, K.; Soytong, M.; Soytong, K.; Yahuafai, J.; Siripong, P.; Kanokmedhakul, S. Meroterpenoid pyrones, alkaloid and bicyclic brasiliamide from the fungus Neosartorya hiratsukae. Fitoterapia 2020, 142, 104485. [Google Scholar] [CrossRef]
- Eamvijarn, A.; Kijjoa, A.; Bruyere, C.; Mathieu, V.; Manoch, L.; Lefranc, F.; Silva, A.; Kiss, R.; Herz, W. Secondary metabolites from a culture of the fungus Neosartorya pseudofischeri and their in vitro cytostatic activity in human cancer cells. Planta Med. 2012, 78, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.G.; Bao, L.; Liu, H.W.; Ren, J.W.; Wang, W.Z.; Wang, L.; Li, W.; Yin, W.B. Chemical diversity from the Tibetan Plateau fungi Penicillium kongii and P. brasilianum. Mycology 2018, 9, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Liu, D.; Guan, X.; Yan, Y.; Zhang, J.; Zhang, Y.; Yang, D.; Ma, M.; Lin, W. Piperazine ring formation by a single-module NRPS and cleavage by an alpha-KG-dependent nonheme iron dioxygenase in brasiliamide biosynthesis. Appl. Microbiol. Biotechnol. 2020, 104, 6149–6159. [Google Scholar] [CrossRef]
- Kang, H.H.; Zhong, M.J.; Ma, L.Y.; Rong, X.G.; Liu, D.S.; Liu, W.Z. Iizukines C-E from a saline soil fungus Aspergillus iizukae. Bioorg. Chem. 2019, 91, 103167. [Google Scholar] [CrossRef]
