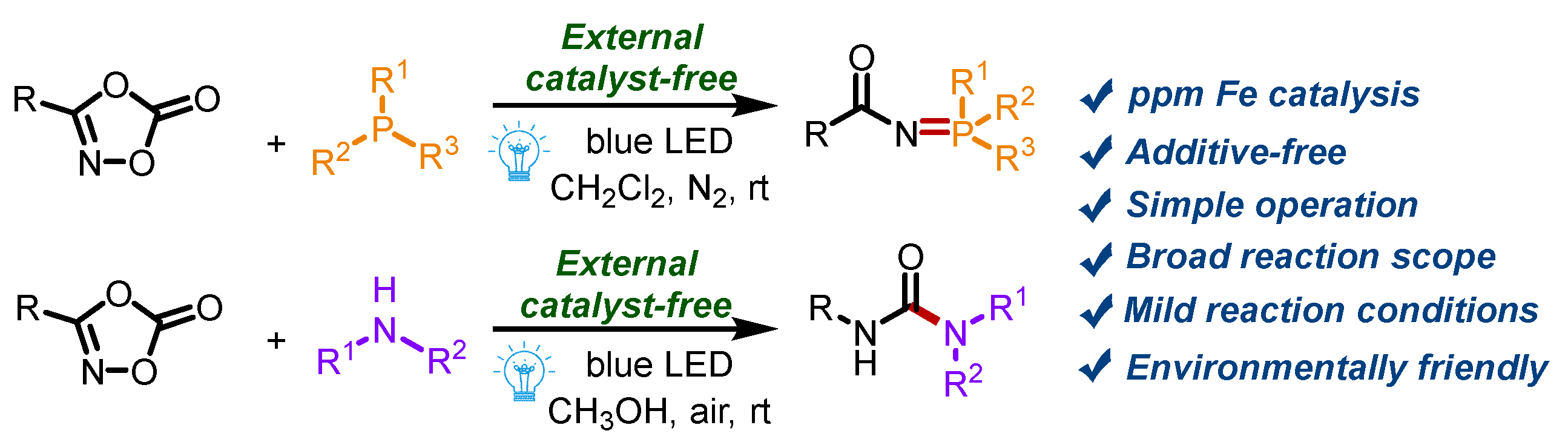
Abstract
A visible-light-induced external catalyst-free decarboxylation of dioxazolones was realized for the bond formation of N=P and N–C bonds to access phosphinimidic amides and ureas. Various phosphinimidic amides and ureas (47 examples) were synthesized with high yields (up to 98%) by this practical strategy in the presence of the system’s ppm Fe.
1. Introduction
Nowadays, the development of clean, environmentally friendly, and efficient chemical processes has become one of the goals of sustainable chemistry [1,2,3,4]. Visible light, as a safe, abundant, and renewable natural energy source, has promoted many feasible and valuable transformations [5,6,7,8]. Photocatalytic strategies were widely recognized as an attractive “green synthesis pathway” in organic transformations, which are promising from the standpoint of an environmentally friendly and sustainable perspective [9,10,11,12,13,14,15,16,17,18]. Despite the simple operation and mild reaction conditions, a precious metal complex or a synthetically elaborate organic dye is usually required [19,20,21,22]. It is of great significance to develop cleaner and greener photochemical pathways in the external catalyst-free protocol [23,24,25,26,27].
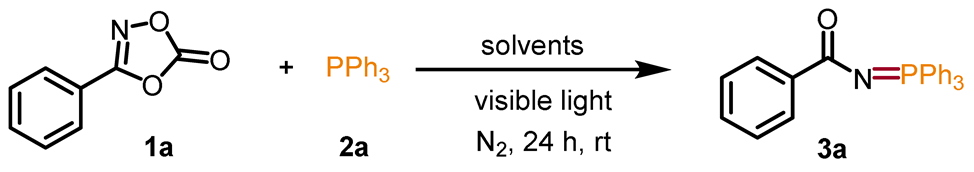
Nitrene intermediates have attracted great interest from chemists, due to their high reactivity [28,29,30,31,32,33,34,35,36]. Nitrene-based transformations allow the direct installation of nitrogen-containing building blocks into molecular backbones to build structurally complex compounds [37,38,39,40]. In the past few decades, a series of nitrene precursors were reported, such as organic azides [41,42], iminoiodinanes [43,44], amide N-O compounds [45,46], dioxazolones [47,48,49,50], and so on. Among them, dioxazolones are highly attractive because of their high activity, stability, convenience, and high coordination ability [51,52,53]. Herein, we present a visible-light-induced strategy to build N=P and N–C bonds for the generation of phosphinimidic amides and ureas from the reaction of dioxazolones and triarylphosphines or secondary amines (Scheme 1). The transformations are realized without any other catalyst or additive at room temperature. The ppm Fe in the reactants, confirmed by ICP-MS, might play an important role in this reaction.
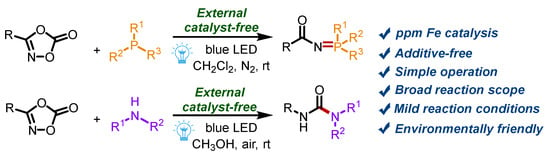
Scheme 1.
The construction of N=P and N–C bonds from dioxazolones.
2. Results and Discussion
Optimization of the Reaction Conditions
We commenced our study with the model reaction between 3-phenyl-1,4,2-dioxazol-5-one (1a) and triphenylphosphine (2a) under visible light and N2 atmosphere. The results are shown in Table 1. Initially, the reaction was carried out by employing DCE as the solvent under irradiation of 10 W 430 nm blue LED at room temperature, and the desired product N-(triphenyl-λ5-phosphinylidene)benzamide (3a) could be detected in 11% yield (entry 1). Afterwards, the solvent effect on the yield was investigated (entries 2–8). Different solvents, such as 1,4-dioxane, CH3OH, acetone, CH3CN, DMF, THF, and CH2Cl2, were surveyed, and the reaction exhibited excellent reaction performance in CH2Cl2 to provide the target product in 81% yield (entry 8). Further examination of the wavelengths of LED and substrate ratios showed no more positive results (entries 9–14). Control reactions confirmed that nearly no amidation product 3a was detected at room temperature in the absence of visible light (entry 15). Moreover, when the reaction was carried out in the air, only a trace amount of the product 3a was detected (entry 16). Therefore, the optimized reaction conditions were illustrated as follows: 1a (0.1 mmol); 2a (0.1 mmol); and CH2Cl2 (1 mL) in a N2 atmosphere under the irradiation of 430 nm blue LED (10 W) for 24 h at room temperature (entry 8).

Table 1.
Optimization of reaction conditions a.
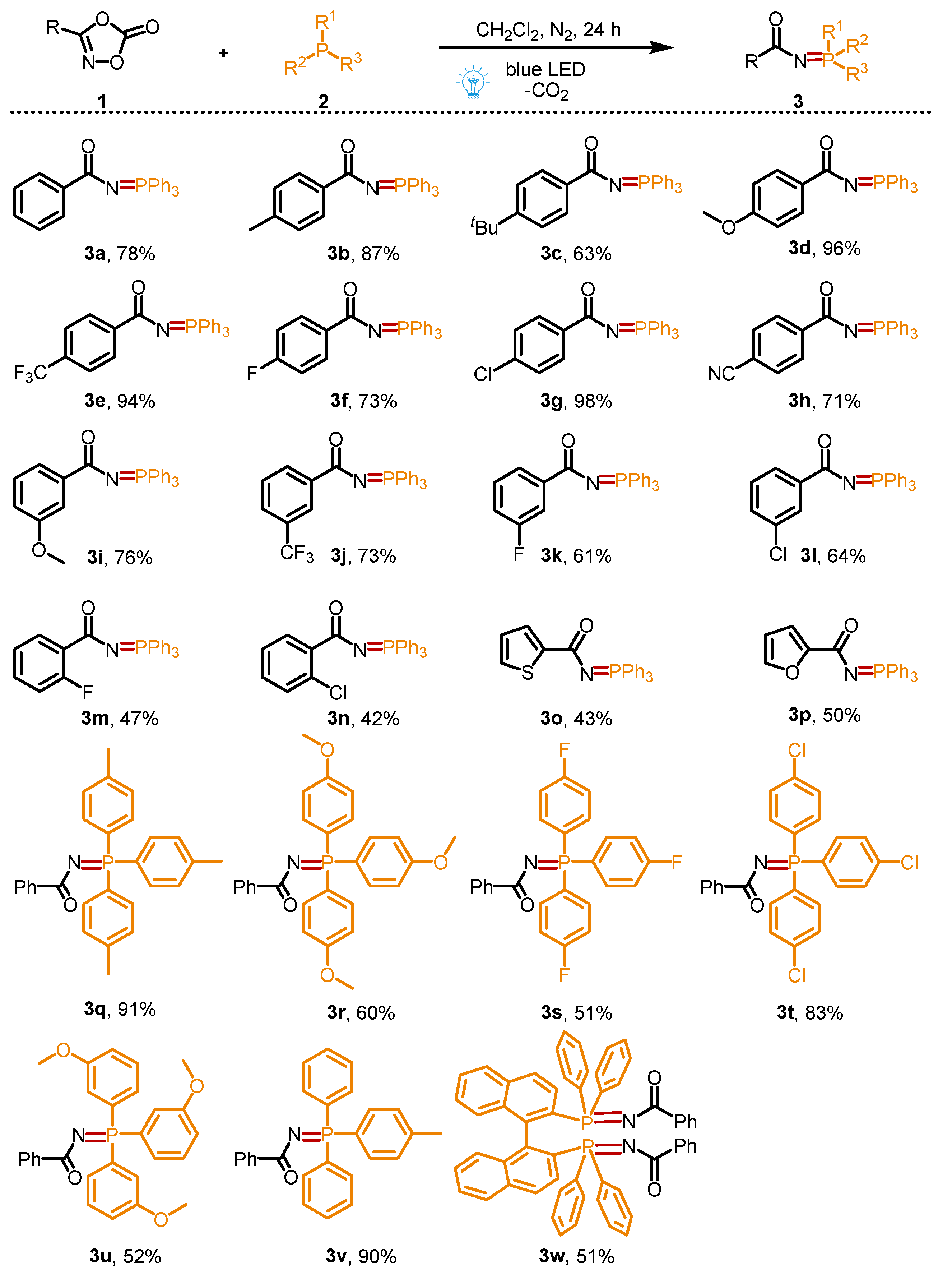
With the optimized conditions in hand, the scope of the organophosphorus compounds and dioxazolones 1 was investigated (Scheme 2). To our delight, various 3-phenyl dioxazolones bearing different electron-donating groups (-CH3, -tBu, and -OCH3) or electron-withdrawing groups (-CF3, -F, -Cl, and -CN) on the phenyl ring at different positions could react smoothly with 2a to produce the desired products (3a–3n) in moderate to excellent yields (42–98%). Among these cases, a slight steric hindrance effect was observed, and para-substituted 3-phenyl dioxazolones (3a–3h, 63–98%) showed higher reaction reactivities than those of ortho-substituted 3-phenyl dioxazolones (3m–3n, 42–47%). Moreover, the desired products 3o and 3p, which contain the skeletons of thiophene and furan, could also be successfully obtained in 43% and 50% of the yields, respectively. Additionally, electron-poor and electron-rich triphenylphosphine derivatives were all applicable to this transformation to access the desired products (3q–3v) in 51–91% yields. In addition, the phosphorus ligand, 1.1′-binaphthyl-2.2′-diphenylphosphine (BINAP), was also a suitable substrate to react with 1a, providing the corresponding product 3w in 51% yield.
