
Pyridazinones and Structurally Related Derivatives with Anti-Inflammatory Activity


 ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Screening of Compounds for Anti-Inflammatory Activity
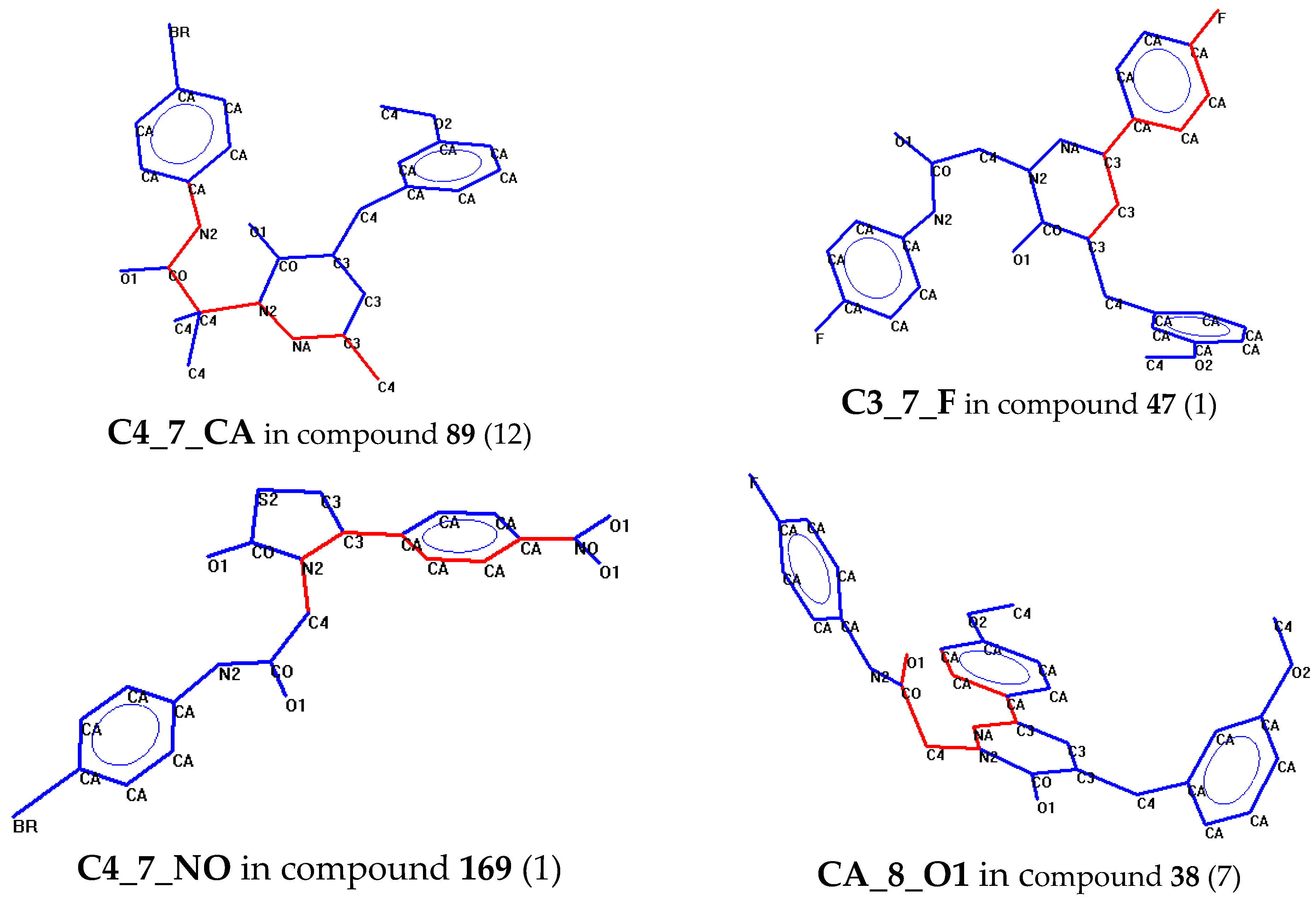
2.2. Classification Tree Analysis
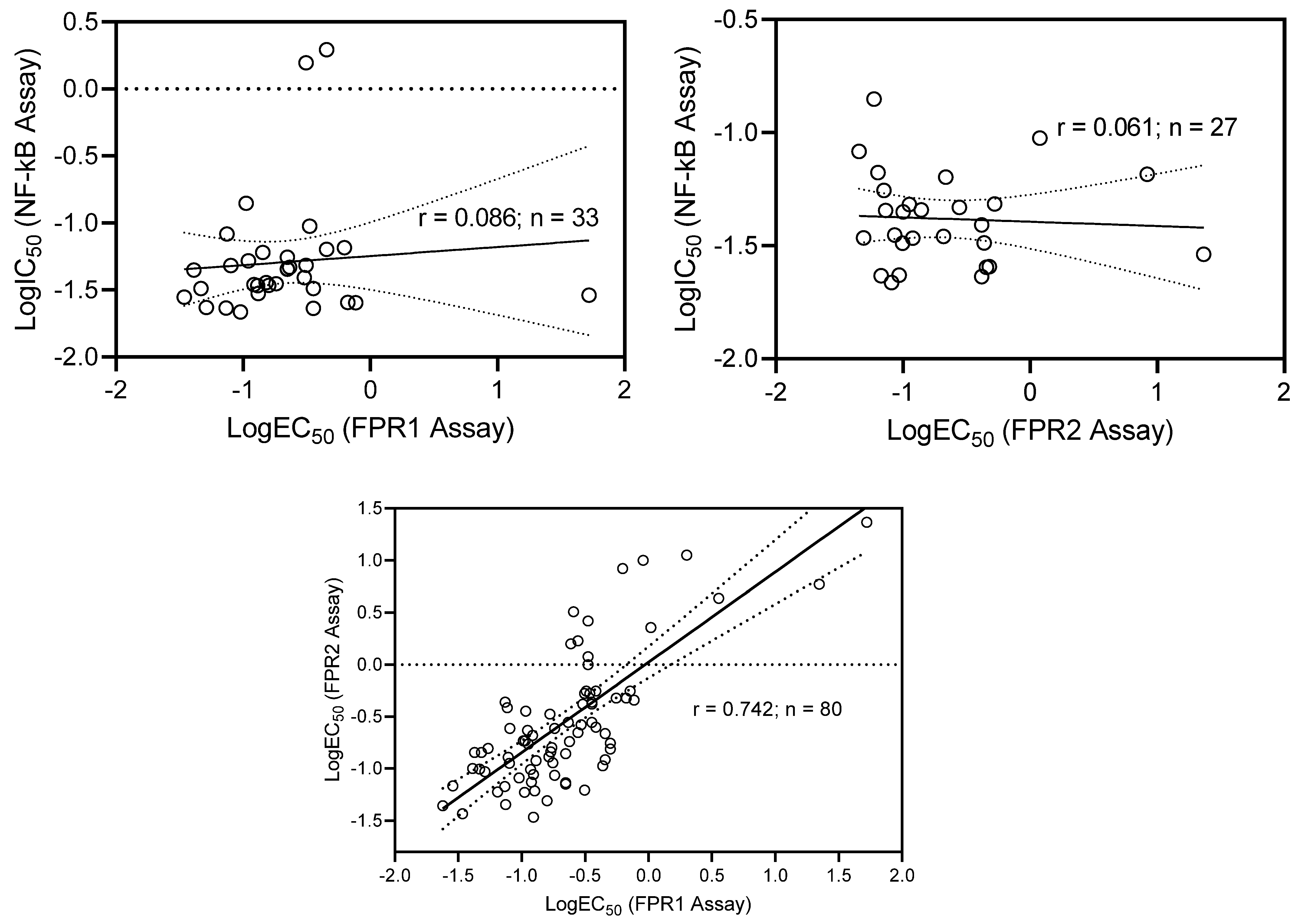
2.3. Comparative Analysis
3. Materials and Methods
3.1. Materials
3.2. Compounds
3.3. Cell Culture
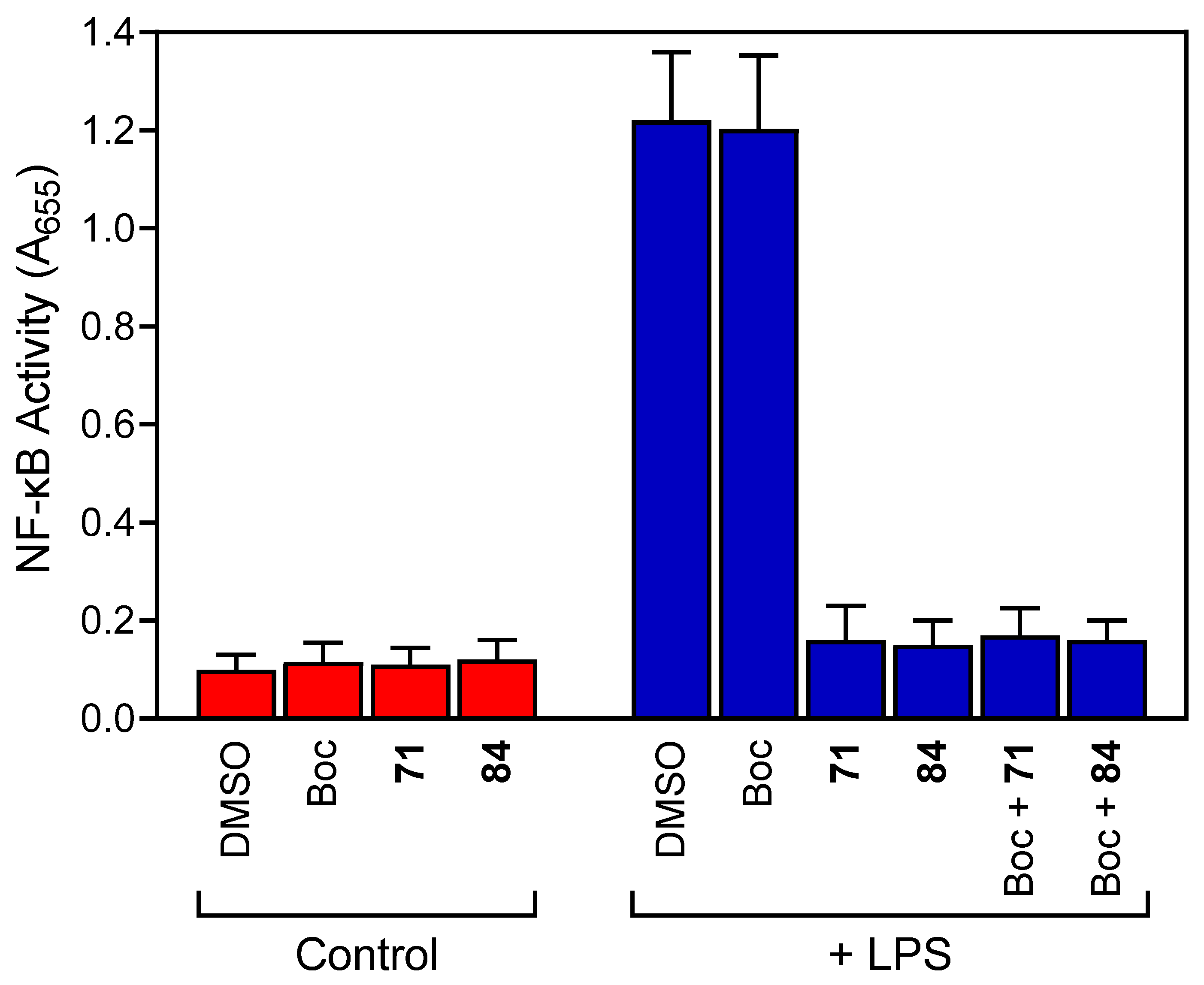
3.4. Analysis of NF-κB Activation
3.5. IL-6 Analysis
3.6. Cytotoxicity Assay
3.7. Ca2+ Mobilization Assay
3.8. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug. Targets. Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Woods, C.R.; Mora, A.L.; Xu, J.; Brigham, K.L. Endotoxin-induced lung injury in mice: Structural, functional, and biochemical responses. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2005, 288, L333–L341. [Google Scholar] [CrossRef] [Green Version]
- Munn, L.L. Cancer and inflammation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Lee, Y.-W.; Kim, P.H.; Lee, W.-H.; Hirani, A.A. Interleukin-4, Oxidative Stress, Vascular Inflammation and Atherosclerosis. Biomol. Ther. 2010, 18, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; González-Billault, C.; Núñez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- Saini, M.; Mehta, D.K.; Das, R.; Saini, G. Recent Advances in Anti-inflammatory Potential of Pyridazinone Derivatives. Mini-Reviews Med. Chem. 2016, 16, 996–1012. [Google Scholar] [CrossRef]
- Pau, A.; Catto, M.; Pinna, G.; Frau, S.; Murineddu, G.; Asproni, B.; Curzu, M.M.; Pisani, L.; Leonetti, F.; Loza, M.I.; et al. Multitarget-Directed Tricyclic Pyridazinones as G Protein-Coupled Receptor Ligands and Cholinesterase Inhibitors. ChemMedChem 2015, 10, 1054–1070. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Kassab, A.E.; El-Malah, A.A.; Hassan, M.S. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2019, 171, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Peregrym, K.; Szczukowski, L.; Wiatrak, B.; Potyrak, K.; Czyznikowska, Z.; Swiatek, P. In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone as Promising Cyclooxygenase Inhibitors. Int. J. Mol. Sci. 2021, 22, 9130. [Google Scholar] [CrossRef] [PubMed]
- Wakulik, K.; Wiatrak, B.; Szczukowski, Ł.; Bodetko, D.; Szandruk-Bender, M.; Dobosz, A.; Świątek, P.; Gąsiorowski, K. Effect of Novel Pyrrolo[3,4-d]pyridazinone Derivatives on Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2020, 21, 2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potyrak, K.; Wiatrak, B.; Krzyżak, E.; Szczukowski, Ł.; Świątek, P.; Szeląg, A. Effect of pyrrolo[3,4-d]pyridazinone derivatives in neuroinflammation induced by preincubation with lipopolysaccharide or coculturing with microglia-like cells. Biomed. Pharmacother. 2021, 141, 111878. [Google Scholar] [CrossRef] [PubMed]
- Allart-Simon, I.; Moniot, A.; Bisi, N.; Ponce-Vargas, M.; Audonnet, S.; Laronze-Cochard, M.; Sapi, J.; Hénon, E.; Velard, F.; Gérard, S. Pyridazinone derivatives as potential anti-inflammatory agents: Synthesis and biological evaluation as PDE4 inhibitors. RSC Med. Chem. 2021, 12, 584–592. [Google Scholar] [CrossRef]
- Barberot, C.; Moniot, A.; Allart-Simon, I.; Malleret, L.; Yegorova, T.; Laronze-Cochard, M.; Bentaher, A.; Médebielle, M.; Bouillon, J.-P.; Hénon, E.; et al. Synthesis and biological evaluation of pyridazinone derivatives as potential anti-inflammatory agents. Eur. J. Med. Chem. 2018, 146, 139–146. [Google Scholar] [CrossRef]
- Elagawany, M.; Ibrahim, M.A.; Ahmed, H.E.A.; El Etrawy, A.A.; Ghiaty, A.; Abdel-Samii, Z.K.; El-Feky, S.A.; Bajorath, J. Design, synthesis, and molecular modelling of pyridazinone and phthalazinone derivatives as protein kinases inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 2007–2013. [Google Scholar] [CrossRef]
- Woods, K.W.; Lai, C.; Miyashiro, J.M.; Tong, Y.; Florjancic, A.S.; Han, E.K.; Soni, N.; Shi, Y.; Lasko, L.; Leverson, J.D.; et al. Aminopyrimidinone Cdc7 Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1940–1943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, L.; Tian, H.; Liu, M.; Jiang, S.; Shen, J.; Wang, K.; Cao, Z. Discovery of pyridazinone analogs as potent transient receptor potential canonical channel 5 inhibitors. Bioorg. Med. Chem. Lett. 2022, 61, 128612. [Google Scholar] [CrossRef] [PubMed]