- Fukuda, T.; Furukawa, T.; Kobayashi, K.; Nagai, K.; Uchida, R.; Tomoda, H. Helvamide, a new inhibitor of sterol O-acyltransferase produced by the fungus Aspergillus nidulans BF-0142. J. Antibiot. 2019, 72, 8–14. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, A.A.; Khalil, Z.G.; Elbanna, A.H.; Wu, T.; Capon, R.J. Methods in microbial biodiscovery. Mar. Drugs 2021, 19, 503. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]

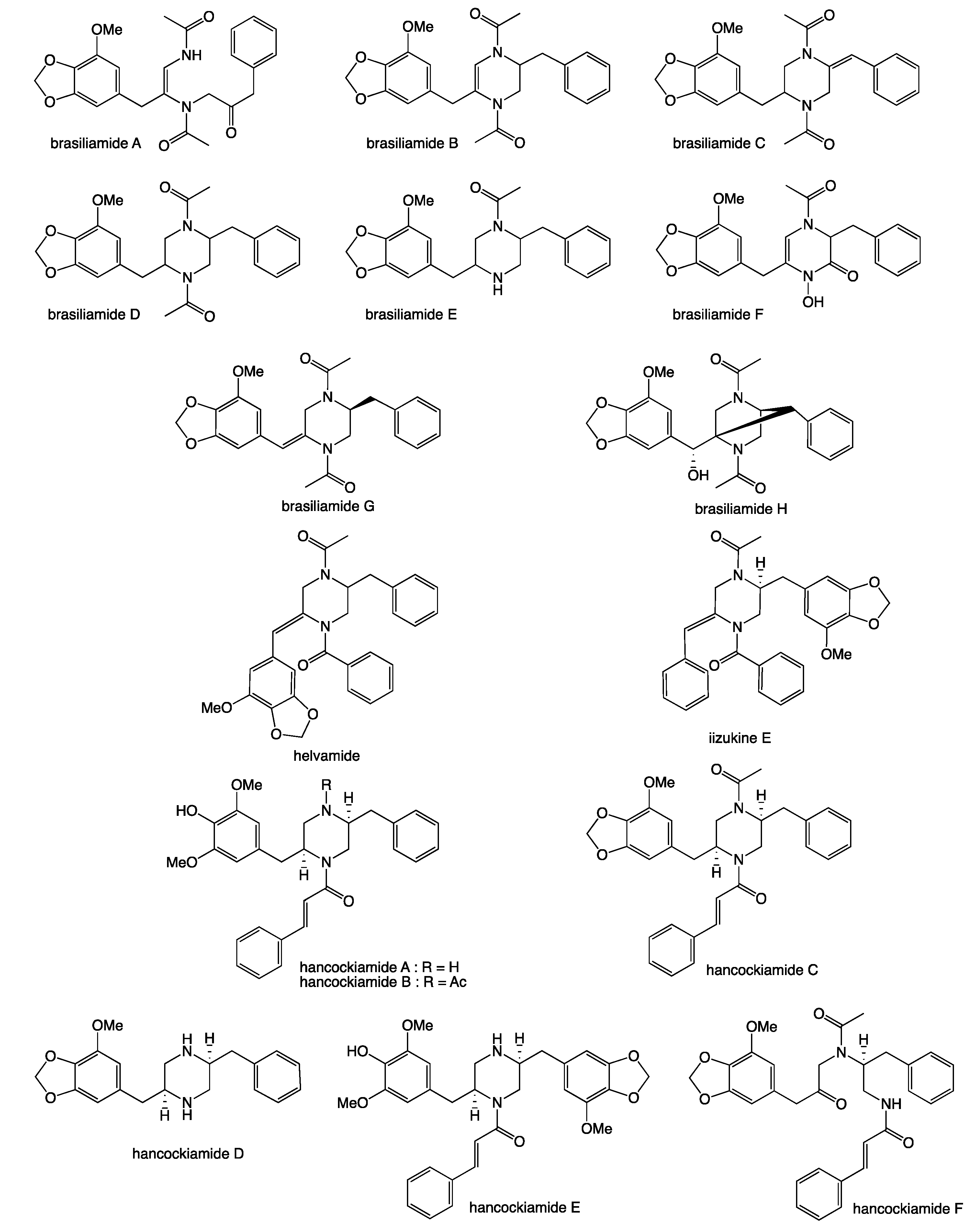
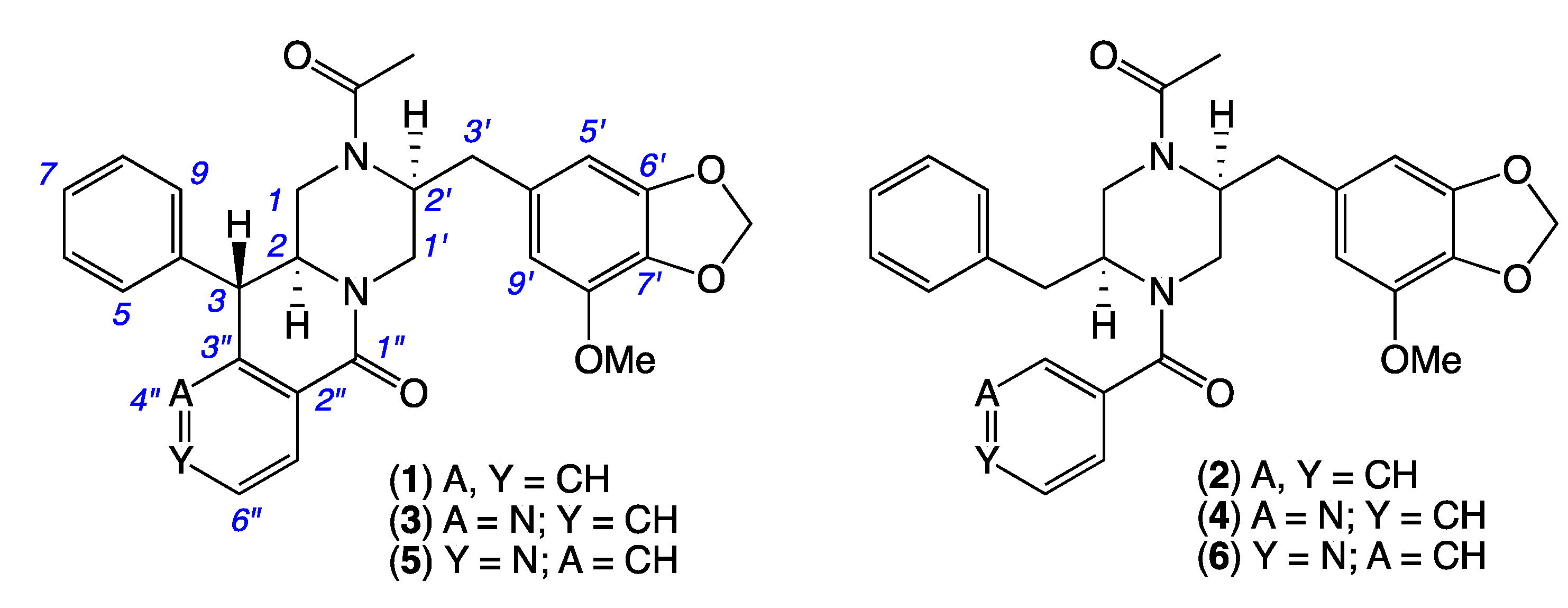
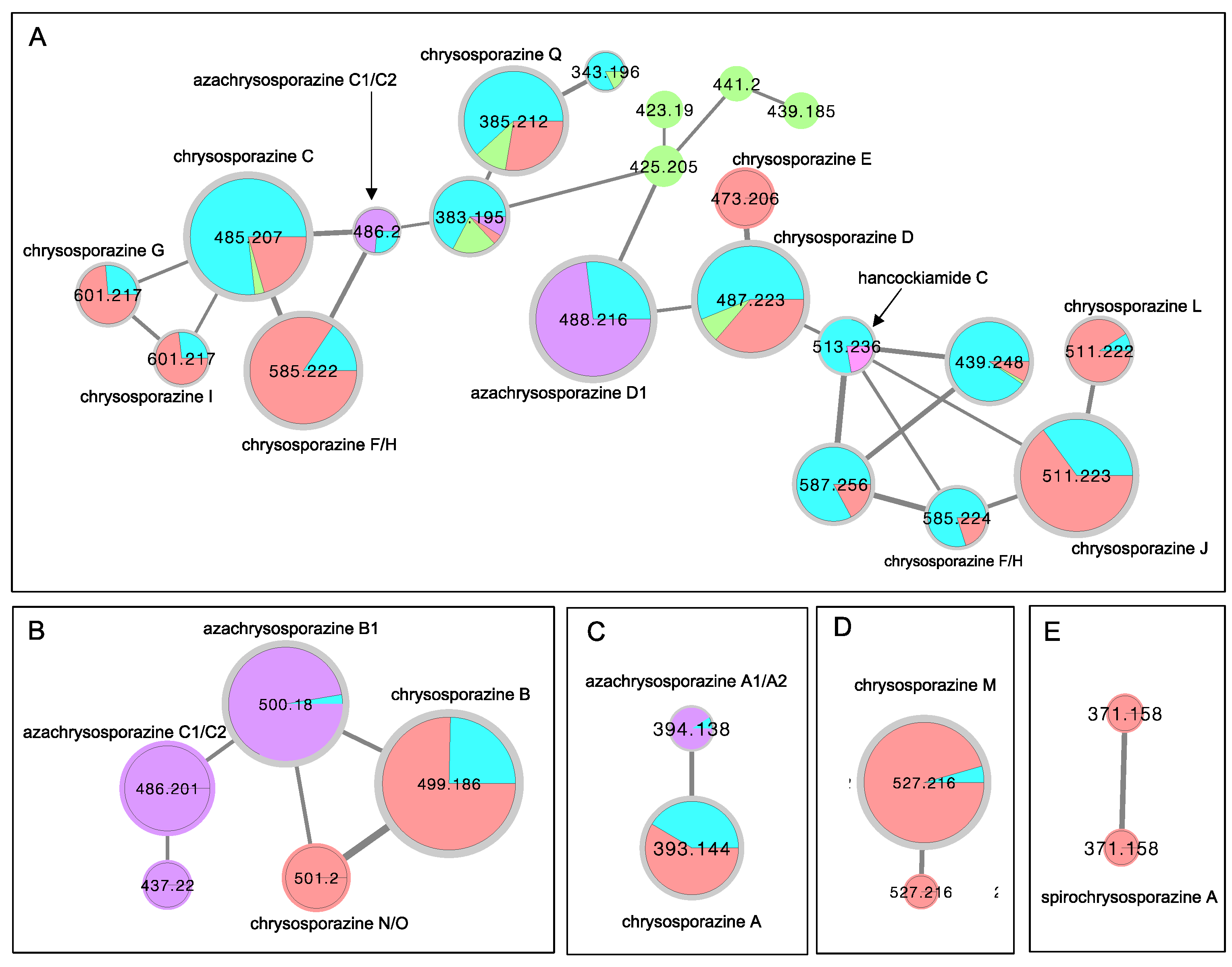


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | (1) δH, Multi (J in Hz) | (3) δH, Multi (J in Hz) | (5) δH, Multi (J in Hz) |
|---|---|---|---|
| 1 | a. 4.14, dd (13.8, 3.9) | a. 4.25, dd (13.3, 3.8) | a. 4.19, dd (14.5, 4.0) |
| b. 2.94, m | b. 3.03, m | b. 2.98, m | |
| 2 | 3.84, ddd (10.6, 10.6, 3.9) | 3.87, ddd (11.2, 8.8, 3.8) | 3.92, ddd (14.5, 10.3, 3.7) |
| 3 | 4.47, d (10.6) | 4.56, d (8.8) | 4.56, d (10.3) |
| 5/9 | 7.35, m | 7.26, m | 7.40, m |
| 6/8 | 7.44, m | 7.35, m | 7.46, m |
| 7 | 7.37, m | 7.29, m | 7.40, m |
| 1′ | a. 4.57, dd (13.3, 1.2) | a. 4.56, m | a. 4.55, dd (13.4, 1.3) |
| b. 2.95, m | b. 3.01, m | b. 2.99, m | |