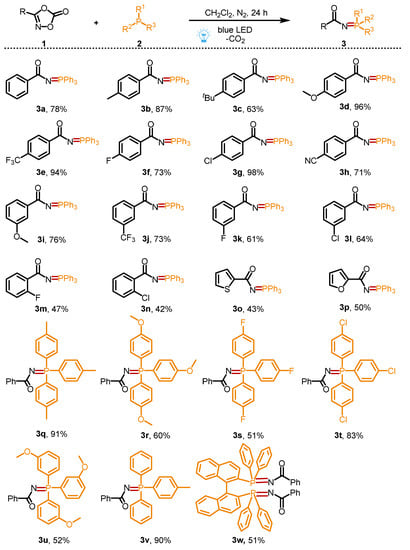
Scheme 2.
Substrate scope for the synthesis of phosphinimidic amides.
Reaction conditions: 1a (0.2 mmol); 2a (0.2 mmol) in solvent (2 mL) at room temperature for 24 h under the irradiation of 10 W 430 nm blue LED. Isolated yields were given.
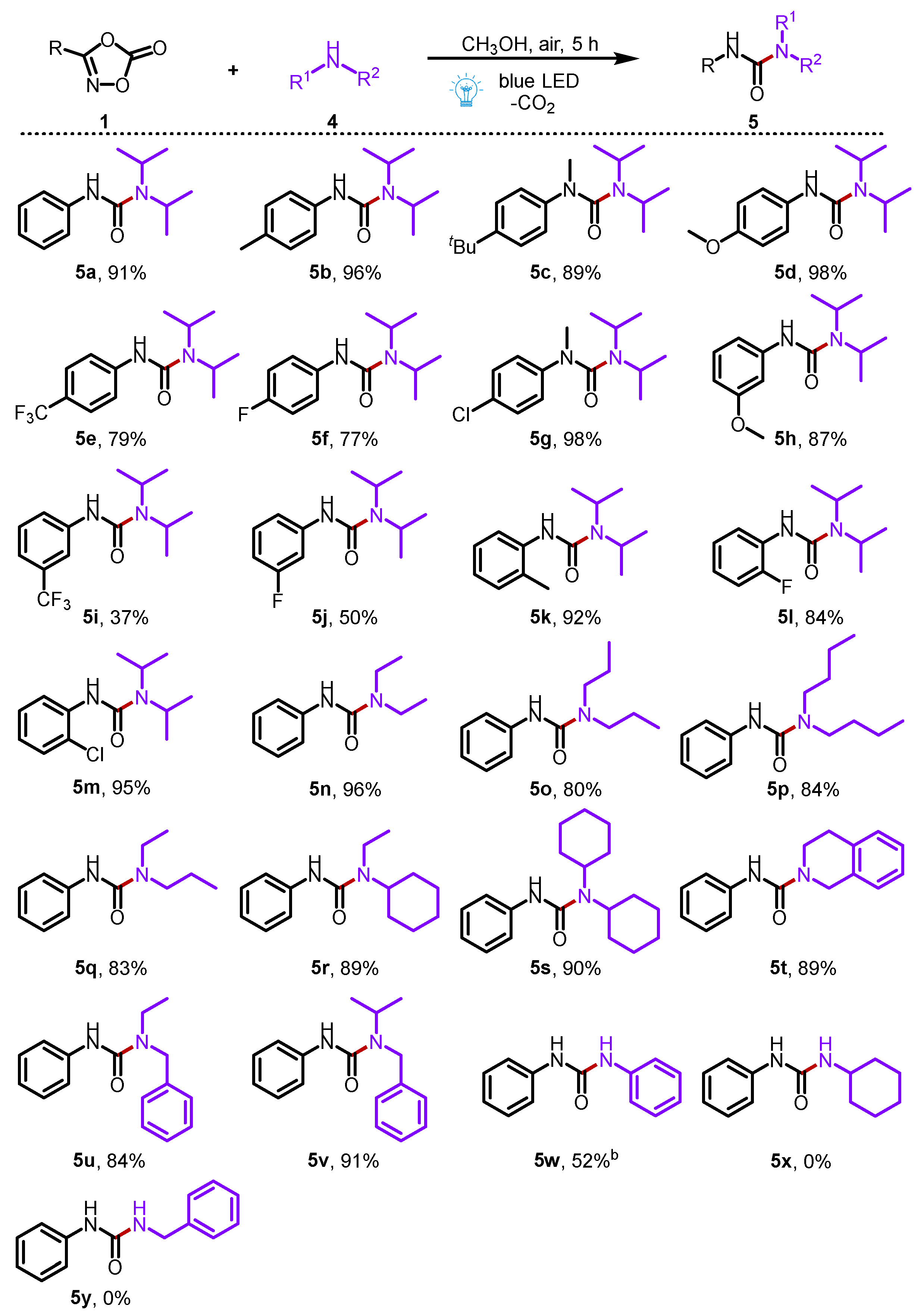
Then, we expanded the photocatalytic decarboxylation reaction of dioxazolones to the synthesis of unsymmetrical urea compounds (Scheme 3). To our delight, a wide range of 3-phenyl dioxazolones all reacted efficiently with diisopropylamine 4a to furnish the corresponding aryl ureas (5a–5m) in moderate to excellent yields (37–98%). In these cases, 3-phenyl dioxazolones bearing electron-donating groups (-CH3, -tBu, and -OCH3) showed a better reaction efficiency than those of 3-phenyl dioxazolones bearing electron-withdrawing groups (-CF3, -F, -Cl). Moreover, the broad scope of the commercially available secondary amines all reacted smoothly in this transformation, adding to the formation of desired ureas (5n–5v) in good to excellent yields (80–96%). In addition, the primary amine, such as aniline, was also a suitable substrate for reaction with 1a, providing the corresponding product 5w in 52% yield. However, cyclohexylamine (4l) and benzylamine (4m) were not suitable in this transformation to react with 1a to access the corresponding products 5x and 5y. Compared with the previous report [32], our method effectively avoids the harsh conditions of high temperature, showing good sustainability.
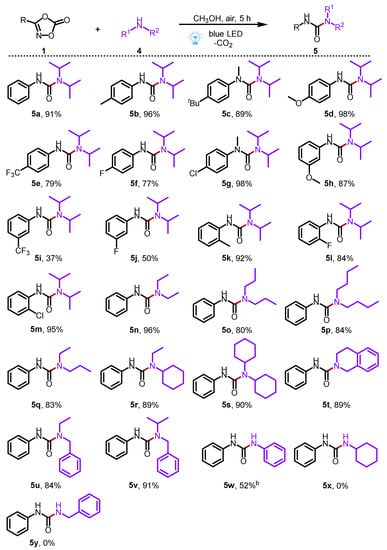
Scheme 3.
Substrate scope for the synthesis of ureas. Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol) in solvent (2 mL) at room temperature for 5 h under the irradiation of 10 W 430 nm blue LED. Isolated yields were given; b 20 h.
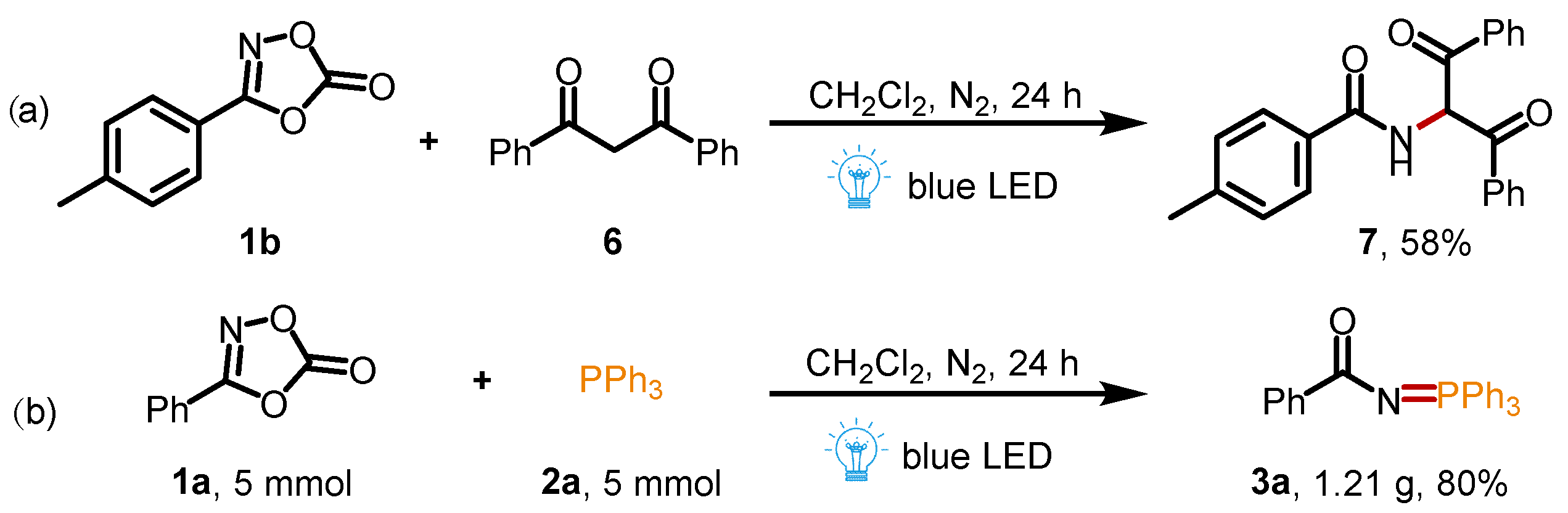
To our satisfaction, this method is also suitable for the reaction between 3-(p-tolyl)-1,4,2-dioxazol-5-one 1b and 1,3-diphenylpropane-1,3-dione 6 to give the corresponding amide product in 58% yield (Scheme 4a), which was previously reported in the presence of additional FeCl3 catalyst [29]. To verify the practicability of this synthetic protocol, the gram-scale synthesis of 3a was carried out (for details, see the Supplementary Materials). When the reaction was performed at a 5 mmol scale, the desired product 3a was isolated in 80% yield, indicating that this approach has a good practicability and application prospect (Scheme 4b).
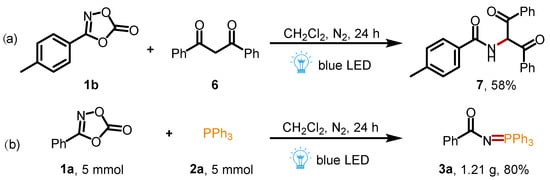
Scheme 4.
(a) Preparation of amide compounds; (b) gram-scale synthesis of 3a.
Furthermore, we also evaluated the sensitivity of the reaction of 1a and 2a. Compared with the standard conditions, the changes in concentration, temperature, oxygen level, water level, light intensity, and scale were measured. The yields were measured by 31P NMR and the yield deviation was calculated (for details, see the Supplementary Materials). Among them, light intensity and oxygen levels are important parameters for the reaction. Moreover, this transformation is moderately sensitive to water. Other parameters, such as concentration and temperature, can be regarded as random errors, which have a negligible impact on reaction efficiency (Figure S4, Supplementary Materials).
Next, we calculated the E-factor [54,55] and EcoScale scores [56,57] of the chemical process to evaluate the safety, economic, and ecological properties of the method. The results are summarized in Tables S2–S5, Supplementary Materials. As can be seen, the E-factor is extremely low at 0.38 and 0.82, respectively, and the EcoScale penalty is also low, at 21.5 and 15.5. Both parameters reflect the excellent green chemistry metrics of the protocol.