- Cilibrizzi, A.; Quinn, M.T.; Kirpotina, L.N.; Schepetkin, I.A.; Holderness, J.; Ye, R.D.; Rabiet, M.-J.; Biancalani, C.; Cesari, N.; Graziano, A.; et al. 6-Methyl-2,4-Disubstituted Pyridazin-3(2H)-ones: A Novel Class of Small-Molecule Agonists for Formyl Peptide Receptors. J. Med. Chem. 2009, 52, 5044–5057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, M.P.; Schepetkin, I.A.; Cilibrizzi, A.; Crocetti, L.; Khlebnikov, A.I.; Dahlgren, C.; Graziano, A.; Dal Piaz, V.; Kirpotina, L.N.; Zerbinati, S.; et al. Further studies on 2-arylacetamide pyridazin-3(2H)-ones: Design, synthesis and evaluation of 4,6-disubstituted analogs as formyl peptide receptors (FPRs) agonists. Eur. J. Med. Chem. 2013, 64, 512–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergelli, C.; Schepetkin, I.A.; Ciciani, G.; Cilibrizzi, A.; Crocetti, L.; Giovannoni, M.P.; Guerrini, G.; Iacovone, A.; Kirpotina, L.N.; Khlebnikov, A.I.; et al. 2-Arylacetamido-4-phenylamino-5-substituted pyridazinones as formyl peptide receptors agonists. Bioorganic Med. Chem. 2016, 24, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Vergelli, C.; Schepetkin, I.A.; Ciciani, G.; Cilibrizzi, A.; Crocetti, L.; Giovannoni, M.P.; Guerrini, G.; Iacovone, A.; Kirpotina, L.N.; Ye, R.D.; et al. Synthesis of Five- and Six-MemberedN-Phenylacetamido Substituted Heterocycles as Formyl Peptide Receptor Agonists. Drug Dev. Res. 2017, 78, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Crocetti, L.; Vergelli, C.; Guerrini, G.; Cantini, N.; Kirpotina, L.N.; Schepetkin, I.A.; Quinn, M.T.; Parisio, C.; Mannelli, L.D.C.; Ghelardini, C.; et al. Novel formyl peptide receptor (FPR) agonists with pyridinone and pyrimidindione scaffolds that are potentially useful for the treatment of rheumatoid arthritis. Bioorganic Chem. 2020, 100, 103880. [Google Scholar] [CrossRef]
- Cilibrizzi, A.; Crocetti, L.; Giovannoni, M.P.; Graziano, A.; Vergelli, C.; Bartolucci, G.; Soldani, G.; Quinn, M.T.; Schepetkin, I.A.; Faggi, C. Synthesis, HPLC enantioresolution, and X-ray analysis of a new series of C5-methyl pyridazines as N-formyl peptide eceptor (FPR) agonists. Chirality 2013, 25, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Crocetti, L.; Vergelli, C.; Cilibrizzi, A.; Graziano, A.; Khlebnikov, A.I.; Kirpotina, L.N.; Schepetkin, I.A.; Quinn, M.T.; Giovannoni, M.P. Synthesis and Pharmacological Evaluation of New Pyridazin-Based Thioderivatives as Formyl Peptide Receptor (FPR) Agonists. Drug Dev. Res. 2013, 74, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Vergelli, C.; Khlebnikov, A.I.; Crocetti, L.; Guerrini, G.; Cantini, N.; Kirpotina, L.N.; Schepetkin, I.A.; Cilibrizzi, A.; Quinn, M.T.; Rossi, P.; et al. Synthesis, biological evaluation, molecular modeling, and structural analysis of new pyrazole and pyrazolone derivatives as N-formyl peptide receptors (FPRs) agonists. Chem. Biol. Drug. Des. 2021, 98, 582–603. [Google Scholar] [CrossRef]
- Deora, G.S.; Qin, C.X.; Vecchio, E.A.; Debono, A.J.; Priebbenow, D.L.; Brady, R.M.; Beveridge, J.; Teguh, S.C.; Deo, M.; May, L.T.; et al. Substituted Pyridazin-3(2H)-ones as Highly Potent and Biased Formyl Peptide Receptor Agonists. J. Med. Chem. 2019, 62, 5242–5248. [Google Scholar] [CrossRef]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef] [PubMed]
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Tylek, K.; Trojan, E.; Regulska, M.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Formyl peptide receptor 2, as an important target for ligands triggering the inflammatory response regulation: A link to brain pathology. Pharmacol. Rep. 2021, 73, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Trojan, E.; Bryniarska, N.; Leśkiewicz, M.; Regulska, M.; Chamera, K.; Szuster-Głuszczak, M.; Leopoldo, M.; Lacivita, E.; Basta-Kaim, A. The Contribution of Formyl Peptide Receptor Dysfunction to the Course of Neuroinflammation: A Potential Role in the Brain Pathology. Curr. Neuropharmacol. 2020, 18, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Maciuszek, M.; Cacace, A.; Brennan, E.; Godson, C.; Chapman, T.M. Recent advances in the design and development of formyl peptide receptor 2 (FPR2/ALX) agonists as pro-resolving agents with diverse therapeutic potential. Eur. J. Med. Chem. 2021, 213, 113167. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Lee, M.; Bae, Y. Formyl Peptide Receptors in Cellular Differentiation and Inflammatory Diseases. J. Cell. Biochem. 2017, 118, 1300–1307. [Google Scholar] [CrossRef]
- Marshall, S.A.; Qin, C.X.; Jelinic, M.; O’Sullivan, K.; Deo, M.; Walsh, J.; Li, M.; Parry, L.J.; Ritchie, R.H.; Leo, C.H. The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice. Int. J. Mol. Sci. 2020, 21, 1384. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; May, L.T.; Li, R.; Cao, N.; Rosli, S.; Deo, M.; Alexander, A.E.; Horlock, D.; Bourke, J.; Yang, Y.H.; et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 2017, 8, 14232. [Google Scholar] [CrossRef]
