3.2.3. General Procedure for the Synthesis of 4a–k
To a solution of appropriate 3-(alkyloxy)-1-phenyl-1H-pyrazole-4-carbaldehyde (3a–d) (1 mmol) in EtOH (96%, 5 mL), NaOH (0.2 g, 5 mmol) and appropriate acetophenone (1.1 mmol) were added. The reaction mixture was stirred at 55 °C for 30 min., cooled to room temperature, diluted with H2O, and extracted with EtOAc (3 × 10 mL). The organic layers were combined, dried with sodium sulphate, filtrated off and concentrated. The obtained residue was purified by column chromatography (SiO2, eluent: ethyl acetate/n-hexane, 1:6, v/v) to provide the desired product 4a–k.
(2E)-3-(3-Methoxy-1-phenyl-1H-pyrazol-4-yl)-1-phenylprop-2-en-1-one (4a)
Yellow solid; yield 61% (186 mg); m.p. 127–131 °C; Rf = 0.47 (EtOAc/Hex 1/4, v/v). IR (νmax, KBr, cm−1): 3142, 3111, 3040, 2951, 1660 (C=O), 1599, 1592, 1506, 1416, 1219, 1022, 973, 746, 681, 638. 1H NMR (700 MHz, CDCl3): δH ppm 4.14 (s, 3H, OCH3), 7.24–7.27 (m, 1H, NPh 4-H), 7.42–7.46 (m, 2H, NPh 3,5-H), 7.48–7.51 (m, 2H, CPh 3,5-H), 7.55–7.57 (m, 1H, CPh 4-H), 7.60 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.62–7.64 (m, 2H, NPh 2,6-H), 7.74 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.96 (s, 1H, Pz 5-H), 8.02–8.03 (m, 2H, CPh 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 56.6 (OCH3), 107.0 (Pz C-4), 118.1 (NPh C-2,6), 120.5 (CHCHC(O)Ph), 126.2 (NPh C-4), 128.4 (CPh C-2,6), 128.5 (CPh C-3,5), 129.0 (Pz C-5), 129.5 (NPh C-3,5), 132.4 (CPh C-4), 133.7 (CHCHC(O)Ph), 138.6 (CPh C-1), 139.4 (NPh C-1), 163.3 (Pz C-3), 190.7 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −183.7 (N-1), −118.7 (N-2). HRMS (ESI+) for C19H16N2NaO2 ([M + Na]+) calcd 327.1104, found 327.1101.
(2E)-1-(4-Methoxyphenyl)-3-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (4b)
Yellowish white solid; yield 73% (244 mg); m.p. 167–169 °C; Rf = 0.22 (EtOAc/Hex 1/6, v/v). IR (νmax, KBr, cm−1): 3137, 3098, 3072, 2948, 1656 (C=O), 1510, 1422, 1253, 1227, 1176, 1020, 974, 833, 754, 689, 603. 1H NMR (700 MHz, CDCl3): δH ppm 3.88 (s, 3H, PhOCH3), 4.15 (s, 3H, PzOCH3), 6.97–6.99 (m, 2H, CPh 3,5-H), 7.25–7.27 (m, 1H, NPh 4-H), 7.43–7.45 (m, 2H, NPh 3,5-H), 7.61 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.63–7.64 (m, 2H, NPh 2,6-H), 7.72 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.96 (s, 1H, Pz 5-H), 8.04–8.05 (m, 2H, CPh 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 55.6 (PhOCH3), 56.7 (PzOCH3), 107.3 (Pz C-4), 113.8 (CPh C-3,5), 118.2 (NPh C-2,6), 120.5 (CHCHC(O)Ph), 126.2 (NPh C-4), 129.0 (Pz C-5), 129.6 (NPh C-3,5), 130.8 (CPh C-2,6), 131.6 (CPh C-1), 133.0 (CHCHC(O)Ph), 139.6 (NPh C-1), 163.3 (CPh C-4), 163.4 (Pz C-3), 189.1 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −185.2 (N-1), −118.5 (N-2). HRMS (ESI+) for C20H18N2NaO3 ([M + Na]+) calcd 357.1210, found 357.1212.