| 2′ | 4.21, m | 4.21, m | 4.23, m |
| 3′ | a. 2.90, dd (13.5, 8.1) | a. 2.88, dd (13.5, 8.3) | a. 2.90, dd (13.4, 8.6) |
| b. 2.86, dd (13.5, 6.6) | b. 2.83, dd (13.5, 6.3) | b. 2.85, dd (13.4, 6.4) | |
| 4′ | - | - | - |
| 5′ | 6.54, d (1.2) | 6.54, d (1.3) | 6.54, d (1.4) |
| 9′ | 6.55, d (1.2) | 6.55, d (1.3) | 6.55, d (1.4) |
| 4″ | 6.60, d (7.8) | - | 7.89, s |
| 5″ | 7.44, m | 8.57, dd (4.7, 1.8) | - |
| 6″ | 7.39, m | 7.44, ddd (7.8, 4.7) | 8.64, d (4.7) |
| 7″ | 8.04, dd (7.7, 1.4) | 8.34, dd (7.8, 1.8) | 7.88, d (4.7) |
| 1-NCOCH3 | - | - | - |
| 1-NCOCH3 | 1.70, s | 1.68, s | 1.70, s |
| 6′-OCH2 | 5.94/5.93, ABq | 5.95/5.93, ABq | 5.95/5.94, ABq |
| 8′-OCH3 | 3.79, s | 3.78, s | 3.78, s |
| Position | (1) δC, Type | (3) δC, Type | (5) δC, Type |
|---|---|---|---|
| 1 | 40.1, CH2 | 40.4, CH2 | 40.1, CH2 |
| 2 | 57.8, CH | 58.6, CH | 58.2, CH |
| 3 | 46.3, CH | 48.8, CH | 43.7, CH |
| 4 | 140.4 A, C | 140.7, C | 139.4, C |
| 5/9 | 129.3, CH | 129.3, CH | 129.2A, CH |
| 6/8 | 129.1, CH | 128.6, CH | 129.1A, CH |
| 7 | 127.6, CH | 127.1, CH | 127.9, CH |
| 1′ | 44.6, CH2 | 45.0, CH2 | 44.8, CH2 |
| 2′ | 54.6, CH | 54.9, CH | 54.6, CH |
| 3′ | 34.9, CH2 | 35.0, CH2 | 34.8, CH2 |
| 4′ | 132.5, C | 132.6, C | 132.6, C |