- Crocetti, L.; Vergelli, C.; Guerrini, G.; Giovannoni, M.P.; Kirpotina, L.N.; Khlebnikov, A.I.; Ghelardini, C.; Mannelli, L.D.C.; Lucarini, E.; Schepetkin, I.A.; et al. Pyridinone Derivatives as Interesting Formyl Peptide Receptor (FPR) Agonists for the Treatment of Rheumatoid Arthritis. Molecules 2021, 26, 6583. [Google Scholar] [CrossRef]
- Bürli, R.W.; Xu, H.; Zou, X.; Muller, K.; Golden, J.; Frohn, M.; Adlam, M.; Plant, M.H.; Wong, M.; McElvain, M.; et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorganic Med. Chem. Lett. 2006, 16, 3713–3718. [Google Scholar] [CrossRef]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-Inflammatory Role of the Murine Formyl-Peptide Receptor 2: Ligand-Specific Effects on Leukocyte Responses and Experimental Inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sogawa, Y.; Ohyama, T.; Maeda, H.; Hirahara, K. Inhibition of neutrophil migration in mice by mouse formyl peptide receptors 1 and 2 dual agonist: Indication of cross-desensitization in vivo. Immunology 2011, 132, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Odobasic, D.; Jia, Y.; Kao, W.; Fan, H.; Wei, X.; Gu, R.; Ngo, D.; Kitching, A.R.; Holdsworth, S.R.; Morand, E.; et al. Formyl peptide receptor activation inhibits the expansion of effector T cells and synovial fibroblasts and attenuates joint injury in models of rheumatoid arthritis. Int. Immunopharmacol. 2018, 61, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kao, W.; Gu, R.; Jia, Y.; Wei, X.; Fan, H.; Harris, J.; Zhang, Z.; Quinn, J.; Morand, E.; Yang, Y.H. A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis. J. Cereb. Blood Flow Metab. 2014, 171, 4087–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, R.A.; Ito, B.R.; Lupisella, J.; Carson, N.A.; Hsu, M.-Y.; Fernando, G.; Heroux, M.; Bouvier, M.; Dierks, E.; Kick, E.; et al. Preservation of Post-Infarction Cardiac Structure and Function via Long-Term Oral Formyl Peptide Receptor Agonist Treatment. JACC: Basic Transl. Sci. 2019, 4, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.; Wurtz, N.R.; Arakawa, K.; Carson, N.; Fujii, K.; Fukuchi, K.; Garcia, R.; Hsu, M.Y.; Ishiyama, J.; Ito, B.; et al. Discovery of BMS-986235/LAR-1219: A Potent Formyl Peptide Receptor 2 (FPR2) Selective Agonist for the Prevention of Heart Failure. J. Med. Chem. 2020, 63, 9003–9019. [Google Scholar] [CrossRef]
- Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. Int. J. Mol. Sci. 2020, 21, 379. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Karin, M. Nuclear factor-κB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Hanks, T.S.; Kochetkova, I.; Pascual, D.W.; Jutila, M.A.; Quinn, M.T. Identification and Characterization of a Novel Class of c-Jun N-terminal Kinase Inhibitors. Mol. Pharmacol. 2012, 81, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Chang, Y.-P.; Hsiao, C.-C.; Wu, C.-C.; Wang, Y.-H.; Chao, T.-Y.; Leung, S.-Y.; Fang, W.-F.; Lee, C.-P.; Wang, T.-Y.; et al. Blood M2a monocyte polarization and increased formyl peptide receptor 1 expression are associated with progression from latent tuberculosis infection to active pulmonary tuberculosis disease. Int. J. Infect. Dis. 2020, 101, 210–219. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, M.C.; Chin, C.H.; Lin, I.C.; Hsu, P.Y.; Liou, C.W.; Huang, K.T.; Wang, T.Y.; Lin, Y.Y.; Zheng, Y.X.; et al. Formyl peptide receptor 1 up-regulation and formyl peptide receptor 2/3 down-regulation of blood immune cells along with defective lipoxin A4/resolvin D1 production in obstructive sleep apnea patients. PLoS ONE 2019, 14, e0216607. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.F.; Hachicha, M.; Takano, T.; Petasis, N.; Fokin, V.V.; Serhan, C.N. Lipoxin A4 Stable Analogs Are Potent Mimetics That Stimulate Human Monocytes and THP-1 Cells via a G-protein-linked Lipoxin A4 Receptor. J. Biol. Chem. 1997, 272, 6972–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floresta, G.; Crocetti, L.; Giovannoni, M.P.; Biagini, P.; Cilibrizzi, A. Repurposing strategies on pyridazinone-based series by pharmacophore- and structure-driven screening. J. Enzym. Inhib. Med. Chem. 2020, 35, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Linari, G.; Spanò, R. Substituted anilides of 1-(p-chlorobenzoyl)-5-methoxy-2-methyl-indole-3-acetic acid. Arzneimittelforschung 1973, 23, 89–91. [Google Scholar]