(2E)-1-(4-Fluorophenyl)-3-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (4c)
Yellow solid; yield 60% (193 mg); m.p. 144–147 °C; Rf = 0.3 (EtOAc/Hex 1/6, v/v). IR (νmax, KBr, cm−1): 3138, 3106, 3061, 2950, 1660 (C=O), 1585, 1514, 1504, 1421, 1218, 1014, 973, 826, 750, 591. 1H NMR (700 MHz, CDCl3): δH ppm 4.15 (s, 3H, CH3), 7.15–7.18 (m, 2H, CPh 3,5-H), 7.26–7.28 (m, 1H, NPh 4-H), 7.44–7.46 (m, 2H, NPh 3,5-H), 7.57 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.63–7.65 (m, 2H, NPh 2,6-H), 7.74 (d, J = 15.6 Hz, 1H, CHCHC(O)Ph), 7.97 (s, 1H, Pz 5-H), 8.04–8.07 (m, 2H, CPh 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 56.8 (CH3), 107.1 (Pz C-4), 115.7 (d, 2J = 21.8 Hz, CPh C-3,5), 118.2 (NPh C-2,6), 120.1 (CHCHC(O)Ph), 126.4 (NPh C-4), 129.2 (Pz C-5), 129.6 (NPh C-3,5), 131.1 (d, 3J = 9.2 Hz, CPh C-2,6), 134.1 (CHCHC(O)Ph), 135.0 (d, 4J = 3.1 Hz, CPh C-1), 139.5 (NPh C-1), 163.5 (Pz C-3), 165.6 (d, 1J = 253.6 Hz, CPh C-4), 189.1 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −184.3 (N-1), −118.5 (N-2). HRMS (ESI+) for C19H15FN2NaO2 ([M + Na]+) calcd 345.1010, found 345.1007.
(2E)-1-(4-Chlorophenyl)-3-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (4d)
Yellow solid; yield 66% (224 mg); m.p. 179–182 °C; Rf = 0.37 (EtOAc/Hex 1/8, v/v). IR (νmax, KBr, cm−1): 3139, 3108, 3071, 2950, 1659 (C=O), 1592, 1514, 1505, 1421, 1219, 1013, 973, 823, 750, 685, 655. 1H NMR (700 MHz, CDCl3): δH ppm 4.15 (s, 3H, CH3), 7.26–7.29 (m, 1H, NPh 4-H), 7.44–7.48 (m, 4H, NPh 3,5-H, CPh 3,5-H), 7.55 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.63–7.65 (m, 2H, NPh 2,6-H), 7.74 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.96–7.97 (m, 3H, Pz 5-H, CPh 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 56.8 (CH3), 107.1 (Pz C-4), 118.3 (NPh C-2,6), 120.1 (CHCHC(O)Ph), 126.4 (NPh C-4), 129.0 (CPh C-3,5), 129.3 (Pz C-5), 129.7 (NPh C-3,5), 130.0 (CPh C-2,6), 134.4 (CHCHC(O)Ph), 137.0 (CPh C-1), 138.9 (CPh C-4), 139.5 (NPh C-1), 163.5 (Pz C-3), 189.5 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −183.9 (N-1), −119.0 (N-2). HRMS (ESI+) for C19H15ClN2NaO2 ([M + Na]+) calcd 361.0714, found 361.0716.