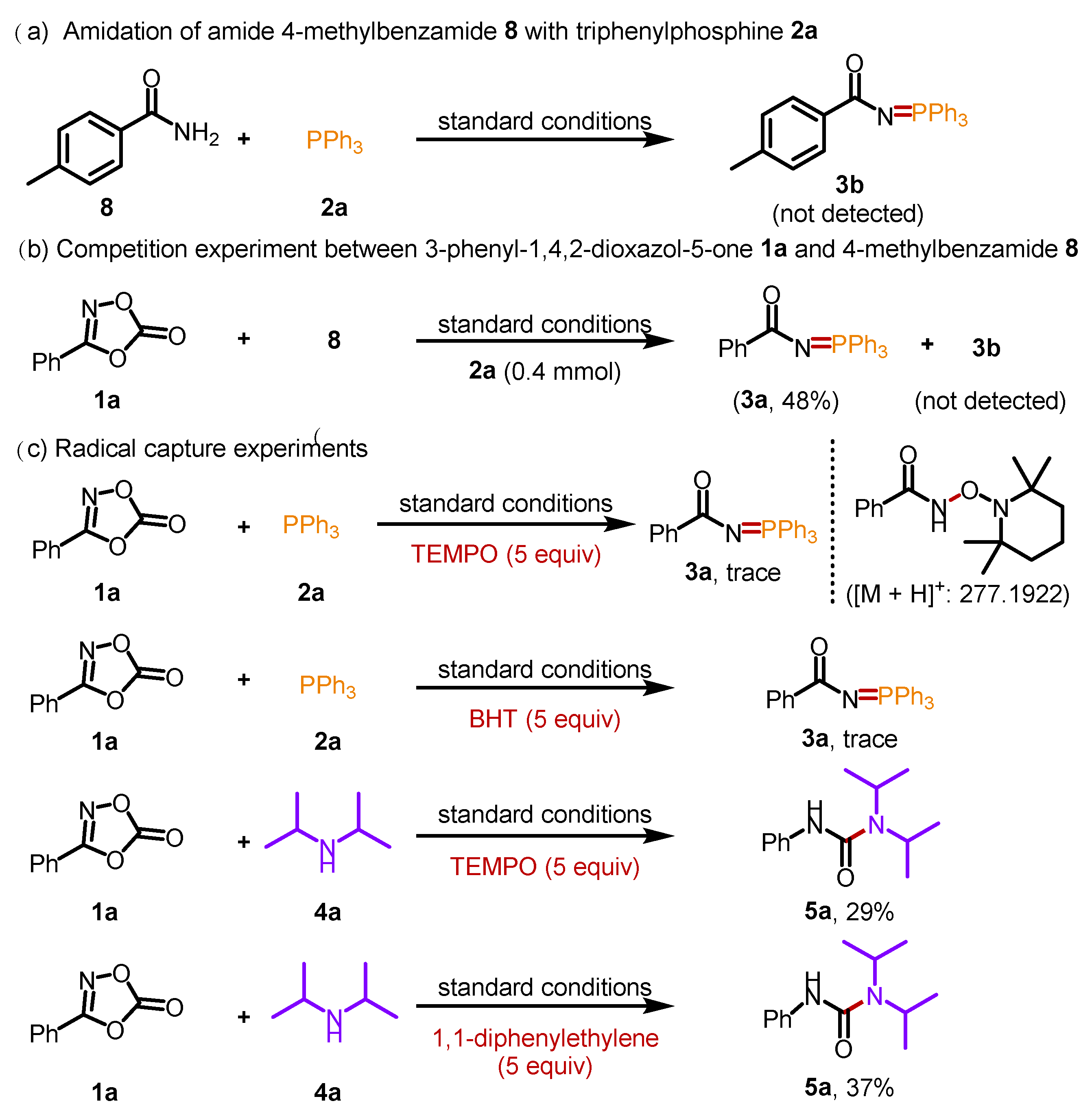
To understand the mechanism of this transformation, a set of control experiments were performed (Scheme 5). The phosphorylation of 4-methylbenzamide 8 with triphenylphosphine 2a was performed to determine whether the N=P bond was formed through the amide intermediate. However, 4-methyl-N-(triphenyl-λ5-phosphaneylidene) benzamide 3b was not detected (Scheme 5a). Moreover, intermolecular competition experiments of 1a and 8 were conducted, and only product 3a was obtained with 48% yield (Scheme 5b). These results demonstrated that the phosphorylation of dioxazolones was not conducted through amide intermediates. Furthermore, various radical trapping experiments were conducted (Scheme 5c). When (2,2,6,6-tetramethylpiperidine-1-yl)oxidanyl (TEMPO) was added to the model reaction under standard conditions, the reaction was significantly inhibited. The TEMPO-trapped acyl nitrene adducts were detected by high-resolution mass spectrometry (HRMS), with peaks at 277.1922 m/z. Subsequently, when another radical scavenger, 2,6-di-tert-butyl-4-methylphenol (BHT), was subjected under standard conditions, the reaction was also severely suppressed, indicating a radical process in the phosphorylation of dioxazolone with triphenylphosphine. Then, the radical trapping experiments of 1a and 4a were conducted (Scheme 5c). The decreased yields of product 5a indicated that the transformation also involved a radical process.
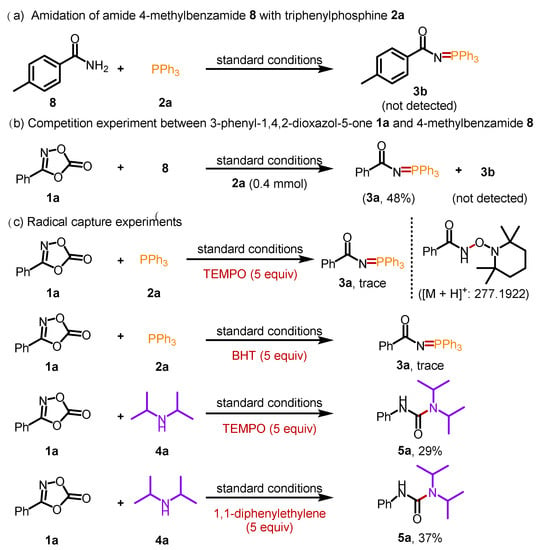
Scheme 5.
Control experiments.
In 2021, Yu and Bao et al., disclosed that FeCl3 (15 mol%) was required for the imidization of phosphines with dioxazolones under visible light irradiation [29]. While in our case, the transformations worked very well without any other additives. Considering the contamination issues in coupling reactions [58], we reasoned that some iron contamination might be possible in the manufacture of the starting materials. Therefore, the model reaction mixture was analyzed with inductively coupled plasma mass spectrometry (ICP-MS). Consequently, it is found that the Fe content of the reactions for the preparation of phosphinimidic amide (3a) and urea (5a) is approximately 27 ppm and 3 ppm, respectively (for details, see the Supplementary Materials). ICP-MS experiments were also performed on the starting materials of the model reactions (dioxazolone, PPh3, and amine), and the results showed that the iron contents of the dioxazolone, PPh3, and amine were 123 ppm, 420 ppm, and 0.9 ppm, respectively (for details, see the Supplementary Materials). It is reasoned that iron contamination issues in commercial chemicals are unavoidable during the production and transportation processes. When additional iron catalyst FeCl3 (5 mol%) was added to the model reaction under standard conditions, the reaction time was shortened and the yield was increased. These results confirmed that this reaction could be facilitated by iron catalysis (for details, see the Supplementary Materials). These results suggest that, although it is not a real transition-meta-free system, it is still a synthetically useful procedure for the synthesis of phosphinimidic amides and ureas, especially from an industrial chemistry standpoint.
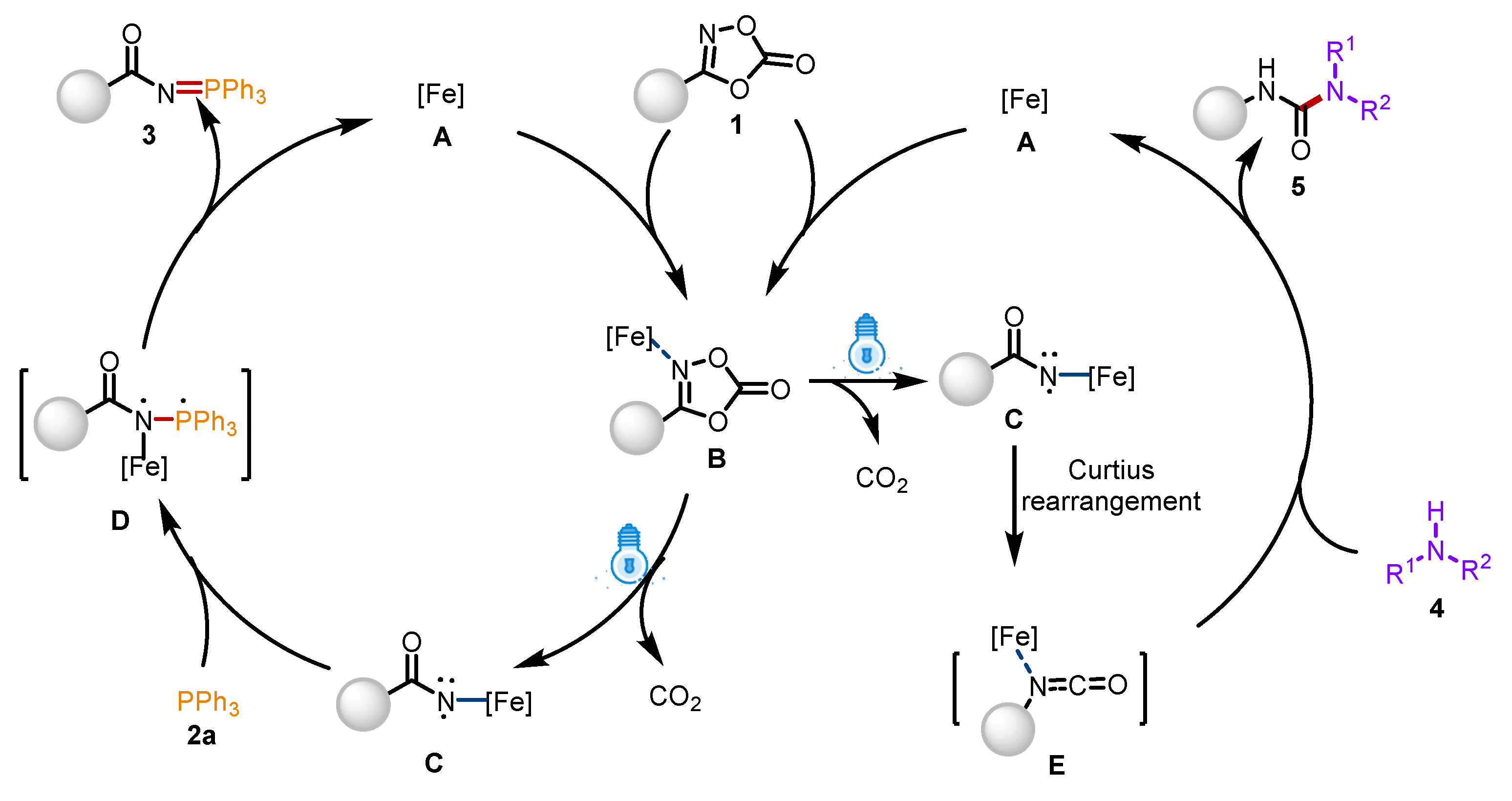
Based on these control experiments and previous literature reports, a plausible reaction pathway is proposed in Scheme 6. Initially, the N atom of dioxazolones 1 coordinates with the Fe center to form complex B, which is excited by visible light to generate the highly active iron-aminyl radical C with the release of CO2. Subsequently, radical C reacts with triphenylphosphine 2a to form the complex D, followed by a reduction and elimination process to obtain product 3. On the other hand, intermediate C underwent Curtius rearrangement to form intermediate E, which further reacts with secondary amines 4 to obtain product 5.