| 5′ | 103.3, CH | 103.4, CH | 103.3, CH |
| 6′ | 148.3, C | 148.2, C | 148.3, C |
| 7′ | 133.0, C | 133.4, C | 133.3, C |
| 8′ | 143.0, C | 143.1, C | 143.0, C |
| 9′ | 109.0, CH | 109.1, CH | 109.0, CH |
| 1″ | 163.9, C | 163.2, C | 162.3, C |
| 2″ | 127.4, C | 122.9 A, C | 134.4, C |
| 3″ | 140.3 A, C | 158.4, C | 133.7, C |
| 4″ | 126.9 B, CH | - | 148.6, CH |
| 5″ | 132.3, CH | 152.6, CH | - |
| 6″ | 127.0 B, CH | 122.8 A, CH | 148.3, CH |
| 7″ | 127.6, CH | 135.6, CH | 120.2, CH |
| 1-NCOCH3 | 168.4, C | 168.6, C | 168.5, C |
| 1-NCOCH3 | 20.8, CH3 | 20.8, CH3 | 20.8, CH3 |
| 6′-OCH2 | 101.0, CH2 | 101.0, CH2 | 101.0, CH2 |
| 8′-OCH3 | 56.2, CH3 | 56.3, CH3 | 56.2, CH3 |
| SW620 Ad300 | ||||
|---|---|---|---|---|
| Treatment | IC50 (μM) | FR | GS | FI |
| doxorubicin | 5.75 | 57.5 | 1.0 | 0.12 |
| verapamil | >30 | ND | ND | - |
| + verapamil (2.5 μM) | 0.71 | 7.1 | 8.1 | 1.00 |
| + chrysosporazine T (1) | 0.97 | 9.7 | 5.9 | 0.61 |
| + chrysosporazine C (9) | 0.31 | 3.1 | 18.5 | 2.28 |
| + chrysosporazine U (2) | 2.76 | 27.6 | 2.0 | 0.25 |
| + chrysosporazine D (7) | 4.36 | 43.6 | 1.32 | 0.16 |
| + azachrysosporazine T1 (3) | 0.89 | 8.9 | 6.4 | 0.80 |
| + azachrysosporazine C1 (11) | 0.27 | 2.7 | 21.3 | 2.63 |