(2E)-1-Phenyl-3-(1-phenyl-3-propoxy-1H-pyrazol-4-yl)prop-2-en-1-one (4e)
Yellow solid; yield 58% (156 mg); m.p. 133–135 °C; Rf = 0.49 (EtOAc/Toluene 1/12, v/v). IR (νmax, KBr, cm−1): 3136, 3105, 3071, 2964, 1655 (C=O), 1588, 1574, 1502, 1417, 1215, 1021, 1010, 998, 748, 685, 643. 1H NMR (700 MHz, CDCl3): δH ppm 1.16 (t, J = 7.5 Hz, 3H, CH3), 1.93–1.98 (m, 2H, OCH2CH2CH3), 4.42 (t, J = 6.5 Hz, 2H, OCH2CH2CH3), 7.25–7.28 (m, 1H, NPh 4-H), 7.43–7.46 (m, 2H, NPh 3,5-H), 7.49–7.53 (m, 2H, CPh 3,5-H), 7.57–7.59 (m, 1H, CPh 4-H), 7.63–7.66 (m, 2H, NPh 2,6-H), 7.69 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.78 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.98 (s, 1H, Pz 5-H), 8.04–8.06 (m, 2H, CPh 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 10.7 (CH3), 22.6 (OCH2CH2CH3), 71.0 (OCH2CH2CH3), 107.3 (Pz C-4), 118.1 (NPh C-2,6), 120.4 (CHCHC(O)Ph), 126.1 (NPh C-4), 128.4 (CPh C-2,6), 128.6 (CPh C-3,5), 128.8 (Pz C-5), 129.5 (NPh C-3,5), 132.5 (CPh C-4), 133.9 (CHCHC(O)Ph), 138.7 (CPh C-1), 139.5 (NPh C-1), 163.1 (Pz C-3), 190.6 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −184.4 (N-1), −119.3 (N-2). HRMS (ESI+) for C21H20N2NaO2 ([M + Na]+) calcd 355.1417, found 355.1416.
(2E)-3-[3-(2-Methoxyethoxy)-1-phenyl-1H-pyrazol-4-yl]-1-phenylprop-2-en-1-one (4f)
Yellow solid; yield 62% (216 mg); m.p. 116–117 °C; Rf = 0.28 (EtOAc/Hex 1/6, v/v). IR (νmax, KBr, cm−1): 3138, 3101, 3063, 3040, 2931, 1655 (C=O), 1594, 1503, 1413, 1219, 1051, 976, 848, 743, 683, 640. 1H NMR (700 MHz, CDCl3): δH ppm 3.52 (s 3H, CH3), 3.87 (t, J = 4.6 Hz, 2H, OCH2CH2OCH3), 4.59 (t, J = 4.6 Hz, 2H, OCH2CH2OCH3), 7.25–7.27 (m, 1H, NPh 4-H), 7.43–7.45 (m, 2H, NPh 3,5-H), 7.48–7.50 (m, 2H, CPh 3,5-H), 7.55–7.57 (m, 1H, CPh 4-H), 7.61–7.63 (m, 2H, NPh 2,6-H), 7.71 (d, J = 14.6 Hz, 1H, CHCHC(O)Ph), 7.75 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.97 (s, 1H, Pz 5-H), 8.03–8.05 (m, 2H, CPh 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 59.3 (CH3), 68.7 (OCH2CH2OCH3), 71.1 (OCH2CH2OCH3), 107.4 (Pz C-4), 118.2 (NPh C-2,6), 120.8 (CHCHC(O)Ph), 126.3 (NPh C-4), 128.57 (CPh C-2,6), 128.63 (CPh C-3,5), 128.8 (Pz C-5), 129.6 (NPh C-3,5), 132.6 (CPh C-4), 133.6 (CHCHC(O)Ph), 138.7 (CPh C-1), 139.5 (NPh C-1), 162.8 (Pz C-3), 190.6 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −184.2 (N-1), −118.3 (N-2). HRMS (ESI+) for C21H20N2NaO3 ([M + Na]+) calcd 371.1366, found 371.1364.