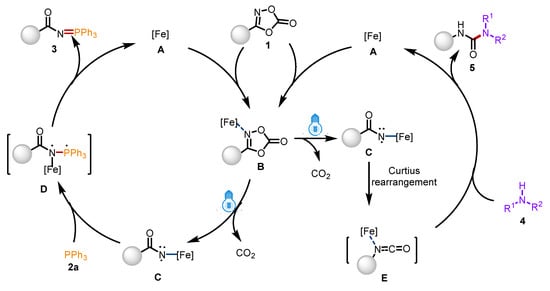
Scheme 6.
Proposed reaction mechanism.
3. Experimental Section
3.1. General Information
All nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 400 MHz in CDCl3 at room temperature (20 ± 3 °C), by using tetramethylsilane as the internal standard. High-resolution mass spectra (HRMS) were conducted on a 3000-mass spectrometer, using Waters Q-Tof MS/MS system with the ESI technique.
Photochemical reactions were carried out under visible light irradiation by a blue LED at 25 °C. The RLH-18 8-position Photo Reaction System manufactured by Beijing Roger Tech Ltd. was used in this system (Figure S1, Supplementary Materials). Eight 10 W blue LEDs were equipped in this photochemical reactor. The wavelength for blue LED is 430 nm, peak width at half-height is 18.4 nm (Figure S2, Supplementary Materials). The distance from the light source to the irradiation vessel was approximately 15 mm.
3.2. General Experimental Procedures for the Synthesis of (3a–3w)
In a 25 mL reaction tube, dioxazolones 1 (0.2 mmol, 1.0 equiv), organic phosphine substrate 2 (0.2 mmol, 1.0 equiv) in 1 mL CH2Cl2 were allowed to stir with irradiation of 10 W blue LED under N2 atmosphere at room temperature for 24 h. After the reaction, the solvent was evaporated under vacuum, and the residue was purified by column chromatography on silica gel to afford the desired products 3a–3w.
N-(triphenyl-λ5-phosphanylidene)benzamide (3a):
White solid (59.7 mg, 78%). 1H NMR (400 MHz, Chloroform-d) δ 8.45–8.38 (m, 2H), 7.94–7.85 (m, 6H), 7.62–7.55 (m, 3H), 7.54–7.41 (m, 9H); 13C NMR (101 MHz, Chloroform-d) δ 176.3 (d, JC-P = 8.0 Hz), 138.7 (d, JC-P = 20.6 Hz), 133.2 (d, JC-P = 10.0 Hz), 132.3 (d, JC-P = 2.9 Hz), 130.7, 129.6 (d, JC-P = 2.6 Hz), 128.7 (d, JC-P = 12.4 Hz), 128.4 (d, JC-P = 99.7 Hz), 127.7; 31P NMR (162 MHz, Chloroform-d) δ 20.71;
4-methyl-N-(triphenyl-λ5-phosphanylidene)benzamide (3b):
White solid (69.0 mg, 87%). 1H NMR (400 MHz, Chloroform-d) δ 8.30 (d, J = 8.2 Hz, 2H), 7.92–7.85 (m, 6H), 7.62–7.55 (m, 3H), 7.53–7.47 (m, 6H), 7.24 (d, J = 7.9 Hz, 2H), 2.42 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 176.5 (d, JC-P = 8.1 Hz), 140.9, 135.9 (d, JC-P = 20.5 Hz), 133.2 (d, JC-P = 9.7 Hz), 132.2 (d, JC-P = 2.9 Hz), 129.6 (d, JC-P = 2.6 Hz), 128.7 (d, JC-P = 12.1 Hz), 128.5 (d, JC-P = 99.6 Hz), 128.4, 21.6; 31P NMR (162 MHz, Chloroform-d) δ 20.47;
4-(tert-butyl)-N-(triphenyl-λ5-phosphanylidene)benzamide (3c):
White solid (55.0 mg, 63%). 1H NMR (400 MHz, Chloroform-d) δ 8.33 (d, J = 8.5 Hz, 2H), 7.92–7.84 (m, 6H), 7.61–7.55 (m, 3H), 7.53–7.45 (m, 8H), 1.38 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 176.4 (d, JC-P = 8.1 Hz), 153.9, 136.0 (d, JC-P = 20.5 Hz), 133.2 (d, JC-P = 10.1 Hz), 132.2 (d, JC-P = 2.9 Hz), 129.4 (d, JC-P = 2.6 Hz), 128.7 (d, JC-P = 12.4 Hz), 128.5 (d, JC-P = 99.3 Hz), 124.6, 34.9, 31.4; 31P NMR (162 MHz, Chloroform-d) δ 20.24;
4-methoxy-N-(triphenyl-λ5-phosphanylidene)benzamide (3d):
White solid (79 mg, 96%). 1H NMR (400 MHz, Chloroform-d) δ 8.35 (d, J = 8.8 Hz, 2H), 7.91–7.84 (m, 6H), 7.60–7.54 (m, 3H), 7.52–7.46 (m, 6H), 6.94 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 176.1 (d, JC-P = 7.8 Hz), 161.8, 133.2 (d, JC-P = 9.9 Hz), 132.2 (d, JC-P = 2.9 Hz), 131.5, 131.4 (d, JC-P = 2.5 Hz), 128.7 (d, JC-P = 12.2 Hz), 128.5 (d, JC-P = 99.6 Hz), 112.8, 55.3; 31P NMR (162 MHz, Chloroform-d) δ 20.30;
4-(trifluoromethyl)-N-(triphenyl-λ5-phosphanylidene)benzamide (3e):
White solid (87.6 mg, 94%). 1H NMR (400 MHz, Chloroform-d) δ 8.49 (d, J = 8.1 Hz, 2H), 7.91–7.84 (m, 6H), 7.69 (d, J = 8.1 Hz, 2H), 7.64–7.57 (m, 3H), 7.56–7.49 (m, 6H); 13C NMR (101 MHz, Chloroform-d) δ 174.8 (d, JC-P = 7.7 Hz), 142.0 (d, JC-P = 21.0 Hz), 133.2 (d, JC-P = 10.3 Hz), 132.5 (d, JC-P = 2.9 Hz), 132.2 (q, JC-F = 32.0 Hz), 129.8 (d, JC-P = 2.5 Hz), 128.8 (d, JC-P = 12.4 Hz), 127.9 (d, JC-P = 99.7 Hz), 124.7 (q, JC-F = 3.8 Hz), 124.3 (q, JC-F = 272.4 Hz); 31P NMR (162 MHz, Chloroform-d) δ 21.40; 19F NMR (376 MHz, Chloroform-d) δ −62.47;
4-fluoro-N-(triphenyl-λ5-phosphanylidene)benzamide (3f):
White solid (57.5 mg, 73%). 1H NMR (400 MHz, Chloroform-d) δ 8.38 (dd, J = 8.7, 5.9 Hz, 2H), 7.90–7.82 (m, 6H), 7.61–7.56 (m, 3H), 7.55–7.46 (m, 6H), 7.08 (t, J = 8.8 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ 175.3 (d, JC-P = 8.0 Hz), 164.7 (d, JC-F = 249.4 Hz), 134.9 (d, JC-P = 20.1 Hz), 133.2 (d, JC-P = 10.1 Hz), 132.3 (d, JC-P = 2.9 Hz), 131.8 (dd, JC-F = 8.9, JC-P = 2.3 Hz), 128.7 (d, JC-P = 12.3 Hz), 128.2 (d, JC-P = 99.7 Hz), 114.4 (d, JC-F = 21.4 Hz); 31P NMR (162 MHz, Chloroform-d) δ 20.87; 19F NMR (376 MHz, Chloroform-d) δ −110.66;
4-chloro-N-(triphenyl-λ5-phosphanylidene)benzamide (3g):
White solid (81.8 mg, 98%). 1H NMR (400 MHz, Chloroform-d) δ 8.33 (d, J = 8.5 Hz, 2H), 7.91–7.82 (m, 6H), 7.62–7.55 (m, 3H), 7.54–7.48 (m, 6H), 7.39 (d, J = 8.5 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ 175.2 (d, JC-P = 7.9 Hz), 137.2 (d, JC-P = 21.2 Hz), 136.8, 133.2 (d, JC-P = 9.7 Hz), 132.4 (d, JC-P = 2.9 Hz), 131.1 (d, JC-P = 2.3 Hz), 128.8 (d, JC-P = 12.4 Hz), 128.1 (d, JC-P = 99.7 Hz), 127.8; 31P NMR (162 MHz, Chloroform-d) δ 21.05;
4-cyano-N-(triphenyl-λ5-phosphaneylidene)benzamide (3h):
White solid (57.6 mg, 71%), mp 185.5–187.1 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.44 (d, J = 8.1 Hz, 2H), 7.89–7.80 (m, 6H), 7.70 (d, J = 8.3 Hz, 2H), 7.64–7.57 (m, 3H), 7.56–7.49 (m, 6H); 13C NMR (101 MHz, Chloroform-d) δ 174.2 (d, JC-P = 8.1 Hz), 142.8 (d, JC-P = 21.3 Hz), 133.1 (d, JC-P = 9.7 Hz), 132.6 (d, JC-P = 3.0 Hz), 131.6, 130.0 (d, JC-P = 2.3 Hz), 128.9 (d, JC-P = 12.4 Hz), 127.7 (d, JC-P = 99.8 Hz), 119.1, 113.8; 31P NMR (162 MHz, Chloroform-d) δ 21.79. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H20N2OP, 407.1308; found, 407.1309;
3-methoxy-N-(triphenyl-λ5-phosphanylidene)benzamide (3i):
White solid (62.4 mg, 76%). 1H NMR (400 MHz, Chloroform-d) δ 8.05 (d, J = 7.6 Hz, 1H), 7.93–7.83 (m, 7H), 7.61–7.55 (m, 3H), 7.54–7.47 (m, 6H), 7.35 (t, J = 7.9 Hz, 1H), 7.06–7.01 (m, 1H), 3.88 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 176.1 (d, JC-P = 8.1 Hz), 159.3, 140.2 (d, JC-P = 20.5 Hz), 133.2 (d, JC-P = 9.7 Hz), 132.3 (d, JC-P = 2.9 Hz), 128.7 (d, JC-P = 12.5 Hz), 128.3 (d, JC-P = 99.2 Hz), 122.3 (d, JC-P = 2.6 Hz), 117.4, 113.8 (d, JC-P = 2.9 Hz), 55.4; 31P NMR (162 MHz, Chloroform-d) δ 20.70;
3-(trifluoromethyl)-N-(triphenyl-λ5-phosphanylidene)benzamide (3j):
White solid (65.1 mg, 73%). 1H NMR (400 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.57 (d, J = 7.7 Hz, 1H), 7.92–7.84 (m, 6H), 7.72 (d, J = 8.1 Hz, 1H), 7.64–7.58 (m, 3H), 7.57–7.50 (m, 7H); 13C NMR (101 MHz, Chloroform-d) δ 174.7 (d, JC-P = 7.7 Hz), 139.5 (d, JC-P = 21.2 Hz), 133.2 (d, JC-P = 10.2 Hz), 132.8, 132.4 (d, JC-P = 3.0 Hz), 130.1 (q, JC-F = 32.2 Hz), 128.8 (d, JC-P = 12.4 Hz), 128.2, 128.0 (d, JC-P = 99.8 Hz), 127.1 (q, JC-F = 3.8 Hz), 126.5 (q, JC-F = 3.5 Hz), 123.0 (q, JC-F = 272.2 Hz); 31P NMR (162 MHz, Chloroform-d) δ 21.62; 19F NMR (376 MHz, Chloroform-d) δ −62.33;
3-fluoro-N-(triphenyl-λ5-phosphanylidene)benzamide (3k):
White solid (48.7 mg, 61%), mp 154.6–156.5 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.14 (d, J = 7.7 Hz, 1H), 8.10–8.05 (m, 1H), 7.91–7.82 (m, 6H), 7.63–7.56 (m, 3H), 7.55–7.48 (m, 6H), 7.42–7.34 (m, 1H), 7.19–7.13 (m, 1H); 13C NMR (101 MHz, Chloroform-d) δ 175.0 (d, JC-P = 7.4 Hz), 162.6 (d, JC-F = 244.8 Hz), 141.2 (d, JC-F = 21.3 Hz), 133.2 (d, JC-P = 10.0 Hz), 132.4 (d, JC-F = 2.9 Hz), 129.1 (d, JC-P = 7.7 Hz), 128.8 (d, JC-P = 12.4 Hz), 128.1 (d, JC-P = 99.8 Hz), 125.1 (d, JC-P = 2.8 Hz), 117.5 (d, JC-P = 21.6 Hz), 116.4 (dd, JC-F = 22.2, JC-P = 2.6 Hz); 31P NMR (162 MHz, Chloroform-d) δ 21.13; 19F NMR (376 MHz, Chloroform-d) δ −114.38. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H20FNOP, 400.1261; found, 400.1261;
