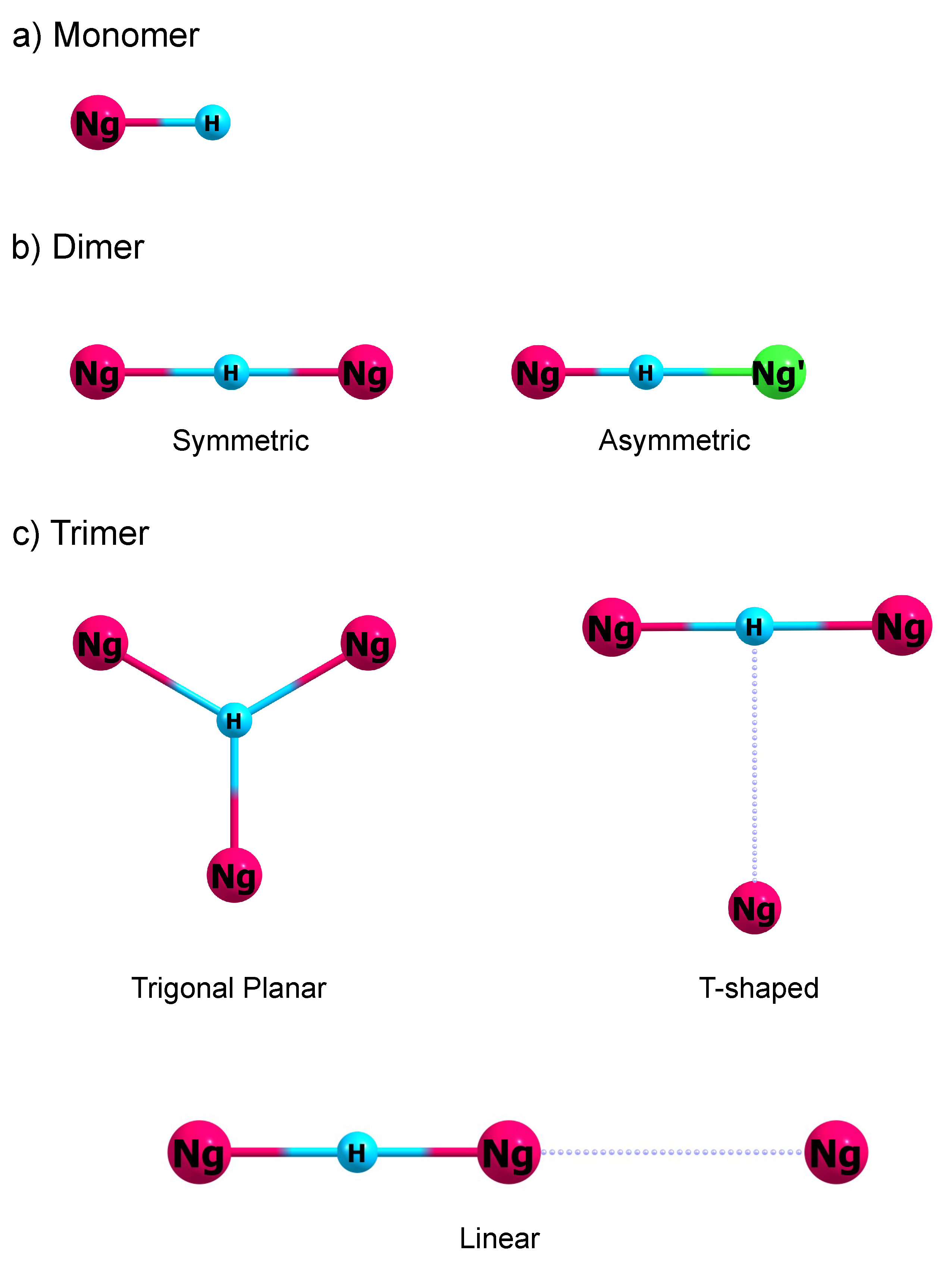
3.1. Proton-Bound Noble Gas Monomers
Among the proton-bound noble gas clusters, the protonated noble gas monomers (NgH
+) are the simplest. These species have a linear structure as shown in
Figure 1a. The Cartesian coordinates for these structures are given in
Section A of the Supplementary Material. Experimental studies have shown that the Ng-H
+ internuclear distance increases from NeH
+ to XeH
+ [
26,
27,
28,
29]. As shown in
Table 1, the experimental internuclear distances for NeH
+, ArH
+, KrH
+, and XeH
+are 0.9912, 1.2804, 1.4212, and 1.6028 Å, respectively. Such a trend is easily rationalized as the Ng’s size increases from Ne to Xe.