- Cilibrizzi, A.; Schepetkin, I.A.; Bartolucci, G.; Crocetti, L.; Piaz, V.D.; Giovannoni, M.P.; Graziano, A.; Kirpotina, L.N.; Quinn, M.T.; Vergelli, C. Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones as potent N-formyl peptide receptor agonists. Bioorganic Med. Chem. 2012, 20, 3781–3792. [Google Scholar] [CrossRef]
- Cilibrizzi, A.; Floresta, G.; Abbate, V.; Giovannoni, M.P. iVS analysis to evaluate the impact of scaffold diversity in the binding to cellular targets relevant in cancer. J. Enzym. Inhib. Med. Chem. 2019, 34, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Schepetkin, I.A.; Kirpotina, L.N.; Tian, J.; Khlebnikov, A.I.; Ye, R.D.; Quinn, M.T. Identification of Novel Formyl Peptide Receptor-Like 1 Agonists That Induce Macrophage Tumor Necrosis Factor α Production. Mol. Pharmacol. 2008, 74, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Leopoldo, M.; Lucente, E.; Lacivita, E.; De Giorgio, P.; Quinn, M.T. 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: Molecular modeling of chiral recognition by FPR2. Biochem. Pharmacol. 2013, 85, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Khlebnikov, A.I.; Schepetkin, I.A.; Quinn, M.T. Computational structure–activity relationship analysis of small-molecule agonists for human formyl peptide receptors. Eur. J. Med. Chem. 2010, 45, 5406–5419. [Google Scholar] [CrossRef] [Green Version]
- Khlebnikov, A.I.; Schepetkin, I.A.; Quinn, M.T. Structure–activity relationship analysis of N-benzoylpyrazoles for elastase inhibitory activity: A simplified approach using atom pair descriptors. Bioorganic Med. Chem. 2008, 16, 2791–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khlebnikov, A.I.; Schepetkin, I.A.; Kirpotina, L.N.; Quinn, M.T. Computational structure–activity relationship analysis of non-peptide inducers of macrophage tumor necrosis factor-α production. Bioorganic Med. Chem. 2008, 16, 9302–9312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giner, B.; Lafuente, C.; Lapena, D.; Errazquin, D.; Lomba, L. QSAR study for predicting the ecotoxicity of NADES towards Aliivibrio fischeri. Exploring the use of mixing rules. Ecotox. Env. Safe 2020, 191, 110004. [Google Scholar] [CrossRef] [PubMed]
- Matsson, P.; Kihlberg, J. How big is too big for cell permeability? J. Med. Chem. 2017, 60, 1662–1664. [Google Scholar] [CrossRef] [Green Version]
- Chung, N.C.; Miasojedow, B.; Startek, M.; Gambin, A. Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinform. 2019, 20, 644. [Google Scholar] [CrossRef]
- Stama, M.L.; Lacivita, E.; Kirpotina, L.N.; Niso, M.; Perrone, R.; Schepetkin, I.A.; Quinn, M.T.; Leopoldo, M. Functional N -Formyl Peptide Receptor 2 (FPR2) Antagonists Based on the Ureidopropanamide Scaffold Have Potential To Protect Against Inflammation-Associated Oxidative Stress. ChemMedChem 2017, 12, 1839–1847. [Google Scholar] [CrossRef]
- Capelli, A.M.; Castelletti, L.; Salvagno, C.; Oliosi, B.; Di Lenarda, E.; Virginio, C.; Lightfoot, A.; Kew, J.N.; Teague, S. Identification of novel α7 nAChR positive allosteric modulators with the use of pharmacophore in silico screening methods. Bioorganic Med. Chem. Lett. 2010, 20, 4561–4565. [Google Scholar] [CrossRef]
- Papke, R.L.; Bagdas, D.; Kulkarni, A.R.; Gould, T.; AlSharari, S.D.; Thakur, G.A.; Damaj, M.I. The analgesic-like properties of the α7 nAChR silent agonist NS6740 is associated with non-conducting conformations of the receptor. Neuropharmacology 2015, 91, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.K.; Wang, J.; Papke, R.L. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: Advantages and limitations. Biochem. Pharmacol. 2011, 82, 915–930. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Chu, Z.; Shan, H.; Zhangsun, D.; Zhu, X.; Luo, S. Inflammation regulation via an agonist and antagonists of α7 nicotinic acetylcholine receptors in RAW264.7 macrophages. Mar. Drugs 2022, 20, 200. [Google Scholar] [CrossRef]
- Richter, K.; Papke, R.L.; Stokes, C.; Roy, D.C.; Espinosa, E.S.; Wolf, P.M.K.; Hecker, A.; Liese, J.; Singh, V.K.; Padberg, W.; et al. Comparison of the anti-inflammatory properties of two nicotinic acetylcholine receptor ligands, phosphocholine and pCF3-diEPP. Front. Cell. Neurosci. 2022, 16, 779081. [Google Scholar] [CrossRef] [PubMed]
- van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. The cholinergic anti-inflammatory pathway: Towards innovative treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Khattab, S.N.; Bekhit, A.A.; El-Faham, A.; El Massry, A.M.; Amer, A. Synthesis of Some Pyridazinylacetic Acid Derivatives as a Novel Class of Monoamine Oxidase-A Inhibitors. Chem. Pharm. Bull. 2008, 56, 1717–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, Z.; Alagöz, M.A.; Uslu, H.; Karakurt, A.; Erikci, A.; Ucar, G.; Uysal, M. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol. Rep. 2020, 72, 692–704. [Google Scholar] [CrossRef]