3-chloro-N-(triphenyl-λ5-phosphanylidene)benzamide (3l):
Colorless liquid (56.7 mg, 64%). 1H NMR (400 MHz, Chloroform-d) δ 8.40–8.36 (m, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.90–7.82 (m, 6H), 7.63–7.57 (m, 3H), 7.55–7.48 (m, 6H), 7.46–7.41 (m, 1H), 7.37–7.32 (m, 1H); 13C NMR (101 MHz, Chloroform-d) δ 174.9 (d, JC-P = 8.0 Hz), 140.6 (d, JC-P = 21.2 Hz), 133.7, 133.2 (d, JC-P = 10.2 Hz), 132.4 (d, JC-P = 2.6 Hz), 130.6, 129.8 (d, JC-P = 2.7 Hz), 129.0, 128.8 (d, JC-P = 12.0 Hz), 128.0 (d, JC-P = 99.8 Hz), 127.6 (d, JC-P = 2.2 Hz); 31P NMR (162 MHz, Chloroform-d) δ 21.40. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H20ClNOP, 416.0966; found, 416.0963;
2-fluoro-N-(triphenyl-λ5-phosphanylidene)benzamide (3m):
White solid (37.5 mg, 47%). 1H NMR (400 MHz, Chloroform-d) δ 8.22–8.16 (m, 1H), 7.93–7.84 (m, 6H), 7.61–7.55 (m, 3H), 7.53–7.47 (m, 6H), 7.41–7.34 (m, 1H), 7.18–7.06 (m, 2H); 13C NMR (101 MHz, Chloroform-d) δ 174.1 (dd, JC-P = 7.9 Hz, JC-F = 3.6 Hz), 161.9 (d, JC-F = 255.2 Hz), 133.2 (d, JC-P = 10.1 Hz), 132.4 (dd, JC-P = 2.3 Hz, JC-F = 2.2 Hz), 132.3 (d, JC-P = 2.9 Hz), 131.7 (d, JC-F = 8.8 Hz), 128.7 (d, JC-P = 12.4 Hz), 128.0 (d, JC-P = 99.3 Hz), 127.5 (dd, JC-P = 21.4 Hz, JC-F = 9.9 Hz), 123.3 (d, JC-F = 3.8 Hz), 116.5 (d, JC-F = 23.4 Hz); 31P NMR (162 MHz, Chloroform-d) δ 20.39; 19F NMR (376 MHz, Chloroform-d) δ −111.60;
2-chloro-N-(triphenyl-λ5-phosphanylidene)benzamide (3n):
White solid (35.0 mg, 42%), mp 196.6–197.8 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.43–8.36 (m, 2H), 7.92–7.84 (m, 6H), 7.62–7.56 (m, 3H), 7.54–7.48 (m, 6H), 7.46–7.41 (m, 2H); 13C NMR (101 MHz, Chloroform-d) δ 176.4 (d, JC-P = 8.6 Hz), 138.6 (d, JC-P = 20.6 Hz), 133.2 (d, JC-P = 9.7 Hz), 132.3 (d, JC-P = 2.9 Hz), 130.7, 129.6 (d, JC-P = 2.3 Hz), 128.7 (d, JC-P = 12.3 Hz), 128.4 (d, JC-P = 99.5 Hz), 127.7; 31P NMR (162 MHz, Chloroform-d) δ 20.72. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H20ClNOP, 416.0966; found, 416.0969;
N-(triphenyl-λ5-phosphanylidene)thiophene-2-carboxamide (3o):
Brown solid (33.3 mg, 43%). 1H NMR (400 MHz, Chloroform-d) δ 7.89–7.82 (m, 6H), 7.80 (dd, J = 3.6, 1.2 Hz, 1H), 7.62–7.56 (m, 3H), 7.53–7.47 (m, 6H), 7.40 (dd, J = 5.0, 1.2 Hz, 1H), 7.10–7.01 (m, 1H); 13C NMR (101 MHz, Chloroform-d) δ 171.1 (d, JC-P = 7.0 Hz), 145.2 (d, JC-P = 24.3 Hz), 133.2 (d, JC-P = 9.7 Hz), 132.3 (d, JC-P = 2.9 Hz), 130.2 (d, JC-P = 2.8 Hz), 129.6, 128.7 (d, JC-P = 12.4 Hz), 128.1 (d, JC-P = 99.7 Hz), 127.3; 31P NMR (162 MHz, Chloroform-d) δ 19.47;
N-(triphenyl-λ5-phosphanylidene)furan-2-carboxamide (3p):
White solid (37.2 mg, 50%). 1H NMR (400 MHz, Chloroform-d) δ 7.87–7.79 (m, 6H), 7.59–7.53 (m, 3H), 7.51–7.45 (m, 7H), 7.17 (d, J = 3.3 Hz, 1H), 6.44 (dd, J = 3.2, 1.7 Hz, 1H); 13C NMR (101 MHz, Chloroform-d) δ 168.0 (d, JC-P = 6.7 Hz), 152.8 (d, JC-P = 26.0 Hz), 144.0, 133.2 (d, JC-P = 10.1 Hz), 132.4, 128.7 (d, JC-P = 12.4 Hz), 127.9 (d, JC-P = 100.2 Hz), 114.3, 111.3; 31P NMR (162 MHz, Chloroform-d) δ 21.86;
N-(tri-p-tolyl-λ5-phosphanylidene)benzamide (3q):
White solid (77.1 mg, 91%). 1H NMR (400 MHz, Chloroform-d) δ 8.42 (d, J = 7.8 Hz, 2H), 7.79 (dd, J = 12.2, 7.7 Hz, 6H), 7.48–7.41 (m, 3H), 7.32 (dd, J = 8.3, 2.8 Hz, 6H), 2.43 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 176.2 (d, JC-P = 8.0 Hz), 142.7 (d, JC-P = 2.9 Hz), 138.9 (d, JC-P = 20.8 Hz), 133.2 (d, JC-P = 10.3 Hz), 130.6, 129.6 (d, JC-P = 2.3 Hz), 129.4 (d, JC-P = 12.9 Hz), 127.6, 125.4 (d, JC-P = 102.0 Hz), 21.7; 31P NMR (162 MHz, Chloroform-d) δ 20.63;
N-(tri-p-methoxyphenyl-λ5-phosphanylidene)benzamide (3r):
White solid (56.7 mg, 60%). 1H NMR (400 MHz, Chloroform-d) δ 8.40–8.34 (m, 2H), 7.78 (dd, J = 11.7, 8.8 Hz, 6H), 7.46–7.38 (m, 3H), 7.00 (dd, J = 8.9, 2.3 Hz, 6H), 3.85 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 176.1 (d, JC-P = 7.4 Hz), 162.6 (d, JC-P = 2.9 Hz), 139.0 (d, JC-P = 20.4 Hz), 135.0 (d, JC-P = 11.1 Hz), 130.5, 129.5 (d, JC-P = 2.3 Hz), 127.6, 119.9 (d, JC-P = 106.5 Hz), 114.3 (d, JC-P = 13.3 Hz), 55.4; 31P NMR (162 MHz, Chloroform-d) δ 19.68;
N-(tris(4-fluorophenyl)-λ5-phosphanylidene)benzamide (3s):
White solid (44.2 mg, 51%). 1H NMR (400 MHz, Chloroform-d) δ 8.34–8.29 (m, 2H), 7.90–7.81 (m, 6H), 7.50–7.41 (m, 3H), 7.26–7.19 (m, 6H); 13C NMR (101 MHz, Chloroform-d) δ 176.5 (d, JC-P = 8.1 Hz), 165.4 (dd, JC-F = 255.1 Hz, JC-P = 3.2 Hz), 138.1 (d, JC-P = 20.6 Hz), 135.6 (dd, JC-F = 11.7 Hz, JC-P = 8.8 Hz), 131.0, 129.5 (d, JC-P = 2.7 Hz), 127.8, 123.9 (dd, JC-P = 103.9 Hz, JC-F = 3.3 Hz), 116.4 (dd, JC-F = 21.6 Hz, JC-P = 13.6 Hz); 31P NMR (162 MHz, Chloroform-d) δ 18.98; 19F NMR (376 MHz, Chloroform-d) δ −105.30;
N-(tris(4-chlorophenyl)-λ5-phosphanylidene)benzamide (3t):
White solid (80.5 mg, 83%). 1H NMR (400 MHz, Chloroform-d) δ 8.36–8.30 (m, 2H), 7.79 (dd, J = 12.0, 8.4 Hz, 6H), 7.54–7.47 (m, 7H), 7.43 (dd, J = 8.1, 6.2 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ 176.7 (d, JC-P = 8.0 Hz), 139.5 (d, JC-P = 3.6 Hz), 137.9 (d, JC-P = 20.5 Hz), 134.4 (d, JC-P = 11.0 Hz), 131.1, 129.5 (d, JC-P = 2.5 Hz), 129.4 (d, JC-P = 12.8 Hz), 127.8, 126.2 (d, JC-P = 102.0 Hz); 31P NMR (162 MHz, Chloroform-d) δ 19.49;
N-(tris(3-methoxyphenyl)-λ5-phosphaneylidene)benzamide (3u):
White solid (48.5 mg, 52%), mp 146.3–147.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.41–8.36 (m, 2H), 7.52–7.47 (m, 3H), 7.46–7.34 (m, 9H), 7.12–7.07 (m, 3H), 3.79 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 176.2 (d, JC-P = 7.9 Hz), 159.6 (d, JC-P = 15.4 Hz), 138.7 (d, JC-P = 20.6 Hz), 130.7, 129.9 (d, JC-P = 14.6 Hz), 129.6 (d, JC-P = 99.1 Hz), 129.5 (d, JC-P = 2.4 Hz), 127.7, 125.4 (d, JC-P = 9.7 Hz), 118.4 (d, JC-P = 11.0 Hz), 118.1 (d, JC-P = 2.9 Hz), 55.4; 31P NMR (162 MHz, Chloroform-d) δ 21.38. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C28H27NO4P, 472.1672; found, 472.1677;
N-(diphenyl(p-tolyl)-λ5-phosphanylidene)benzamide (3v):
White solid (71.1 mg, 90%). 1H NMR (400 MHz, Chloroform-d) δ 8.45–8.37 (m, 2H), 7.93–7.85 (m, 4H), 7.81–7.74 (m, 2H), 7.61–7.55 (m, 2H), 7.53–7.42 (m, 7H), 7.32 (dd, J = 8.3, 2.9 Hz, 2H), 2.43 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 176.3 (d, JC-P = 8.0 Hz), 142.9 (d, JC-P = 2.9 Hz), 138.7 (d, JC-P = 20.6 Hz), 133.3 (d, JC-P = 10.5 Hz), 133.2 (d, JC-P = 9.5 Hz), 132.2 (d, JC-P = 3.0 Hz), 130.7, 129.6, 129.5 (d, JC-P = 10.0 Hz), 128.7 (d, JC-P = 12.3 Hz), 128.6 (d, JC-P = 99.8 Hz), 127.7, 124.8 (d, JC-P = 101.3 Hz), 21.7; 31P NMR (162 MHz, Chloroform-d) δ 20.67;
N,N’-([1,1′-binaphthalene]-2,2′-diylbis(diphenyl-λ5-phosphaneylylidene))dibenzamide (3w):
White solid (87.7 mg, 51%), mp 184.5–185.8 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.18 (d, J = 7.1 Hz, 4H), 7.78–7.69 (m, 6H), 7.57–7.46 (m, 7H), 7.44–7.29 (m, 13H), 7.28–7.21 (m, 3H), 7.20–7.07 (m, 7H), 6.41 (dd, J = 8.3, 5.0 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ 175.9 (d, JC-P = 8.0 Hz), 159.6 (d, JC-P = 2.0 Hz), 138.8 (d, JC-P = 21.2 Hz), 135.1 (d, JC-P = 7.1 Hz), 134.1 (d, JC-P = 2.6 Hz), 133.3 (d, JC-P = 10.5 Hz), 132.9 (d, JC-P = 10.3 Hz), 132.1 (d, JC-P = 3.0 Hz), 131.6 (d, JC-P = 2.4 Hz), 130.4, 129.4 (d, JC-P = 2.7 Hz), 128.5 (d, JC-P = 12.5 Hz), 128.2 (d, JC-P = 12.6 Hz), 127.8 (d, JC-P = 104.7 Hz), 127.50, 127.45, 123.9 (d, JC-P = 11.4 Hz), 121.0 (d, JC-P = 6.8 Hz), 119.2 (d, JC-P = 100.4 Hz); 31P NMR (162 MHz, Chloroform-d) δ 19.95. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C58H43N2O2P2, 861.2794; found, 861.2791.