Table 1 also shows the CCSD(T) equilibrium internuclear distance for NgH
+. The predicted bond distance at this level of theory not only captures the trend, but also agrees well with the reported experimental values. Furthermore, our CCSD(T) internuclear distances have reproduced the reported calculations of Borocci et al. [
51] Furthermore, our calculated internuclear distances for NeH
+ and ArH
+ agrees well with those reported by de Oca-Estévez et al., which were calculated at the CCSD(T)/CBS[56] and MRCI+Q/aug-cc-pV6Z levels of theory and basis [
31].
As for the nature of the Ng-H
+ bond, Borocci et al. have performed an extensive bonding analysis [
51]. Their findings indicate that the nature of the Ng-H
+ bond is dominantly covalent.
The dissociation of NgH
+ can occur in two distinct pathways. The energetics for these pathways are compiled in
Table 2. On the one hand the dissociation may lead to Ng and H
+. The dissociation energy for this pathway is very high (>50 kcal/mol) [
51,
57,
58,
60]. The CCSD(T)/CBS//CCSD(T)/aug-cc-pVTZ dissociation energies are 53.03 kcal/mol for NeH
+, 93.64 kcal/mol for ArH
+, 104.79 kcal/mol for KrH
+, and 120.01 kcal/mol for XeH
+[
60]. The increasing binding energy from NeH
+ to XeH
+ is attributed to the increasing polarizability of the Ng from Ne to Xe. We note that these values agree well with our previous dissociation energy calculations at the CCSD(T)/aug-cc-pVQZ//MP2/aug-cc-pVQZ level of theory and basis [
58]. In our previous work, these dissociation energies are 53.43 kcal/mol for NeH
+, 93.96 kcal/mol for ArH
+, 105.94 kcal/mol for KrH
+, and 121.43 kcal/mol for XeH
+. The discrepancies between the present and previous results are attributed to the difference in reference geometry used and basis set size. The previous study used the MP2/aug-cc-pVQZ geometries, while the present study used the CCSD(T)/aug-cc-pVTZ geometries. Moreover, the present dissociation energy calculations are based on the CBS limit.
On the other hand, the dissociation could also lead to Ng+ and H. The dissociation energies at the CCSD(T)/CBS//CCSD(T)/aug-cc-pVTZ level for this pathway are 237.46 kcal/mol for NeH+, 144.95 kcal/mol for ArH+, 118.21 kcal/mol for KrH+, and 94.35 kcal/mol for XeH+. Notice that from NeH+to XeH+, the trends in dissociation energy between these two pathways are opposite. In the NgH+→ Ng + H+ pathway, the dissociation energy increases from NeH+ to XeH+, while in the NgH+→ Ng+ + H, the dissociation energy decreases from NeH+ to XeH+. We interpret these opposing trends as follows: In the NgH+→ Ng + H+ pathway, the polarizability of the Ng atom dictates the trend. However, in the NgH+→ Ng+ + H, the Ng atom’s ionization energy dictates the trend.
A comparison of the dissociation energy between these two pathways suggests that XeH
+ might dissociate differently from the rest of the NgH
+ complexes. As shown in
Table 2, the NgH
+→ Ng + H
+ pathway is energetically favored for NeH
+, ArH
+, and KrH
+, while the NgH
+→ Ng
+ + H pathway is more favored for XeH
+. As a result, the XeH
+ dissociates to Xe
+ and H, while the rest of the NgH
+ dissociates to Ng and H
+. To understand this unusual behavior of XeH
+, one needs to examine the ionization energies of H and Ng. The ionization energies in decreasing order are as follows: Ne (21.56 eV), Ar (15.76 eV), Kr (14.00 eV), H (13.59 eV), and Xe (12.13 eV) [
91]. Notice that Ne, Ar, and Kr have a higher ionization energy than H, while the ionization energy of Xe is lower than that of H. Hence, in NeH
+, ArH
+, and KrH
+, one would expect that the charge is more localized at the H atom, but in XeH
+, the charge would be more localized on the Xe atom as the dissociation proceeds.
To investigate further, we examined the atomic charges as the internuclear distance is varied. Since the goal is to investigate the qualitative behavior of the atomic charges with respect to bond elongation, we have performed Mulliken population analysis [
92] and examine the Mulliken charges at the MP2 level. For NeH
+, ArH
+, and KrH
+, we found that at 10 Å, the H atom carries the positive charge, while the Ng atom has a zero Mulliken charge. Meanwhile for XeH
+, the positive charge is more localized to the Xe atom when the internuclear distance is at 10 Å. In addition to the Mulliken population analysis, we have also performed natural population analysis (NPA) [
93]. Both of these population analyses have predicted that at large internuclear distances (10 Å) the postive charge is localized at the H atom for NeH
+, ArH
+, KrH
+, and XeH
+. However, for XeH
+, the positive charge tends to localize on the Xe atom. A comparison of the atomic charges from these population analyses can be found in
Section B of the Supplementary Material.