- Ostadkarampour, M.; Putnins, E.E. Monoamine Oxidase Inhibitors: A Review of Their Anti-Inflammatory Therapeutic Potential and Mechanisms of Action. Front. Pharmacol. 2021, 12, 676239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Wang, X.; Yu, J.; Ma, F.; Li, Z.; Zhou, Y.; Zeng, S.; Ma, X.; Li, Y.-R.; Neal, A.; et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat. Commun. 2021, 12, 3530. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees; Routledge: New York, NY, USA, 1984. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Preti, D.; Tabrizi, M.A.; Fruttarolo, F.; Saponaro, G.; Baraldi, S.; Romagnoli, R.; Moorman, A.R.; Gessi, S.; Varani, K.; et al. N(6)-[(hetero)aryl/(cyclo)alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5′-N-ethylca rboxamido-adenosines: The first example of adenosine-related structures with potent agonist activity at the human A(2B) adenosine receptor. Bioorg. Med. Chem. 2007, 15, 2514–2527. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
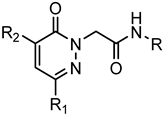 Series A1: Pyridazinones | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R | R1 | R2 | FPR1 | FPR2 | Ref. | |
| EC50 (µM) | |||||||
| 1 | p-Br-Ph | CH3 | m-(OCH3)-Bn | 3.4 | 3.8 | [22] | |
| 2 | m-Br-Ph | CH3 | m-(OCH3)-Bn | N.A. | N.A.a | [22] | |
| 3 | o-Br-Ph | CH3 | m-(OCH3)-Bn | N.A. | N.A. | [22] | |
| 4 | p-Cl-Ph | CH3 | m-(OCH3)-Bn | 2.6 | 4.0 | [22] | |
| 5 | p- NO2-Ph | CH3 | m-(OCH3)-Bn | 10.5 | 12.3 | [22] | |
| 6 | p,m-(OCH3)-Ph | CH3 | m-(OCH3)-Bn | 15.5 | 16.8 | [22] | |
| 7 | p-CF3-Ph | CH3 | m-(OCH3)-Bn | 5.7 | 8.8 | [22] | |
| 8 | p-Br-Ph | Ph | m-(OCH3)-Bn | 9.0 | 4.3 | [23] | |
| 9 | p-Br-Ph | CH3 | CH2-3-thienyl | 4.5 | 14.1 | [23] | |
| 10 | p-Br-Ph | iPr | m-(OCH3)-Bn | 4.5 | 7.2 | [23] | |
| 11 | p-Br-Ph | CH3 | CH2-2-thienyl | 8.1 | 11.4 | [23] | |
| 12 | p-Br-Ph | H | m-(OCH3)-Bn | 6.1 | 7.7 | [23] | |
| 13 | p-Br-Ph | CH3 | CH2-1-naphtyl | 13.8 | N.A. | [23] | |
| 14 | p-(tBu)-Ph | CH3 | m-(OCH3)-Bn | N.A. | N.A. | [22] | |
| 15 | p-F-Ph | CH3 | m-(OCH3)-Bn | 7.6 | N.A. | [22] | |
| 16 | Ph | CH3 | m-(OCH3)-Bn | N.A. | N.A. | [22] | |
| 17 | p-CN-Ph | CH3 | m-(OCH3)-Bn | N.A. | N.A. | [22] | |
| 18 | p-Br-Ph | C6H11 | m-(OCH3)-Bn | 10.8 | N.A. | [23] | |
| 19 | p-Br-Ph | CH3 | p-(OCH3)-Ph | 11.2 | N.A. | [23] | |
| 20 | p-I-Ph | Et | m-(OCH3)-Bn | 4.2 | 5.5 | [23] | |
| 21 | p-I-Ph | CH3 | p-(SCH3)-Bn | 2.3 | 9.4 | [28] | |
| 22 | p-I-Ph | CH3 | m,m-(OCH3)-Bn | 7.6 | N.A. | [23] | |
| 23 | p-I-Ph | CH3 | m-Cl-Bn | 9.5 | 16.9 | [23] | |
| 24 | p-(SCH3)-Ph | CH3 | m-(OCH3)-Bn | 2.2 | 8.2 | [28] | |
| 25 | p-I-Ph | CH3 | H | N.A. | N.A. | [23] | |
| 26 | p-Br-Ph | CH3 | NH2 | 8.1 | 29.4 | [23] | |
| 27 | p-Br-Ph | CH3 | NHCO-p-Br-Ph | N.A. | N.A. | [23] | |
| 28 | p-F-Ph | C6H11 | H | N.A. | N.A. | [53] | |
| 29 | p-F-Ph | CH3 | Bn | N.A. | N.A. | [23] | |
| 30 | p-Br-Ph | CH3 | Bn | 5.5 | 11.6 | [23] | |
| 31 | 5-benzo[d][1,3]dioxole | C6H11 | H | N.A. | N.A. | [53] | |
| 32 | 5-benzo[d][1,3]dioxole | CH3 | Bn | 6.9 | N.A. | [23] | |
| 33 | 5-benzo[d][1,3]dioxole | Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 34 | p-F-Ph | Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 35 | 5-benzo[d][1,3]dioxole | 2-thienyl | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 36 | p-F-Ph | 2-thienyl | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 37 | p-Br-Ph | 2-thienyl | p-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 38 | p-F-Ph | p-(OCH3)-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 39 | 5-benzo[d][1,3]dioxole | p-(OCH3)-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 40 | p-Br-Ph | p-(OCH3)-Ph | p-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 41 | p-Br-Ph | p-Cl-Ph | p-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 42 | p-F-Ph | p-Cl-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 43 | 5-benzo[d][1,3]dioxole | p-Cl-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 44 | p-Br-Ph | p-CH3-Ph | p-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 45 | 5-benzo[d][1,3]dioxole | p-CH3-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 46 | p-F-Ph | p-CH3-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 47 | p-F-Ph | p-F-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 48 | 5-benzo[d][1,3]dioxole | p-F-Ph | m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 49 | p-Br-Ph | p-F-Ph | p-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 50 | p-Br-Ph | CH3 | N(p-(OCH3)-Ph)2 | N.A. | N.A. | [53] | |