3.3. General Experimental Procedures for the Synthesis of (5a–5w)
In a 25 mL reaction tube, dioxazolone 1 (0.2 mmol, 1.0 equiv.), and amine 4 (0.4 mmol, 2.0 equiv.) in 1 mL CH3OH were allowed to stir with irradiation of 10 W blue LED at room temperature for 5 h. After the reaction, the solvent was evaporated under vacuum, and the residue was purified by column chromatography on silica gel to afford the desired products 5a–5w.
1,1-diisopropyl-3-phenylurea (5a):
White solid (40.1 mg, 91%). 1H NMR (400 MHz, Chloroform-d) δ 7.39 (dd, J = 8.4, 1.3 Hz, 2H), 7.31–7.26 (m, 2H), 7.06–6.99 (m, 1H), 6.26 (s, 1H), 4.04–3.95 (m, 2H), 1.34 (d, J = 7.0 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.6, 139.4, 128.8, 122.6, 119.7, 45.5, 21.5;
1,1-diisopropyl-3-(p-tolyl)urea (5b):
White solid (44.9 mg, 96%). 1H NMR (400 MHz, Chloroform-d) δ 7.27 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.18 (s, 1H), 4.04–3.94 (m, 2H), 2.30 (s, 3H), 1.33 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.8, 136.8, 132.1, 129.3, 119.9, 45.4, 21.5, 20.7;
3-(4-(tert-butyl)phenyl)-1,1-diisopropylurea (5c):
White solid (49.1 mg, 89%), mp 115.1–116.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.33–7.29 (m, 4H), 6.17 (s, 1H), 4.05–3.96 (m, 2H), 1.34 (d, J = 6.9 Hz, 12H); 1.31 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 154.9, 145.6, 136.7, 125.7, 119.7, 45.4, 34.2, 31.4, 21.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H29N2O, 277.2274; found, 277.2284;
1,1-diisopropyl-3-(4-methoxyphenyl)urea (5d):
White solid (49.0 mg, 98%). 1H NMR (400 MHz, Chloroform-d) δ 7.27 (d, J = 8.9 Hz, 2H), 6.83 (d, J = 9.0 Hz, 2H), 6.13 (s, 1H), 4.01–3.92 (m, 2H), 3.77 (s, 3H), 1.32 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 155.5, 155.1, 132.5, 122.0, 114.1, 55.5, 45.4, 21.5;
1,1-diisopropyl-3-(4-(trifluoromethyl)phenyl)urea (5e):
White solid (44.2 mg, 79%), mp 151.9–153.1 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.57–7.46 (m, 4H), 6.43 (s, 1H), 4.07–3.92 (m, 2H), 1.35 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.0, 142.6, 126.1 (q, JC-F = 3.8 Hz), 124.4 (q, JC-F = 271.1 Hz), 124.2 (q, JC-F = 32.7 Hz), 118.9, 45.7, 21.5; 19F NMR (376 MHz, Chloroform-d) δ −61.81; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20F3N2O, 289.1522; found, 289.1534;
3-(4-fluorophenyl)-1,1-diisopropylurea (5f):
White solid (36.7 mg, 77%), mp 134.5–135.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.35–7.27 (m, 2H), 6.94 (t, J = 8.6 Hz, 2H), 6.31 (s, 1H), 4.03–3.86 (m, 2H), 1.30 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 158.6 (d, JC-F = 241.2 Hz), 154.8, 135.4 (d, JC-F = 2.9 Hz), 121.8 (d, JC-F = 7.9 Hz), 115.2 (d, JC-F = 22.1 Hz), 45.6, 21.4; 19F NMR (376 MHz, Chloroform-d) δ -120.90; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H20FN2O, 239.1554; found, 239.1567;
3-(4-chlorophenyl)-1,1-diisopropylurea (5g):
White solid (49.8 mg, 98%). 1H NMR (400 MHz, Chloroform-d) δ 7.32 (d, J = 8.9 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 6.29 (s, 1H), 4.03–3.90 (m, 2H), 1.32 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.4, 138.0, 128.7, 127.4, 121.0, 45.6, 21.5;
1,1-diisopropyl-3-(3-methoxyphenyl)urea (5h):
White solid (43.6 mg, 87%). 1H NMR (400 MHz, Chloroform-d) δ 7.20–7.14 (m, 2H), 6.85 (dd, J = 8.0, 1.2 Hz, 1H), 6.57 (dd, J = 8.2, 1.7 Hz, 1H), 6.27 (s, 1H), 4.05–3.95 (m, 2H), 3.81 (s, 3H), 1.33 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 160.2, 154.5, 140.7, 129.4, 111.7, 108.6, 105.1, 55.3, 45.4, 21.5;
1,1-diisopropyl-3-(3-(trifluoromethyl)phenyl)urea (5i):
White solid (21.3 mg, 37%), mp 151.2–152.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.67 (s, 1H), 7.62–7.55 (m, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.29–7.25 (m, 1H), 6.37 (s, 1H), 4.07–3.94 (m, 2H), 1.36 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.2, 139.9, 131.2 (q, JC-F = 271.6 Hz), 129.3, 122.7, 119.1 (q, JC-F = 3.7 Hz), 116.1 (q, JC-F = 4.0 Hz), 45.7, 21.5; 19F NMR (376 MHz, Chloroform-d) δ -62.64. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20F3N2O, 289.1522; found, 289.1536;
3-(3-fluorophenyl)-1,1-diisopropylurea (5j):
White solid (23.9 mg, 50%), mp 118.5–119.8 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.40–7.32 (m, 1H), 7.23–7.14 (m, 1H), 7.00 (dd, J = 8.3, 2.1 Hz, 1H), 6.72–6.64 (m, 1H), 6.39 (s, 1H), 4.03–3.89 (m, 2H), 1.32 (d, J = 6.6 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 163.2 (d, JC-F = 243.5 Hz), 154.2, 141.1 (d, JC-F = 11.2 Hz), 129.7 (d, JC-F = 9.6 Hz), 114.7 (d, JC-F = 2.9 Hz), 109.0 (d, JC-F = 21.3 Hz), 106.9 (d, JC-F = 26.4 Hz), 45.6, 21.5; 19F NMR (376 MHz, Chloroform-d) δ -112.35. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H20FN2O, 239.1554; found, 239.1564;
1,1-diisopropyl-3-(o-tolyl)urea (5k):
White solid (43.0 mg, 92%), mp 136.8–138.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.77 (dd, J = 8.1, 1.2 Hz, 1H), 7.23–7.14 (m, 2H), 7.04–6.96 (m, 1H), 6.07 (s, 1H), 4.11–3.99 (m, 2H), 2.28 (s, 3H), 1.36 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.8, 137.6, 130.3, 127.6, 126.7, 123.2, 122.3, 45.4, 21.5, 18.3. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H23N2O, 235.1805; found, 235.1816;
3-(2-fluorophenyl)-1,1-diisopropylurea (5l):
White solid (40.0 mg, 84%), mp 109.1–110.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.24–8.16 (m, 1H), 7.13–7.02 (m, 2H), 6.99–6.90 (m, 1H), 6.58 (s, 1H), 4.12–4.03 (m, 2H), 1.35 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.1, 152.3 (d, JC-F = 239.8 Hz), 128.0 (d, JC-F = 9.5 Hz), 124.5 (d, JC-F = 3.6 Hz), 122.0 (d, JC-F = 7.5 Hz), 121.1, 114.3 (d, JC-F = 19.2 Hz), 45.3, 21.4; 19F NMR (376 MHz, Chloroform-d) δ -133.39. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H20FN2O, 239.1554; found, 239.1564;
3-(2-chlorophenyl)-1,1-diisopropylurea (5m):
White solid (49.6 mg, 95%). 1H NMR (400 MHz, Chloroform-d) δ 7.41–7.37 (m, 2H), 7.29 (d, J = 8.6 Hz, 1H), 7.06–6.98 (m, 1H), 6.27 (s, 1H), 4.05–3.95 (m, 2H), 1.34 (d, J = 6.9 Hz, 12H); 13C NMR (101 MHz, Chloroform-d) δ 154.6, 139.4, 128.8, 122.6, 119.7, 45.5, 21.5;
1,1-diethyl-3-phenylurea (5n):
White solid (36.8 mg, 96%). 1H NMR (400 MHz, Chloroform-d) δ 7.43–7.39 (m, 2H), 7.31–7.25 (m, 2H), 7.02 (t, J = 7.3 Hz, 1H), 6.40 (s, 1H), 3.41–3.35 (m, 4H), 1.22 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, Chloroform-d) δ 154.7, 139.4, 128.8, 122.8, 119.9, 41.6, 13.9;
3-phenyl-1,1-dipropylurea (5o):
White solid (35 mg, 80%). 1H NMR (400 MHz, Chloroform-d) δ 7.43–7.37 (m, 2H), 7.31–7.25 (m, 2H), 7.06–6.97 (m, 1H), 6.41 (s, 1H), 3.31–3.24 (m, 4H), 1.71–1.61 (m, 4H), 0.96 (t, J = 7.4 Hz, 6H); 13C NMR (101 MHz, Chloroform-d) δ 155.0, 139.4, 128.8, 122.7, 119.8, 49.4, 21.9, 11.4;
1,1-dibutyl-3-phenylurea (5p):
White solid (41.6 mg, 84%). 1H NMR (400 MHz, Chloroform-d) δ 7.44–7.38 (m, 2H), 7.30–7.25 (m, 2H), 7.05–6.99 (m, 1H), 6.38 (s, 1H), 3.34–3.28 (m, 4H), 1.65–1.58 (m, 4H), 1.43–1.34 (m, 4H), 0.98 (t, J = 7.3 Hz, 6H); 13C NMR (101 MHz, Chloroform-d) δ 155.0, 139.4, 128.8, 122.7, 119.7, 47.5, 30.8, 20.2, 13.9;
1-ethyl-3-phenyl-1-propylurea (5q):
Colorless liquid (34.0 mg, 83%), mp 56.3–57.6 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.43–7.37 (m, 2H), 7.31–7.25 (m, 2H), 7.02 (t, J = 7.4 Hz, 1H), 6.39 (s, 1H), 3.42–3.34 (m, 2H), 3.30–3.24 (m, 2H), 1.71–1.61 (m, 2H), 1.22 (t, J = 7.1 Hz, 3H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ 154.9, 139.4, 128.8, 122.7, 119.8, 48.8, 42.1, 22.0, 13.8, 11.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H19N2O, 207.1492; found, 207.1499;
1-cyclohexyl-1-ethyl-3-phenylurea (5r):
White solid (44.0 mg, 89%), mp 123.5–124.7 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.44–7.39 (m, 2H), 7.31–7.25 (m, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.41 (s, 1H), 4.14–4.03 (m, 1H), 3.33–3.25 (m, 2H), 1.85–1.76 (m, 4H), 1.72–1.64 (m, 1H), 1.47–1.33 (m, 4H), 1.25 (t, J = 7.2 Hz, 3H), 1.18–1.06 (m, 1H); 13C NMR (101 MHz, Chloroform-d) δ 154.8, 139.5, 128.8, 122.7, 119.9, 54.8, 36.9, 31.5, 26.0, 25.6, 16.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H23N2O, 247.1805; found, 247.1815;
1,1-dicyclohexyl-3-phenylurea (5s):