Although it is worthwhile to examine the dipole moment function (DMF) with respect to internuclear distance, one should note that for charged systems the dipole moment function is not unique. In other words, it depends on the choice of coordinate’s origin [
94].
We now examine the pure vibrational spectroscopy of NgH
+.
Table 1 also has the harmonic and anharmonic frequencies for NgH
+. Examining the harmonic frequencies
, it is evident that from NeH
+ to XeH
+the harmonic frequencies
decreases. Inclusion of anharmonicity leads to a lower oscillation frequency. Comparing our calculated anharmonic frequencies with the experimental frequencies, we note that these frequencies agree within 50 cm
−1. There are several factors for such discrepancy. These include basis set truncation error, neglect of the rotational-vibrational interactions, as well as other factors in the experiment that were not considered in the present Hamiltonian. To assess the basis set effects, we have reconstructed the PES using the aug-cc-pVQZ and aug-cc-pV5Z basis sets for H, Ne, and Ar. The corresponding aug-cc-pVQZ-PP and aug-cc-pV5Z-PP basis sets were used for Kr and Xe. We found that the use of larger basis sets leads to a better agreement with the experimental values when compared with the use of aug-cc-pVTZ and aug-cc-pVTZ-PP. In particular, at the aug-cc-pV5Z basis set, the discrepancies between the calculated aharmonic and observed frequencies are 6, 10, 23, and 28 cm
−1 for NeH
+, ArH
+, KrH
+, and XeH
+, respectively. The sensitivity of the anharmonic frequency with the basis set’s size can be found in
Section C of the Supplementary Material.
Table 1.
Comparison of experimental and calculated structural parameter and vibrational frequencies.
Table 1.
Comparison of experimental and calculated structural parameter and vibrational frequencies.
| Species | Method | R (Å) | (cm−1) | (cm−1) | Ref. |
|---|
| NeH+ | Expt. | 0.9912 | 2904 | 2677 a | [26] |
| | CCSD(T)/aug-cc-pVTZ | 0.9923 | 2947 | 2710 | This Work |
| | CCSD(T)/CBS[56] | 0.9913 | 2904 | 2679 | [31] |
| | MRCI+Q/aug-cc-pV6Z | 0.9913 | - | - | [31] |
| | Theory | 0.9918 | 2897 | 2675 | [95] |
| | Theory | 0.9912 | - | 2678 | [96] |
| | MRCI-DKH/ANO | 0.9925 | 2897 | 2674 | [97] |
| ArH+ | Expt. | 1.2804 | 2711 | 2588 a | [27] |
| | CCSD(T)/aug-cc-pVTZ | 1.2821 | 2730 | 2604 | This Work |
| | CCSD(T)/CBS[56] | 1.2810 | 2716 | - | [31] |
| | MRCI+Q/aug-cc-pV6Z | 1.2810 | - | - | [31] |
| | CEPA | 1.286 | 2723 | 2611 a | [20] |
| | MR-AQCC/ANO | 1.279 | 2739 | 2595 a | [98] |
| | Theory | 1.268 | - | - | [99] |
| KrH+ | Expt. | 1.4212 | 2495 | 2398 a | [28] |
| | CCSD(T)/aug-cc-pVTZ | 1.4135 | 2540 | 2436 | This Work |
| | CEPA | 1.419 | 2896 | 2670 a | [21] |
| XeH+ | Expt. | 1.6028 | 2270 | 2187 a | [29] |
| | CCSD(T)/aug-cc-pVTZ | 1.5980 | 2325 | 2237 | This Work |
| | CEPA | 1.611 | 2314 | 2231 | [22] |
Table 2.
CCSD(T)/CBS//CCSD(T)/aug-cc-pVTZ a dissociation and isomerization energies in kcal/mol.
Table 2.