| 51 | p-Br-Ph | CH3 | NH-p-(OCH3)-Ph | 12.8 | 7.8 | [23] | |
| 52 | p-Br-Ph | CH3 | NHCO-m-(OCH3)-Ph | 9.3 | 2.8 | [23] | |
| 53 | p-Br-Ph | CH3 | CO-m-(OCH3)-Ph | 3.0 | 1.0 | [23] | |
| 54 | p-Br-Ph | Bn | p-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 55 | p-Br-Ph | CH3 | m-Br-Bn | N.A. | N.A. | [23] | |
| 56 | p-Br-Ph | CH3 | m,m-(OCH3)-Bn | N.A. | N.A. | [23] | |
| 57 | p-F-Ph | Ph | CH3 | N.A. | N.A. | [27] | |
| 58 | p-Br-Ph | CH3 | p-(SCH3)-Bn | N.A. | N.A. | [28] | |
| 59 | p-Br-Ph | CH3 | CH2-3-furyl | 5.8 | 6.3 | [23] | |
| 60 | p-Br-Ph | CH3 | CH2-3-Pyridyl | 9.3 | 2.8 | [23] | |
| 61 | p-Br-Ph | CH3 | p-Bn-CONH-(p-Br-Ph) | N.A. | N.A. | Suppl.a | |
| 62 | p-Br-Ph | CH3 | p-(CONH2)-Bn | 29.3 | 27.2 | [23] | |
| 63 | p-Br-Ph | CH3 | p-CN-Bn | N.A. | N.A. | [23] | |
| 64 | p-Br-Ph | CH3 | m-F-Bn | 6.6 | N.A. | [23] | |
| 65 | p-Br-Ph | CH3 | m-Cl-Bn | 10.5 | N.A. | [23] | |
| 66 | p-Br-Ph | CH3 | p-CF3-Bn | N.A. | N.A. | [23] | |
| 67 | p-Br-Ph | Ph | CH3 | 21.5 | 10.1 | [27] | |
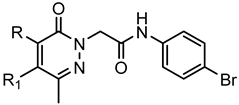 Series A2:Pyridazinones | |||||||
| Compd. | R | R1 | FPR1 | FPR2 | Ref. | ||
| EC50 (µM) | |||||||
| 68 | H | CH3 | 5.7 | 0.51 | [25] | ||
| 69 | m-(OCH3)-Bn | CH3 | 0.019 | 0.043 | [25] | ||
| 70 | H | Ph | N.A. | 0.15 | [25] | ||
| 71 | m-(OCH3)-Bn | Ph | 2.2 | N.A. | [25] | ||
| 72 | NH2 | CO(CH)2-N(CH3)2 | 5.1 | 5.7 | [24] | ||
| 73 | NH-m-(OCH3)-Ph | Ac | 0.045 | 0.17 | [24] | ||
| 74 | NH2 | m-Pirazolyl | 8.4 | 13.5 | [24] | ||
| 75 | NH2 | 1-CH3-3-Pyrazolyl | 2.9 | 1.9 | [24] | ||
| 76 | NH-m-(OCH3)-Ph | 1-CH3-3-Pyrazolyl | 3.6 | 0.59 | [24] | ||
| 77 | NH-p-(OCH3)-Ph | 1-CH3-3-Pyrazolyl | 4.0 | 0.035 | [24] | ||
| 78 | m-(OCH3)-Bn | Et | 3.2 | 1.9 | [25] | ||
| 79 | m-(OCH3)-Bn | nPr | 2.2 | 4.6 | [25] | ||
| 80 | m-(OCH3)-Bn | nBu | N.A. | 15.7 | [25] | ||
| 81 | NH-m-(OCH3)-Ph | nBu | 3.6 | 4.5 | [24] | ||
| 82 | NH-m-(OCH3)-Ph | H | 0.24 | 9.6 | [24] | ||
| 83 | NH-m-(OCH3)-Ph | nPr | N.A. | N.A. | [24] | ||
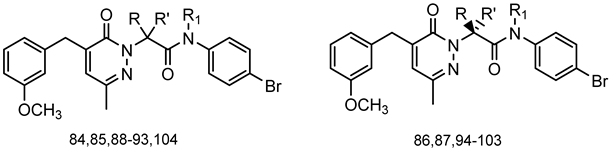 Series A3: Pyridazinones | |||||||
| Compd. | R | R’ | R1 | FPR1 | FPR2 | Ref. | |
| EC50 (µM) | |||||||
| 84 | CH3 | H | H | 3.2 | N.A. | [22] | |
| 85 | H | H | CH3 | N.A. | N.A. | [22] | |
| 86 | H | CH3 | H | 8.5 | 10.2 | [55] | |
| 87 | CH3 | H | H | 3.2 | 16.1 | [55] | |
| 88 | Et | H | H | 1.3 | 2.2 | [55] | |
| 89 | CH3 | CH3 | H | 6.3 | 20.4 | [55] | |
| 90 | Et | CH3 | H | 1.5 | 2.1 | [55] | |
| 91 | nPr | H | H | 2.8 | 3.6 | [55] | |
| 92 | iPr | H | H | 2.0 | 6.5 | [55] | |
| 93 | nBu | H | H | 1.1 | 0.1 | [55] | |
| 94 | H | Et | H | 2.8 | 2.3 | [55] | |
| 95 | Et | H | H | 13.4 | 22.2 | [55] | |
| 96 | H | iPr | H | 9.4 | 5.4 | [55] | |
| 97 | iPr | H | H | N.A. | N.A. | [55] | |
| 98 | H | nPr | H | 3.0 | 0.84 | [55] | |
| 99 | nPr | H | H | N.A. | N.A. | [55] | |
| 100 | H | nBu | H | 0.5 | 0.089 | [55] | |
| 101 | nBu | H | H | 20.8 | 7.0 | [55] | |
| 102 | CH3 | Et | H | 4.5 | 13.7 | [55] | |
| 103 | Et | CH3 | H | 7.0 | N.A. | [55] | |
| 104 | Ph | H | H | 3.1 | 1.8 | [55] | |
 Series A4: Pyridazinones | |||||||
| Compd. | R | R1 | R2 | R3 | FPR1 | FPR2 | Ref. |
| EC50 (µM) | |||||||
| 105 | H | p-(OCH3)-Bn | Cl | p-(OBu)-Ph | N.A. | N.A. | [53] |
| 106 | CH3 | Ph | NH-p-(OBu)-Ph | H | N.A. | N.A. | [22] |
| 107 | H | m-(OCH3)-Bn | Cl | p-(OBu)-Ph | N.A. | N.A. | [53] |
| 108 | H | m-(OCH3)-Bn | p-(OBu)-Ph | OCH3 | N.A. | N.A. | [53] |
| 109 | H | p-(OCH3)-Bn | p-(OBu)-Ph | OCH3 | N.A. | N.A. | [53] |
| 110 | CH3 | CH2CO-N-(CH3)-Pip | m-(OCH3)-Bn | H | N.A. | N.A. | [22] |
| 111 | CH3 | (CH2)2CONH-p-Br-Ph | m-(OCH3)-Bn | H | 9.7 | 5.4 | [22] |
| 112 | CH3 | CH2COO-p-Br-Ph | m-(OCH3)-Bn | H | N.A. | N.A. | [22] |
| 113 | CH3 | (CH2)2NHCONH-p-Br-Ph | m-(OCH3)-Bn | H | N.A. | N.A. | [22] |
| 114 | CH3 | (CH2)2NHCO-p-Br-Ph | m-(OCH3)-Bn | H | N.A. | N.A. | [22] |
| 115 | CH3 | m-(OCH3)-Bn | NHCONH-p-Br-Ph | H | N.A. | N.A. | [22] |
| 116 | CH3 | CH2NHCO-p-Br-Ph | m-(OCH3)-Bn | H | N.A. | N.A. | [22] |
| 117 | CH3 | m-(OCH3)-Bn | NHCO-p-Br-Ph | H | N.A. | N.A. | [22] |
| 118 | CH3 | CH2NHCONH-p- Br-Ph | m-(OCH3)-Bn | H | N.A. | N.A. | [22] |
| 119 | CH3 | CH2-CS-NH-p- Br-Ph | m-(OCH3)-Bn | H | N.A. | N.A. | [28] |
| 120 | CH3 | CH2CONH-p-Br-Ph | NH-p-(OCH3)-Ph | Ac | 13.5 | 1.7 | [23] |
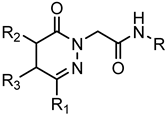 Series B: 4,5-Dihydro-pyridazinones | |||||||
| Compd. | R | R1 | R2 | R3 | FPR1 | FPR2 | Ref. |