(2E)-3-[3-(Benzyloxy)-1-phenyl-1H-pyrazol-4-yl]-1-phenylprop-2-en-1-one (4g)
Yellow solid; yield 66% (253 mg); m.p. 168–169 °C; Rf = 0.36 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3063, 3029, 1654 (C=O), 1591, 1567, 1504, 1359, 1005, 970, 779, 680. 1H NMR (700 MHz, CDCl3): δH ppm 5.50 (s, 2H, CH2), 7.27–7.29 (m, 1H, NPh 4-H), 7.39–7.41 (m, 1H, CH2Ph 4-H), 7.44–7.47 (m, 6H, CH2Ph 3,5-H, C(O)Ph 3,5-H, NPh 3,5-H), 7.54–7.56 (m, 1H, C(O)Ph 4-H), 7.58–7.59 (m, 2H, CH2Ph 2,6-H), 7.65–7.66 (m, 2H, NPh 2,6-H), 7.74 (d, J = 15.5 Hz, 1H, CHCHC(O)Ph), 7.77 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.96–7.97 (m, 2H, C(O)Ph 2,6-H), 7.99 (s, 1H, Pz 5-H). 13C NMR (176 MHz, CDCl3): δC ppm 71.2 (CH2), 107.4 (Pz C-4), 118.3 (NPh C-2,6), 120.8 (CHCHC(O)Ph), 126.4 (NPh C-4), 128.0 (CH2Ph C-2,6), 128.3 (CH2Ph C-4), 128.5 (C(O)Ph C-2,6), 128.6 (C(O)Ph C-3,5), 128.7 (CH2Ph C-3,5), 129.0 (Pz C-5), 129.7 (NPh C-3,5), 132.6 (C(O)Ph C-4), 133.5 (CHCHC(O)Ph), 136.9 (CH2Ph C-1), 138.6 (C(O)Ph C-1), 139.5 (NPh C-1), 162.8 (Pz C-3), 190.4 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −184.29 (Pz N-1), −117.99 (Pz N-2). HRMS (ESI+) for C25H20N2NaO2 ([M + Na]+) calcd 403.1417, found 403.1416.
(2E)-3-[3-(Benzyloxy)-1-phenyl-1H-pyrazol-4-yl]-1-(4-methoxyphenyl)prop-2-en-1-one (4h)
Yellow solid; yield 68% (279 mg); m.p. 164–165 °C; Rf = 0.34 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3064, 2928, 1652 (C=O), 1502, 1417, 1221, 1169, 1010, 972, 826, 700. 1H NMR (700 MHz, CDCl3): δH ppm 3.88 (s, 3H, CH3), 5.50 (s, 2H, CH2), 6.92–6.93 (m, 2H, C(O)Ph 2,6-H), 7.27–7.28 (m, 1H, NPh 4-H), 7.39–7.41 (m, 1H, CH2Ph 4-H), 7.44–7.47 (m, 4H, CH2Ph 3,5-H, NPh 3,5-H), 7.58–7.60 (m, 2H, CH2Ph 2,6-H), 7.64–7.66 (m, 2H, NPh 2,6-H), 7.74 (s, 2H, CHCHC(O)Ph), 7.96–7.97 (m, 2H, C(O)Ph 3,5-H), 7.98 (s, 1H, Pz 5-H). 13C NMR (176 MHz, CDCl3): δC ppm 55.6 (CH3), 71.2 (CH2), 107.5 (Pz C-4), 113.8 (C(O)Ph C-2,6), 118.2 (NPh C-2,6), 120.7 (CHCHC(O)Ph), 126.3 (NPh C-4), 128.0 (CH2Ph C-2,6), 128.3 (CH2Ph C-4), 128.7 (CH2Ph C-3,5), 128.9 (Pz C-5), 129.7 (NPh C-3,5), 130.8 (C(O)Ph C-3,5), 131.6 (C(O)Ph C-1), 132.7 (CHCHC(O)Ph), 137.0 (CH2Ph C-1), 139.6 (NPh C-1), 162.7 (Pz C-3), 163.3 (C(O)Ph C-4), 188.8 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −184.44 (Pz N-1), −117.93 (Pz N-2). HRMS (ESI+) for C26H22N2NaO3 ([M + Na]+) calcd 433.1523, found 433.1525.