| + azachrysosporazine U1 (4) | 2.78 | 27.8 | 2.0 | 0.25 |
| + azachrysosporazine D1 (12) | 3.55 | 35.5 | 1.6 | 0.20 |
| + neochrysosporazine R (5) | 0.58 | 5.8 | 9.9 | 1.22 |
| + neochrysosporazine I (13) | 1.01 | 10.1 | 5.7 | 0.70 |
| + neochrysosporazine S (6) | 1.95 | 19.5 | 2.9 | 0.36 |
| + neochrysosporazine J (14) | 6.18 | 61.8 | 0.9 | 0.11 |
| + brasiliamide A (8) | 5.27 | 52.7 | 1.1 | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agampodi Dewa, A.; Khalil, Z.G.; Elbanna, A.H.; Capon, R.J. Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi. Molecules 2022, 27, 3172. https://doi.org/10.3390/molecules27103172
Agampodi Dewa A, Khalil ZG, Elbanna AH, Capon RJ. Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi. Molecules. 2022; 27(10):3172. https://doi.org/10.3390/molecules27103172
Chicago/Turabian StyleAgampodi Dewa, Amila, Zeinab G. Khalil, Ahmed H. Elbanna, and Robert J. Capon. 2022. "Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi" Molecules 27, no. 10: 3172. https://doi.org/10.3390/molecules27103172
APA StyleAgampodi Dewa, A., Khalil, Z. G., Elbanna, A. H., & Capon, R. J. (2022). Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi. Molecules, 27(10), 3172. https://doi.org/10.3390/molecules27103172