CCSD(T)/CBS//CCSD(T)/aug-cc-pVTZ a dissociation and isomerization energies in kcal/mol.
| Process | Ne | Ar | Kr a | Xe a |
|---|
| Monomer | | | | |
| NgH+→ Ng + H+ | 53.03 b | 93.64 b | 104.79 b | 120.01 b |
| NgH+→ Ng+ + H | 237.46 | 144.95 | 118.21 | 94.35 |
| Dimer | | | | |
| NgH+Ng → Ng + NgH+ | 15.78 | 15.42 c | 15.57 b | 14.27 b |
| NgH+Ng → 2Ng + H+ | 68.81 | 109.06 | 120.36 b | 134.27 b |
| Trimer | | | | |
| Ng3H+ (T-shaped) → Ng + NgH+Ng | 1.29 | 2.05 c | 2.38 | 2.75 |
| Ng3H+ (Linear) → Ng + NgH+Ng | 0.49 | 1.34 c | 1.84 | 2.45 |
| Ng3H+ (T-shaped) → Ng3H+ (Linear) | 0.80 | 0.70 | 0.54 | 0.30 |
3.3. Proton-Bound Noble Gas Trimers
The structure of proton-bound noble gas trimers provides crucial information on the first coordination sphere of the proton. Intuitively, there are three possible structures for Ng
3H
+. These are trigonal planar, T-shaped, and linear structures. These structures are illustrated in
Figure 1c. In the trigonal planar structure, the Ng atoms are all equidistant to the H
+, and their Ng-H
+ distances are 120
apart. Such structure belongs to the
symmetry group. Geometry minimization together with normal mode analysis at the CCSD(T) level reveal that these trigonal planar structures do not correspond to a minimum on the PES. Instead, these structures correspond to a second-order saddle point on the PES and possess two imaginary degenerate modes. These modes correspond to a proton motion towards one side of the equilateral triangle Ng framework. Such finding suggests that when Ng solvates the H
+ in the gas phase, the first coordination sphere is comprised of two Ng atoms only. The succeeding Ng solvating the proton has to occupy the second coordination sphere, which makes the third Ng more distant to the H
+ in comparison with the first two Ng atoms.
Further geometry minimization on the trigonal planar structure’s imaginary mode leads to a T-shaped Ng
3H
+ structure. In the T-shaped structure (
Figure 1c), the distal Ng gas binds on top of the NgH
+Ng structure and is located at the proton’s top. The two proximal Ng atoms are still equidistant from the H
+. This T-shaped structure belongs to the
group. Comparing the structural parameters between NgH
+Ng (
Table 3) and T-shaped Ng
3H
+ (
Table 6) shows that the Ng
proximal-H
+ distance does not change much when the third Ng binds to the NgH
+Ng moiety.
In the linear Ng
3H
+ structure (
Figure 1c), one Ng atom solvates the H
+ on one side, while two well-separated Ng atoms solvate the H
+ on the opposite side. The equilibrium position for the proton is closer towards the side with two Ng atoms. This linear structure belongs to the
symmetry group. The structural parameters for all three isomers of Ng
3H
+are compiled in
Table 6. The Cartesian coordinates for these structures are given in
Sections F and G of the Supplementary Material.
Among these two isomers for Ng
3H
+, the T-shaped isomer is more stable. However, the linear isomer is only within 0.80 kcal/mol higher in energy than the T-shaped isomer.
Table 2 shows that the energetics for the T-shaped to liner isomerization decreases as the Ng gets heavier. The isomerization energies are Ne
3H
+ (0.80 kcal/mol), Ar
3H
+ (0.70 kcal/mol), Kr
3H
+ (0.54 kcal/mol), and Xe
3H
+ (0.30 kcal/mol).
The destruction of Ng
3H
+ to H
+ and three Ng atoms can be examined from a stepwise Ng elimination point of view. The energies involved for these processes are compiled in
Table 2. We first examine the energy required to remove one Ng from Ng
3H
+ (i.e., Ng
3H
+→ Ng + NgH
+Ng). The energy required for this process is only between 1.29 and 2.75 kcal/mol for the T-shaped isomer. This energy goes down in the 0.49 to 2.45 kcal/mol range for the linear isomer. Meanwhile, the energy for the NgH
+Ng → Ng + NgH
+ process requires a much higher energy (14.27–15.78 kcal/mol). Lastly, elimination of Ng from the NgH
+complex would need a much higher energy (53.03 to 120.01 kcal/mol). Based on the relative magnitudes of these energies, one may infer that a NgH
+Ng core-ion is formed when Ng atoms solvate the H
+. The Ng atoms of the core-ion binds much stronger than the additional Ng atoms. In the recent work of McDonald et al., calculations at the MP2/aug-cc-pVQZ level reveal that the ArH
+Ar core-ion has five Ar atoms in its first solvation sphere [
57].