White solid (54.0 mg, 90%). 1H NMR (400 MHz, Chloroform-d) δ 7.40–7.36 (m, 2H), 7.31–7.25 (m, 2H), 7.04–6.98 (m, 1H), 6.32 (s, 1H), 3.55–3.45 (m, 2H), 1.89–1.81 (m, 6H), 1.80–1.75 (m, 6H), 1.69 (d, J = 13.0 Hz, 2H), 1.42–1.31 (m, 4H), 1.22–1.11 (m, 2H); 13C NMR (101 MHz, Chloroform-d) δ 154.9, 139.4, 128.8, 122.5, 119.7, 55.5, 31.9, 26.4, 25.6;
N-phenyl-3,4-dihydroisoquinoline-2(1H)-carboxamide (5t):
White solid (45.1 mg, 89%). 1H NMR (400 MHz, Chloroform-d) δ 7.46–7.42 (m, 2H), 7.32–7.27 (m, 2H), 7.25–7.17 (m, 3H), 7.15–7.10 (m, 1H), 7.05 (t, J = 7.4 Hz, 1H), 6.75 (s, 1H), 4.67 (s, 2H), 3.72 (t, J = 5.9 Hz, 2H), 2.91 (t, J = 5.9 Hz, 2H); 13C NMR (101 MHz, Chloroform-d) δ 155.2, 139.2, 135.0, 133.3, 128.9, 128.4, 126.8, 126.5, 126.4, 123.1, 120.3, 45.8, 41.6, 29.0;
1-benzyl-1-ethyl-3-phenylurea (5u):
Colorless liquid (42.8 mg, 84%). 1H NMR (400 MHz, Chloroform-d) δ 7.42–7.38 (m, 2H), 7.37–7.30 (m, 5H), 7.29–7.24 (m, 2H), 7.06–7.00 (m, 1H), 6.40 (s, 1H), 4.59 (s, 2H), 3.52–3.45 (m, 2H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ 155.4, 139.2, 137.7, 129.0, 128.8, 127.7, 127.1, 122.9, 119.9, 50.3, 42.5, 13.5. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H19N2O, 255.1492; found, 255.1500;
1-benzyl-1-isopropyl-3-phenylurea (5v):
White solid (48.8 mg, 91%), mp 108.8–109.9 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.45–7.37 (m, 4H), 7.37–7.32 (m, 1H), 7.25–7.19 (m, 4H), 7.02–6.96 (m, 1H), 6.36 (s, 1H), 4.86–4.73 (m, 1H), 4.47 (s, 2H), 1.24 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, Chloroform-d) δ 155.8, 139.3, 138.2, 129.2, 128.7, 127.8, 126.4, 122.8, 119.8, 46.4, 45.5, 20.8. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H21N2O, 269.1648; found, 269.1657;
1,3-diphenylurea (5w):
White solid (22.5 mg, 52%). 1H NMR (400 MHz, DMSO-d6) δ 8.7 (s, 2H), 7.5 (dd, J = 8.6, 1.2 Hz, 4H), 7.3–7.2 (m, 4H), 7.0–6.9 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 153.0, 140.2, 129.2, 122.3, 118.6.
4. Conclusions
In conclusion, we have disclosed a visible-light-promoted external catalyst-free procedure for the decarboxylation of dioxazolones to synthesize various phosphinimidic amides and ureas. The method has the advantages of no additional transition metals, economic raw materials, mild reaction conditions, and easy operation. It could be reasoned that the ppm Fe in the reaction mixture played a significant role in this transformation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27123648/s1. References [29,32,34,59,60,61,62,63] are cited in the Supplementary Materials. Synthetic procedure of starting materials, procedure and spectral data of products, copies of 1H-NMR, 13C-NMR, 31P-NMR, 19F-NMR spectra.
Author Contributions
Conceptualization, J.P., H.L. and K.S.; data curation, J.P., H.L. and B.Y.; methodology, J.P. and H.L.; supervision, K.S. and B.Y.; validation, B.Y.; writing—original draft, J.P.; writing—review and editing, K.S. and S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Hunan Provincial Natural Science Foundation of China (2021JJ40432), and the National Natural Science Foundation of China (21971224, 22171249).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not available.
References
- He, X.; Qiu, L.-Q.; Wang, W.-J.; Chen, K.-H.; He, L.-N. Photocarboxylation with CO2: An Appealing and Sustainable Strategy for CO2 Fixation. Green Chem. 2020, 22, 7301–7320. [Google Scholar] [CrossRef]
- Lewis, N.S. Introduction: Solar Energy Conversion. Chem. Rev. 2015, 115, 12631–12632. [Google Scholar] [CrossRef] [Green Version]
- Parisien-Collette, S.; Hernandez-Perez, A.C.; Collins, S.K. Photochemical Synthesis of Carbazoles Using an [Fe(phen)3](NTf2)2/O2 Catalyst System: Catalysis toward Sustainability. Org. Lett. 2016, 18, 4994–4997. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green Chemistry and Resource Efficiency: Towards a Green Economy. Green Chem. 2016, 18, 3180–3183. [Google Scholar] [CrossRef]
- Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev. 2016, 45, 2044–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey, S.; Tehshik, Y. Enabling Chemical Synthesis with Visible Light. Acc. Chem. Res. 2016, 49, 2059–2060. [Google Scholar]
- Fu, X.-Y.; Si, Y.-F.; Qiao, L.-P.; Zhao, Y.-F.; Chen, X.-L.; Yu, B. Visible Light-Promoted Recyclable Carbon Nitride-Catalyzed Dioxygenation of Β,Γ-Unsaturated Oximes. Adv. Synth. Catal. 2022, 364, 574–580. [Google Scholar] [CrossRef]
- Li, G.; Yan, Q.; Gong, X.; Dou, X.; Yang, D. Photocatalyst-Free Regioselective C–H Thiocyanation of 4-Anilinocoumarins under Visible Light. ACS Sustainable Chem. Eng. 2019, 7, 14009–14015. [Google Scholar] [CrossRef]
- Chen, L.; Lin, C.; Lan, Y.; Li, Z.; Huang, D.; Yang, W.; Li, Y. Visible Light-Induced Green Synthesis of 2-Amino-4H-Chromenes. Environ. Chem. Lett. 2020, 18, 2157–2163. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, L.; Yue, H.; Li, J.-S.; Luo, Z.; Wei, W. Catalyst-Free Visible-Light-Initiated Oxidative Coupling of Aryldiazo Sulfones with Thiols Leading to Unsymmetrical Sulfoxides in Air. Green Chem. 2019, 21, 1609–1613. [Google Scholar] [CrossRef]
- Sun, W.; Ma, X.; Pang, Y.; Zhao, L.; Zhong, Q.; Liu, C.; Fan, Q. Straightforward Synthesis of Quinazolin-4(3H)-Ones Via Visible Light-Induced Condensation Cyclization. RSC Adv. 2022, 12, 1494–1498. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, L.; Yue, H.; Bao, P.; Liu, W.; Hu, C.; Wang, H. Metal-Free Visible-Light-Induced C–H/C–H Cross-Dehydrogenative-Coupling of Quinoxalin-2(H)-Ones with Simple Ethers. ACS Sustainable Chem. Eng. 2018, 6, 17252–17257. [Google Scholar] [CrossRef]
- Tang, S.; Yuan, L.; Deng, Y.-L.; Li, Z.-Z.; Wang, L.-N.; Huang, G.-X.; Sheng, R.-L. Visible-Light-Induced Perfluoroalkylation/Arylmigration/Desulfonylation Cascades of Conjugated Tosyl Amides. Tetrahedron Lett. 2017, 58, 329–332. [Google Scholar] [CrossRef]
- Tang, S.; Yuan, L.; Li, Z.-Z.; Peng, Z.-Y.; Deng, Y.-L.; Wang, L.-N.; Sheng, R.-L. Visible-Light-Induced Dearomative Spirocyclization of N-Benzylacrylamides toward Perfluorinated Azaspirocyclic Cyclohexadienones. Tetrahedron Lett. 2017, 58, 2127–2130. [Google Scholar] [CrossRef]
- Yuan, L.; Jiang, S.-M.; Li, Z.-Z.; Zhu, Y.; Yu, J.; Li, L.; Sheng, R.-R. Photocatalyzed Cascade Meerwein Addition/Cyclization of N-Benzylacrylamides toward Azaspirocycles. Org. Biomol. Chem. 2018, 16, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-W.; Yu, J.; Zhou, Q.-Y.; Chen, S.-Y.; Xu, Z.-H.; Tang, S. Visible-Light-Induced Atom Transfer Radical Addition and Cyclization of Perfluoroalkyl Halides with 1, N-Enynes. ACS Sustainable Chem. Eng. 2019, 7, 10154–10162. [Google Scholar] [CrossRef]
- Chen, N.; Lei, J.; Wang, Z.; Liu, Y.; Sun, K.; Tang, S. Construction of Fluoro-Containing Heterocycles Mediated by Free Radicals. Chin. J. Org. Chem. 2022, 42, 1061–1084. [Google Scholar] [CrossRef]
- Liu, T.; Liu, J.; He, J.; Hong, Y.; Zhou, H.; Liu, Y.-L.; Tang, S. Recent Advances in Photoinduced Perfluoroalkylation Using Perfluoroalkyl Halides as the Radical Precursors. Synthesis 2022, 54, 1919–1938. [Google Scholar]
- Cheung, K.P.S.; Sarkar, S.; Gevorgyan, V. Visible Light-Induced Transition Metal Catalysis. Chem. Rev. 2022, 122, 1543–1625. [Google Scholar] [CrossRef]
- Sun, P.; Yang, D.; Wei, W.; Jiang, M.; Wang, Z.; Zhang, L.; Wang, H. Visible Light-Induced C–H Sulfenylation Using Sulfinic Acids. Green Chem. 2017, 19, 4785–4791. [Google Scholar] [CrossRef]
- Yang, B.; Lu, Z. Visible-Light-Promoted Metal-Free Aerobic Hydroxyazidation of Alkenes. ACS Catal. 2017, 7, 8362–8365. [Google Scholar] [CrossRef]
- Liu, X.-C.; Chen, X.-L.; Liu, Y.; Sun, K.; Peng, Y.-Y.; Qu, L.-B.; Yu, B. Visible-Light-Induced Metal-Free Synthesis of 2-Phosphorylated Thioflavones in Water. ChemSusChem. 2020, 13, 298–303. [Google Scholar] [CrossRef]
- Gui, Q.-W.; Wang, B.-B.; Zhu, S.; Li, F.-L.; Zhu, M.-X.; Yi, M.; He, W.-M. Four-Component Synthesis of 3-Aminomethylated Imidazoheterocycles in Etoh under Catalyst-Free, Oxidant-Free and Mild Conditions. Green Chem. 2021, 23, 4430–4434. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, J.-Y.; Ning, J.; Jiang, X.; Deng, J.; Deng, Y.; He, W.-M. Electrochemical Multicomponent Synthesis of 4-Selanylpyrazoles under Catalyst- and Chemical-Oxidant-Free Conditions. Green Chem. 2021, 23, 3950–3954. [Google Scholar] [CrossRef]