| EC50 (µM) | |||||||
| 121 | p-I-Ph | CH3 | H | H | N.A. | N.A. | [23] |
| 122 | p-F-Ph | Ph | CH3 | H | N.A. | N.A. | [27] |
| 123 | 5-benzo[d][1,3]dioxole | Ph | CH3 | H | N.A. | N.A. | [27] |
| 124 | p-Br-Ph | Ph | CH3 | H | 19.5 | 10.7 | [27] |
| 125 | p-Br-Ph | Ph | CH3 | H | 24.4 | 10.0 | [27] |
| 126 | p-F-Ph | Ph | CH3 | H | N.A. | N.A. | [27] |
| 127 | p-F-Ph | Ph | CH3 | H | N.A. | N.A. | [27] |
| 128 | 5-benzo[d][1,3]dioxole | Ph | CH3 | H | N.A. | N.A. | [27] |
| 129 | 5-benzo[d][1,3]dioxole | Ph | CH3 | H | N.A. | N.A. | [27] |
| 130 | p-Br-Ph | Ph | CH3 | H | 23.5 | 7.0 | [27] |
| 131 | p-Br-Ph | CH3 | H | CH3 | 13.0 | 2.6 | [25] |
| 132 | p-Br-Ph | CH3 | H | Ph | 4.1 | 0.63 | [25] |
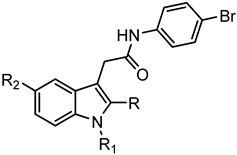 C: Indoles | |||||||
| Compd. | R | R1 | R2 | FPR1 | FPR2 | Ref. | |
| EC50 (µM) | |||||||
| 133 | CH3 | CO-p-Cl-Ph | OCH3 | N.A. | N.A. | [54] | |
| 134 | H | m-(OCH3)-Bn | H | N.A. | N.A. | [56] | |
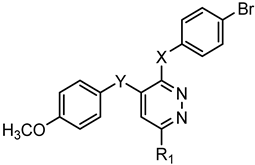 Series D: Pyridazines | |||||||
| Compd. | X | Y | R1 | FPR1 | FPR2 | Ref. | |
| EC50 (µM) | |||||||
| 135 | NHCONH | O | Ph | N.A. | N.A. | [28] | |
| 136 | NHCO | O | Ph | N.A. | N.A. | [28] | |
| 137 | NHCH2CONH | O | Ph | N.A. | N.A. | [28] | |
| 138 | SCH2CONH | CH2 | CH3 | N.A. | N.A. | [28] | |
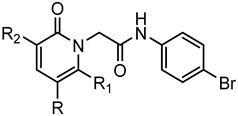 Series E: 2-Pyridinones | |||||||
| Compd. | R | R1 | R2 | FPR1 | FPR2 | Ref. | |
| EC50 (µM) | |||||||
| 139 | 4-Pyridyl | H | NH2 | N.A. | N.A. | Suppl.a | |
| 140 | m,p-(OCH3)-Ph | CH3 | CN | 33.2 | 0.60 | [26] | |
| 141 | m-(OCH3)-Ph | CH3 | CN | 1.6 | 0.12 | [26] | |
| 142 | COOEt | CH3 | CN | 3.0 | 0.38 | [26] | |
| 143 | p-(OCH3)-Ph | CH3 | CN | 1.6 | 0.12 | [26] | |
| 144 | COOEt | CH3 | CONH-m-(OCH3)-Ph | 0.4 | 28.9 | [26] | |
| 145 | COOEt | CH3 | CONH-p-(OCH3)-Ph | 7.9 | 16.4 | [26] | |
| 146 | 4-Pyridyl | CH3 | CONH-m-(OCH3)-Ph | 1.4 | 1.8 | [26] | |
| 147 | CONH-m-(OCH3)-Ph | CH3 | CN | 3.9 | 0.31 | [26] | |
| 148 | CONH-p-(OCH3)-Ph | CH3 | CN | 0.96 | 0.44 | [26] | |
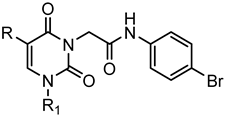 Series F: 2,6-Pyrimidinediones | |||||||
| Compd. | R | R1 | FPR1 | FPR2 | Ref. | ||
| EC50 (µM) | |||||||
| 149 | H | p-CH3-Ph | 13.6 | 14.9 | [26] | ||
| 150 | H | m-(OCH3)-Ph | 4.3 | 3.6 | [26] | ||
| 151 | Ph | p-CH3-Ph | N.A. | N.A. | [26] | ||
| 152 | H | nPr | 5.5 | 4.1 | [26] | ||
| 153 | H | Ph | 12.5 | 8.9 | [26] | ||
| 154 | m-Ph(OCH3) | nPr | 3.3 | 2.4 | [26] | ||
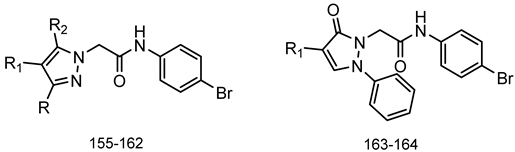 Series G: Pyrazoles and Pyrazolones | |||||||
| Compd. | R | R1 | R2 | FPR1 | FPR2 | Ref. | |
| EC50 (µM) | |||||||
| 155 | H | CN | NH-m-(OCH3)-Ph | N.A. | N.A. | [29] | |
| 156 | H | CN | NH-p-(OCH3)-Ph | N.A. | N.A. | [29] | |
| 157 | Ph | CH3 | NH2 | N.A. | N.A. | [29] | |
| 158 | Ph | CH3 | NH-m-(OCH3)-Ph | N.A. | N.A. | [29] | |
| 159 | Ph | CH3 | NH-p-(OCH3)-Ph | N.A. | N.A. | [29] | |
| 160 | m-(OCH3)-Ph | CN | CH3 | 18.4 | 6.1 | [29] | |
| 161 | p-(OCH3)-Ph | CN | CH3 | N.A. | N.A. | [29] | |
| 162 | H | H | NH-m-(OCH3)-Ph | 13.2 | 23.4 | [29] | |
| 163 | - | H | - | N.A. | 23.1 | [29] | |
| 164 | - | NH-m-(OCH3)-Ph | - | N.A. | N.A. | [29] | |
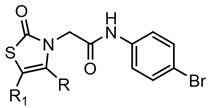 Series H: Thiazol-2-ones | |||||||
| Compd. | R | R1 | FPR1 | FPR2 | Ref. | ||
| EC50 (µM) | |||||||
| 165 | Ph | H | 1.8 | 2.1 | [25] | ||
| 166 | m-(OCH3)-Ph | H | 0.28 | 0.23 | [25] | ||
| 167 | p-(OCH3)-Ph | H | 2.6 | 1.8 | [25] | ||
| 168 | p-Cl-Ph | H | 2.8 | 2.4 | [25] | ||
| 169 | p-NO2-Ph | H | 9.1 | N.A. | [25] | ||
| 170 | m-Cl-Ph | H | 6.0 | 3.0 | [25] | ||
| 171 | p-NH2-Ph | H | 34.9 | 14.7 | [25] | ||
| 172 | CH3 | Ac | 8.9 | 5.8 | [25] | ||
| 173 | CH3 | H | 12.4 | 4.1 | [25] | ||
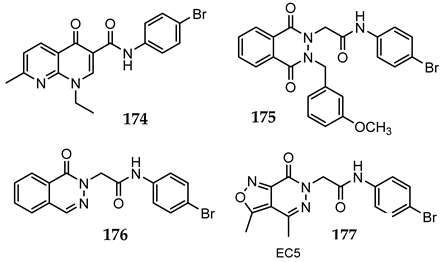 Series I: Bicycliccompounds | |||||||
| Compd. | FPR1 | FPR2 | Ref. | ||||
| EC50 (µM) | |||||||
| 174 | N.A. | N.A. | [56] | ||||
| 175 | N.A. | N.A. | [56] | ||||
| 176 | N.A. | N.A. | [56] | ||||
| 177 | 18.4 | 6.4 | [24] | ||||
| Compd. | Inhibition of NF-κB Activity | Cytotoxicity | Ca2+ Mobilization | |
|---|---|---|---|---|
| FPR1 a | FPR2 a | |||
| IC50 (µM) in THP1-Blue Cells | EC50 (µM) in HL-60 Cells | |||
| 2 | 44.5 ± 2.5 | N.T. | N.A. | N.A. |
| 5 | 46.1 ± 1.0 | N.T. | 10.5 | 12.3 |
| 9 | 18.0 ± 0.7 | N.T. | 4.5 | 14.1 |
| 10 | 22.0 ± 1.7 | N.T. | 4.5 | 7.2 |
| 15 | 33.6 ± 2.1 | N.T. | 7.6 | N.A. |
| 23 | 7.1 ± 2.0 | N.T. | 9.5 | 16.9 |
| 30 | 28.4 ± 5.7 | N.T. | 5.5 | 11.6 |
| 38 | 19.6 ± 4.3 | N.T. | N.A. | N.A. |
| 42 | 22.5 ± 2.3 | N.T. | N.A. | N.A. |
| 46 | 8.2 ± 1.2 | N.T. | N.A. | N.A. |