Before discussing the anharmonic spectra for Ng3H+, we first examine the normal modes of its stable isomers. In addition to the core-ion modes, there are few low-frequency modes in both T-shaped and linear Ng3H+. For the T-shaped isomer, there are two low-frequency modes. These are the frustrated rotation of the NgH+Ng core-ion and the intermolecular stretch between the core-ion and distal Ng. Across all species, the harmonic frequencies for these modes are below 85 cm−1. Meanwhile for the linear isomer, there are three low-frequency modes in addition to the NgH+Ng core-ion modes. These are the doubly-degenerate frustrated rotation of the core-ion and the intermolecular stretch between the core-ion and Ng. The CCSD(T) harmonic frequencies for these modes are less than 60 cm−1.
In this work, we have ignored these low-frequency modes in the anharmonic spectra simulations for three main reasons. First, these frequencies are way too low. Including them in the anharmonic treatment would require us to solve a large number of eigenstates. Second, excited states for these low-frequency modes are often difficult to converge. Third, these low-frequency modes are outside the spectral region of interest. Therefore, we simulate the anharmonic spectrum using only the modes that corresponds to the core-ion.
Figure 4 shows the 4D anharmonic spectra for both T-shaped and linear isomers of Ng
3H
+. In the T-shaped isomer, the core-ion’s symmetric Ng-H
+ stretch
mode belongs to the
representation, while the asymmetric Ng-H
+ stretch
belongs to the
representation. As a result, the
combination bands also belongs to the
representation, which then makes the intensity borrowing between the
and
bands still symmetry allowed. As shown in
Figure 4, the spectra for the T-shaped isomers of Ng
3 still exhibit the
combination bands. Notice that the
and
bands are still very pronounced in the T-shaped Ng
3H
+. Furthermore, the trends in the strengths for these combination bands is similar to that in NgH
+Ng. In other words, the
band’s intensity increases from Ne
3H
+ to Xe
3H
+. Furthermore, the spacing of the combination bands decreases from Ne
3H
+ to Xe
3H
+, which then leads to a more efficient intensity redistribution.
We know turn our attention to the linear isomers of Ng
3H
+. In this case, both the core-ion’s symmetric Ng-H
+ stretch
mode and asymmetric Ng-H
+ stretch
belongs to the
representation. Hence, the
bands will also belong to the
representation. As a result, these combination bands are still allowed to borrow intensity from the bright
band. As shown in
Figure 4, these combination bands are also present in the linear isomers of Ng
3H
+.
In an actual experiment, both T-shaped and linear isomers are present in the sample. The observed spectrum may have peaks which can be attributed to each of these isomers. In the case of the Ar
3H
+, we found that although simulations for the T-shaped isomer can recover most of the experimental bands, simulations for both T-shaped and linear isomers are necessary to explain the observed bands at 1287, 1510, 1759, and 1993 cm
−1 [
57,
59]. Furthermore, the peaks associated with the more stable T-shaped isomer dominate the experimental spectrum [
59]. The observed strong band at 989 cm
−1 agrees well with the predicted 973 cm
−1 band, which then corresponds to the
band of the T-shaped isomer. The experimental spectrum also has a very weak band around 1041 cm
−1. Our previous simulation has attributed this band to the
band of the linear isomer. We note that for Ar
3H
+, the
band (973 cm
−1) for the T-shaped isomer is lower than that of the linear isomer (1002 cm
−1). A similar conclusion is drawn for the
combination band. In the experimental spectrum the
band for the T-shaped isomer is at 1237 cm
−1, while for the linear isomer it is blue shifted to 1287 cm
−1. These relative peak position were also captured in our simulations. In particular, the predicted
peak positions are 1223 cm
−1 for the T-shaped isomer and 1267 cm
−1 for the linear isomer.
Table 7 compiles the predicted anharmonic frequencies for Ng
3H
+. We now compare the predicted
and
peak positions for the linear and T-shaped isomers of the other Ng
3H
+ species. A comparison of the corresponding peaks between these isomers shows that the linear isomer’s peaks are also blue shifted relative to the T-shaped isomer’s peaks. In other words, the relative peak positions for the T-shaped and linear isomers are consistent across all trimer species considered in the present work. Unfortunately, experimental gas phase data for Ne
3H
+, Kr
3H
+, and Xe
3H
+ are not available at the moment. Furthermore, we note that although the present results have used a reasonable size for the basis set, the calculated peaks would be sensitive to the basis set’s size. However, the reported spectral features would be robust with respect to the basis set’s size. We hope that the presented results for Ng
3H
+ would motivate experimentalist to conduct more gas phase measurements in the future.




{kind=link}
{kind=link}
{kind=link}
{kind=link}