- Zeng, F.-L.; Xie, K.-C.; Liu, Y.-T.; Wang, H.; Yin, P.-C.; Qu, L.-B.; Yu, B. Visible-Light-Promoted Catalyst-/Additive-Free Synthesis of Aroylated Heterocycles in a Sustainable Solvent. Green Chem. 2022, 24, 1732–1737. [Google Scholar] [CrossRef]
- Chen, Z.; Xuan, J. Photochemical Synthesis of Aroylated Heterocycles under Catalyst and Additive Free Conditions. Chin. J. Org. Chem. 2022, 42, 923–924. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, Q.-Q.; Tan, F.; Lu, L.-Q.; Xiao, W.-J. Visible-Light-Driven Organic Photochemical Reactions in the Absence of External Photocatalysts. Synthesis 2019, 51, 3021–3054. [Google Scholar] [CrossRef]
- Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Iron-Catalyzed Reactions in Organic Synthesis. Chem. Rev. 2004, 104, 6217–6254. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-J.; Yu, X.; Wang, Y.; Yamamoto, Y.; Bao, M. Interweaving Visible-Light and Iron Catalysis for Nitrene Formation and Transformation with Dioxazolones. Angew. Chem. Int. Ed. 2021, 60, 16426–16435. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.; Schomaker, J.M. Nitrene Transfer Catalysts for Enantioselective C–N Bond Formation. Nat. Rev. Chem. 2021, 5, 580–594. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Lai, X.-J.; Huang, K.; Yadav, S.; Qiu, G.; Zhang, L.; Zhou, H. Unravelling Nitrene Chemistry from Acyclic Precursors: Recent Advances and Challenges. Org. Chem. Front. 2021, 8, 1677–1693. [Google Scholar] [CrossRef]
- Chamni, S.; Zhang, J.; Zou, H. Benign Synthesis of Unsymmetrical Arylurea Derivatives Using 3-Substituted Dioxazolones as Isocyanate Surrogates. Green Chem. Lett. Rev. 2020, 13, 246–257. [Google Scholar] [CrossRef]
- Chen, B.; Peng, J.-B.; Ying, J.; Qi, X.; Wu, X.-F. A Palladium-Catalyzed Domino Procedure for the Synthesis of Unsymmetrical Ureas. Adv. Synth. Catal 2018, 360, 2820–2824. [Google Scholar] [CrossRef]
- Mistry, L.; Mapesa, K.; Bousfield, T.W.; Camp, J.E. Synthesis of Ureas in the Bio-Alternative Solvent Cyrene. Green Chem. 2017, 19, 2123–2128. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Z.; Yan, S.; Xu, S.; Wang, M.-A.; Fu, B.; Zhang, Z. Pd/C Catalyzed Carbonylation of Azides in the Presence of Amines. Org. Lett. 2016, 18, 1736–1739. [Google Scholar] [CrossRef]
- Bizet, V.; Buglioni, L.; Bolm, C. Light-Induced Ruthenium-Catalyzed Nitrene Transfer Reactions: A Photochemical Approach Towards N-Acyl Sulfimides and Sulfoximines. Angew. Chem. Int. Ed. 2014, 53, 5639–5642. [Google Scholar] [CrossRef]
- Darses, B.; Rodrigues, R.; Neuville, L.; Mazurais, M.; Dauban, P. Transition Metal-Catalyzed Iodine(III)-Mediated Nitrene Transfer Reactions: Efficient Tools for Challenging Syntheses. Chem. Commun. 2017, 53, 493–508. [Google Scholar] [CrossRef]
- Das, A.; Chen, Y.-S.; Reibenspies, J.H.; Powers, D.C. Characterization of a Reactive Rh2 Nitrenoid by Crystalline Matrix Isolation. J. Am. Chem. Soc. 2019, 141, 16232–16236. [Google Scholar] [CrossRef]
- Zhang, J.; Shan, C.; Zhang, T.; Song, J.; Liu, T.; Lan, Y. Computational Advances Aiding Mechanistic Understanding of Silver-Catalyzed Carbene/Nitrene/Silylene Transfer Reactions. Coord. Chem. Rev. 2019, 382, 69–84. [Google Scholar] [CrossRef]
- Wentrup, C. Carbenes and Nitrenes: Recent Developments in Fundamental Chemistry. Angew. Chem. Int. Ed. 2018, 57, 11508–11521. [Google Scholar] [CrossRef]
- Shin, K.; Ryu, J.; Chang, S. Orthogonal Reactivity of Acyl Azides in C–H Activation: Dichotomy between C–C and C–N Amidations Based on Catalyst Systems. Org. Lett. 2014, 16, 2022–2025. [Google Scholar] [CrossRef]
- van Vliet, K.M.; de Bruin, B. Dioxazolones: Stable Substrates for the Catalytic Transfer of Acyl Nitrenes. ACS Catal. 2020, 10, 4751–4769. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.W.W.; Ton, T.M.U.; Chan, P.W.H. Transition-Metal-Catalyzed Aminations and Aziridinations of C-H and C-C Bonds with Iminoiodinanes. Chem Record 2011, 11, 331–357. [Google Scholar] [CrossRef]
- Guo, Y.; Pei, C.; Empel, C.; Jana, S.; Koenigs, R.M. Koenigs, Photochemical Nitrene Transfer Reactions of Iminoiodinanes with Sulfides. ChemPhotoChem. 2022, 6, e202100293. [Google Scholar] [CrossRef]
- Guimond, N.; Gouliaras, C.; Fagnou, K. Rhodium(III)-Catalyzed Isoquinolone Synthesis: The N−O Bond as a Handle for C−N Bond Formation and Catalyst Turnover. J. Am. Chem. Soc. 2010, 132, 6908–6909. [Google Scholar] [CrossRef]
- Tan, Y.; Hartwig, J.F. Palladium-Catalyzed Amination of Aromatic C−H Bonds with Oxime Esters. J. Am. Chem. Soc. 2010, 132, 3676–3677. [Google Scholar] [CrossRef]
- Dubé, P.; Nathel, N.F.F.; Vetelino, M.; Couturier, M.; Aboussafy, C.L.; Pichette, S.; Hardink, M. Carbonyldiimidazole-Mediated Lossen Rearrangement. Org. Lett. 2009, 11, 5622–5625. [Google Scholar] [CrossRef]
- Tang, J.-J.; Yu, X.; Yamamoto, Y.; Bao, M. Visible-Light-Promoted Iron-Catalyzed N-Arylation of Dioxazolones with Arylboronic Acids. ACS Catal. 2021, 11, 13955–13961. [Google Scholar] [CrossRef]
- van Vliet, K.M.; Polak, L.H.; Siegler, M.A.; van der Vlugt, J.I.; Guerra, C.F.; de Bruin, B. Efficient Copper-Catalyzed Multicomponent Synthesis of N-Acyl Amidines Via Acyl Nitrenes. J. Am. Chem. Soc. 2019, 141, 15240–15249. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jung, H.; Song, F.; Zhu, S.; Bai, Z.; Chen, D.; He, G.; Chang, S.; Chen, G. Nitrene-Mediated Intermolecular N–N Coupling for Efficient Synthesis of Hydrazides. Nat. Chem. 2021, 13, 378–385. [Google Scholar] [CrossRef]
- Lee, S.; Rovis, T. Rh(III)-Catalyzed Three-Component Syn-Carboamination of Alkenes Using Arylboronic Acids and Dioxazolones. ACS Catal. 2021, 11, 8585–8590. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Lonka, M.R.; Zhang, J.; Zou, H. Rhodium(III)-Catalyzed Cascade Reactions of Benzoic Acids with Dioxazolones: Discovery of 2,5-Substituted Benzoxazinones as Aie Molecules. Chem. Commun. 2019, 55, 11203–11206. [Google Scholar] [CrossRef]
- Park, Y.; Park, K.T.; Kim, J.G.; Chang, S. Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C–H Amination with a New Type of Amidating Reagent. J. Am. Chem. Soc. 2015, 137, 4534–4542. [Google Scholar] [CrossRef]
- Fringuelli, F.; Lanari, D.; Pizzo, F.; Vaccaro, L. An E-Factor Minimized Protocol for the Preparation of Methyl Β-Hydroxy Esters. Green Chem. 2010, 12, 1301–1305. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E Factor 25 Years On: The Rise of Green Chemistry and Sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, Y.-W.; He, W.-M. Microwave-Assisted 6π-Electrocyclization in Water. Chin. Chem. Lett. 2020, 31, 2999–3000. [Google Scholar] [CrossRef]
- Li, X.-Y.; Liu, Y.; Chen, X.-L.; Lu, X.-Y.; Liang, X.-X.; Zhu, S.-S.; Wei, C.-W.; Qu, L.-B.; Yu, B. 6π-Electrocyclization in Water: Microwave-Assisted Synthesis of Polyheterocyclic-Fused Quinoline-2-Thiones. Green Chem. 2020, 22, 4445–4449. [Google Scholar] [CrossRef]
- Leadbeater, N.E. When Is Free Really Free? Nat. Chem. 2010, 2, 1007–1009. [Google Scholar] [CrossRef]
- Ding, J.; Jiang, W.; Bai, H.-Y.; Ding, T.-M.; Gao, D.; Bao, X.; Zhang, S.-Y. Experimental and Computational Studies on H2O- Promoted, Rh-Catalyzed Transient-Ligand-Free Ortho-C(Sp2)–H Amidation of Benzaldehydes with Dioxazolones. Chem. Commun. 2018, 54, 8889–8892. [Google Scholar] [CrossRef]
- Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.G.; Gagne, M.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered Ureas as Masked Isocyanates: Facile Carbamoylation of Nucleophiles under Neutral Conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar] [CrossRef]
- Peron, J.-M.R.; Packman, H.; Peveler, W.J.; Bear, J.C. In Situ Formation of Low Molecular Weight Organogelators for Slick Solidification. RSC Adv. 2020, 10, 13369–13373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Aguiar, L.C.S.; de Mattos, M.C.S.; Sanabria, C.M.; Costa, B.B.S.; Viana, G.M. Efficient Direct Halogenation of Unsymmetrical N-Benzyl- and N-Phenylureas with Trihaloisocyanuric Acids. Synthesis 2018, 50, 1359–1367. [Google Scholar] [CrossRef] [Green Version]
- Gan, Z.; Li, G.; Yan, Q.; Deng, W.; Jiang, Y.-Y.; Yang, D. Visible-Light-Promoted Oxidative Desulphurisation: A Strategy for the Preparation of Unsymmetrical Ureas from Isothiocyanates and Amines Using Molecular Oxygen. Green Chem. 2020, 22, 2956–2962. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).