| 47 | 3.4 ± 0.2 | N.T. | N.A. | N.A. |
| 49 | 34.5 ± 2.1 | N.T. | N.A. | N.A. |
| 50 | 32.8 ± 0.7 | N.T. | N.A. | N.A. |
| 64 | 27.9 ± 2.1 | N.T. | 6.6 | N.A. |
| 66 | 31.5 ± 2.2 | 26.4 ± 4.7 | N.A. | N.A. |
| 67 | 30.9 ± 2.3 | N.T. | 21.5 | 10.1 |
| 69 | 34.5 ± 2.6 | N.T. | 0.019 | 0.043 |
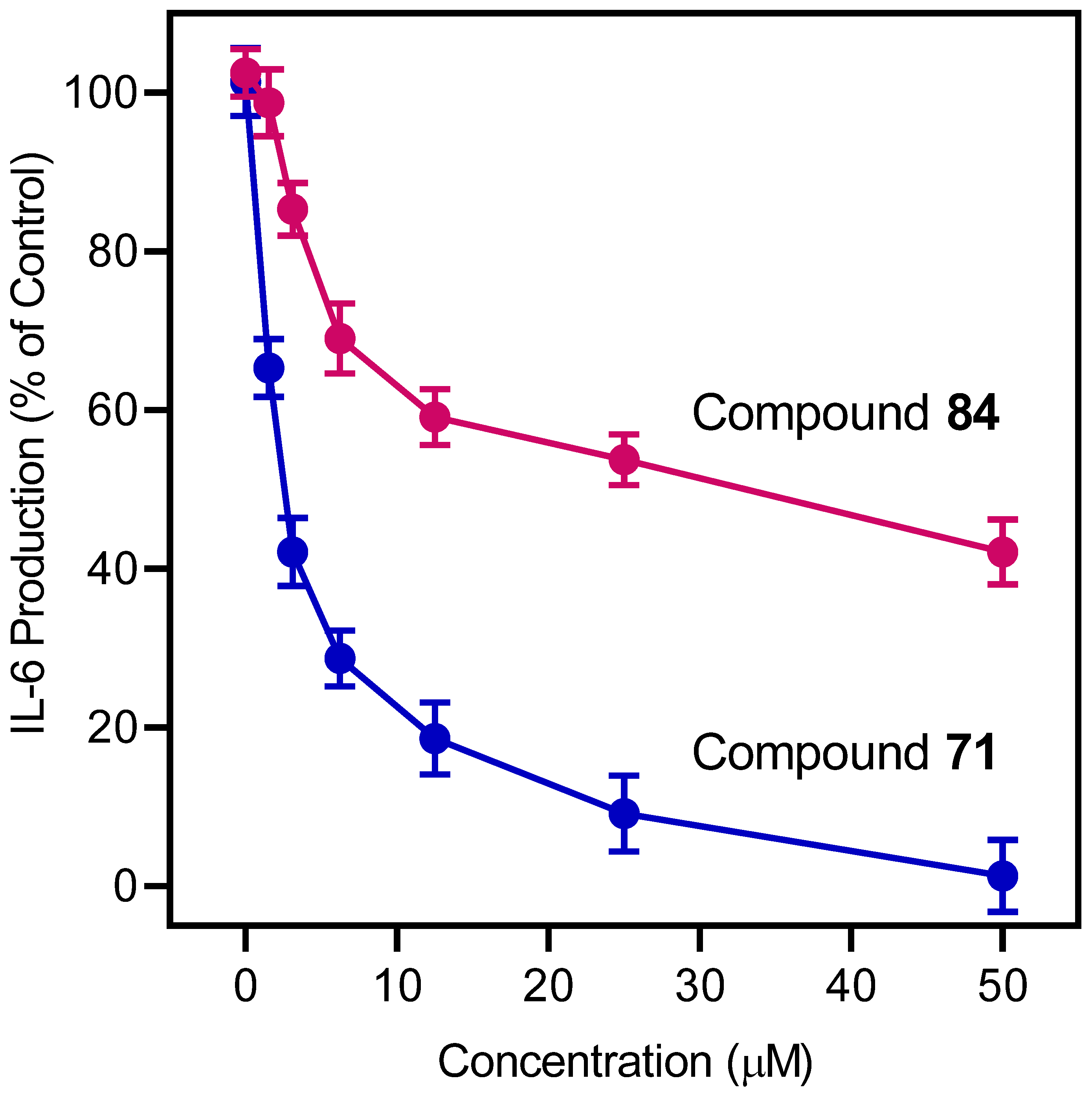
| 71 | 0.5 ± 0.1 | N.T. | 2.2 | N.A. |
| 78 | 20.7 ± 0.7 | N.T. | 3.2 | 1.9 |
| 79 | 15.7 ± 1.4 | N.T. | 2.2 | 4.6 |
| 80 | 15.0 ± 1.5 | N.T. | N.A. | 15.7 |
| 83 | 14.6 ± 1.4 | N.T. | N.A. | N.A. |
| 84 | 0.6 ± 0.1 | N.T. | 3.2 | N.A. |
| 88 | 39.4 ± 4.2 | N.T. | 1.3 | 2.2 |
| 89 | 29.2 ± 1.0 | N.T. | 6.3 | 20.4 |
| 90 | 39.2 ± 6.7 | N.T. | 1.5 | 2.1 |
| 94 | 30.8 ± 6.7 | N.T. | 2.8 | 2.3 |
| 95 | 12.1 ± 2.4 | N.T. | 13.4 | 22.2 |
| 98 | 10.6 ± 2.3 | N.T. | 3.0 | 0.84 |
| 102 | 22.1 ± 1.6 | N.T. | 4.5 | 13.7 |
| 103 | 16.6 ± 0.6 | N.T. | 7.0 | N.A. |
| 109 | 31.3 ± 1.6 | N.T. | N.A. | N.A. |
| 113 | 37.0 ± 0.4 | N.T. | N.A. | N.A. |
| 122 | 46.3 ± 2.4 | N.T. | N.A. | N.A. |
| 124 | 42.7 ± 0.3 | N.T. | 19.5 | 10.7 |
| 125 | 22.4 ± 0.7 | N.T. | 24.4 | 10.0 |
| 141 | 15.3 ± 4.9 | N.T. | 1.6 | 0.12 |
| 149 | 43.0 ± 3.9 | N.T. | 13.6 | 14.9 |
| 150 | 21.4 ± 3.5 | N.T. | 4.3 | 3.6 |
| 153 | 20.8 ± 0.5 | N.T. | 12.5 | 8.9 |
| 154 | 25.6 ± 2.3 | N.T. | 3.3 | 2.4 |
| 156 | 25.0 ± 0.4 | N.T. | N.A. | N.A. |
| 158 | 8.8 ± 1.1 | 22.4 ± 5.2 | N.A. | N.A. |
| 160 | 28.8 ± 2.0 | N.T. | 8.2 | 4.8 |
| 164 | 35.7 ± 1.9 | N.T. | 29 | N.A. |
| 168 | 43.4 ± 4.2 | N.T. | 2.8 | 2.4 |
| 169 | 19.2 ± 0.1 | N.T. | 9.1 | N.A. |
| Cmpd43 | N.A. | N.T. | 0.065 | 0.022 |
| fMLF | N.A. b | N.T. | 0.01 | |
| WKYMVM | N.A. b | N.T. | 0.001 | |
| Compd. | Inhibition of IL-6 Production | Cytotoxicity | Ca2+ Mobilization | |
|---|---|---|---|---|
| FPR1 a | FPR2 a | |||
| IC50 (µM) in MonoMac-6 Cells | EC50 (µM) in HL-60 Cells | |||
| 2 | 30.7 ± 3.6 | N.T. | N.A. | N.A. |
| 9 | 27.5 ± 1.6 | 19.0 ± 3.6 | 4.5 | 14.1 |
| 10 | 8.7 ± 1.6 | N.T. | 4.5 | 7.2 |
| 23 | 13.1 ± 0.3 | N.T. | 9.5 | 16.9 |
| 38 | 20.3 ± 1.8 | N.T. | N.A. | N.A. |
| 42 | 35.6 ± 3.1 | N.T. | N.A. | N.A. |
| 46 | N.A.b | N.T. | N.A. | N.A. |
| 47 | 18.8 ± 2.9 | N.T. | N.A. | N.A. |
| 71 | 2.0 ± 0.3 | N.T. | 2.2 | N.A. |
| 78 | 17.0 ± 0.4 | N.T. | 3.2 | 1.9 |
| 79 | 13.7 ± 1.4 | N.T. | 2.2 | 4.6 |
| 80 | 33.5 ± 2.4 | N.T. | N.A. | 15.7 |
| 83 | 15.2 ± 0.2 | N.T. | N.A. | N.A. |
| 84 | 30.7 ± 1.8 | N.T. | 3.2 | N.A. |
| 89 | 27.9 ± 2.2 | N.T. | 6.3 | 20.4 |
| 94 | 14.3 ± 2.0 | N.T. | 2.8 | 2.3 |
| 95 | 9.8 ± 1.0 | N.T. | 13.4 | 22.2 |
| 98 | 5.6 ± 0.3 | N.T. | 3.0 | 0.84 |
| 102 | 19.0 ± 3.1 | N.T. | 4.5 | 13.7 |
| 103 | 7.3 ± 1.2 | N.T. | 7.0 | N.A. |
| 125 | 7.2 ± 0.03 | N.T. | 24.4 | 10.0 |
| 141 | 24.5 ± 0.8 | N.T. | 1.6 | 0.12 |
| 150 | 4.0 ± 1.2 | N.T. | 4.3 | 3.6 |
| 153 | 10.7 ± 0.1 | N.T. | 12.5 | 8.9 |
| 154 | 2.6 ± 0.3 | N.T. | 3.3 | 2.4 |
| 169 | 3.2 ± 0.4 | N.T. | 9.1 | N.A. |
| fMLF | N.A. b | N.T. d | 0.01 | |
| WKYMVM | N.A. b | N.T. d | 0.001 | |
| Observed | |||
|---|---|---|---|
| Active | Non-Active | ||
| 36 | 111 | ||
| Predicted | Active | 25 | 13 |
| Non-active | 11 | 98 | |
| Activity Classes | Number of Compounds | J (A,B) | |
|---|---|---|---|
| |A ∪ B| | |A ∩ B| | ||
| FPR1 vs. FPR2 | 77 | 64 | 64/77 = 0.831 |
| FPR1 vs. NF-κB | 89 | 21 | 21/89 = 0.236 |
| FPR2 vs. NF-κB | 85 | 18 | 18/85 = 0.212 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantini, N.; Schepetkin, I.A.; Danilenko, N.V.; Khlebnikov, A.I.; Crocetti, L.; Giovannoni, M.P.; Kirpotina, L.N.; Quinn, M.T. Pyridazinones and Structurally Related Derivatives with Anti-Inflammatory Activity. Molecules 2022, 27, 3749. https://doi.org/10.3390/molecules27123749
Cantini N, Schepetkin IA, Danilenko NV, Khlebnikov AI, Crocetti L, Giovannoni MP, Kirpotina LN, Quinn MT. Pyridazinones and Structurally Related Derivatives with Anti-Inflammatory Activity. Molecules. 2022; 27(12):3749. https://doi.org/10.3390/molecules27123749
Chicago/Turabian StyleCantini, Niccolo, Igor A. Schepetkin, Nadezhda V. Danilenko, Andrei I. Khlebnikov, Letizia Crocetti, Maria Paola Giovannoni, Liliya N. Kirpotina, and Mark T. Quinn. 2022. "Pyridazinones and Structurally Related Derivatives with Anti-Inflammatory Activity" Molecules 27, no. 12: 3749. https://doi.org/10.3390/molecules27123749
APA StyleCantini, N., Schepetkin, I. A., Danilenko, N. V., Khlebnikov, A. I., Crocetti, L., Giovannoni, M. P., Kirpotina, L. N., & Quinn, M. T. (2022). Pyridazinones and Structurally Related Derivatives with Anti-Inflammatory Activity. Molecules, 27(12), 3749. https://doi.org/10.3390/molecules27123749