(2E)-3-[3-(Benzyloxy)-1-phenyl-1H-pyrazol-4-yl]-1-(4-chlorophenyl)prop-2-en-1-one (4i)
Yellow solid; yield 97% (411 mg); m.p. 192–193 °C; Rf = 0.56 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3033, 2916, 1600 (C=O), 1501, 1409, 1365, 1215, 1003, 973, 823, 740. 1H NMR (700 MHz, CDCl3): δH ppm 5.49 (s, 2H, CH2), 7.28–7.30 (m, 1H, NPh 4-H), 7.40–7.42 (m, 3H, CH2Ph 4-H, 4ClPh 2,6-H), 7.44–7.48 (m, 4H, CH2Ph 3,5-H, NPh 3,5-H), 7.57–7.58 (m, 2H, CH2Ph 2,6-H), 7.65–7.66 (m, 2H, NPh 2,6-H), 7.68 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.77 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.87–7.89 (m, 2H, 4ClPh 3,5-H), 7.99 (s, 1H, Pz 5-H). 13C NMR (176 MHz, CDCl3): δC ppm 71.3 (CH2), 107.3 (Pz C-4), 118.3 (NPh C-2,6), 120.1 (CHCHC(O)Ph), 126.5 (NPh C-4), 128.1 (CH2Ph C-2,6), 128.4 (CH2Ph C-4), 128.7 (4ClPh C-2,6), 128.9 (CH2Ph C-3,5), 129.1 (Pz C-5), 129.7 (NPh C-3,5), 129.9 (4ClPh C-3,5), 134.1 (CHCHC(O)Ph), 136.9 (CH2Ph C-1), 137.0 (4ClPh C-4), 139.0 (4ClPh C-1), 139.5 (NPh C-1), 162.8 (Pz C-3), 189.0 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −183.94 (Pz N-1), −117.85 (Pz N-2). HRMS (ESI+) for C25H19ClN2NaO3 ([M + Na]+) calcd 437.1027, found 437.1028.
(2E)-3-[3-(Benzyloxy)-1-phenyl-1H-pyrazol-4-yl]-1-(4-fluorophenyl)prop-2-en-1-one (4j)
Yellow solid; yield 79% (318 mg); m.p. 145–146 °C; Rf = 0.54 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3033, 2944, 1654 (C=O), 1596, 1500, 1411, 1364, 1002, 973, 826, 685. 1H NMR (700 MHz, CDCl3): δH ppm 5.49 (s, 2H, CH2), 7.09–7.12 (m, 2H, 4FPh 3,5-H), 7.27–7.30 (m, 1H, NPh 4-H), 7.41–7.42 (m, 1H, CH2Ph 4-H), 7.44–7.48 (m, 4H, CH2Ph 3,5-H, NPh 3,5-H), 7.58–7.59 (m, 2H, CH2Ph 2,6-H), 7.65–7.66 (m, 2H, NPh 2,6-H), 7.70 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.76 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.95–7.98 (m, 2H, 4FPh 2,6-H), 7.99 (s, 1H, Pz 5-H). 13C NMR (176 MHz, CDCl3): δC ppm 71.3 (CH2), 107.3 (Pz C-4), 115.7 (d, 2J = 21.7 Hz, 4FPh C-3,5), 118.3 (NPh C-2,6), 120.3 (CHCHC(O)Ph), 126.4 (NPh C-4), 128.1 (CH2Ph C-2,6), 128.4 (CH2Ph C-4), 128.7 (CH2Ph C-3,5), 129.1 (Pz C-5), 129.7 (NPh C-3,5), 131.1 (d, 3J = 9.1 Hz, 4FPh C-2,6), 133.7 (CHCHC(O)Ph), 135.0 (d, 4J = 3.0 Hz, 4FPh C-1), 136.9 (CH2Ph C-1), 139.5 (NPh C-1), 162.8 (Pz C-3), 164.2 (d, 1J = 253.7 Hz, 4FPh C-4), 188.7 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −184.26 (Pz N-1), −118.00 (Pz N-2). HRMS (ESI+) for C25H19FN2NaO3 ([M + Na]+) calcd 421.1323, found 421.1323.
(2E)-3-[3-(Benzyloxy)-1-phenyl-1H-pyrazol-4-yl]-1-[4-(dimethylamino)phenyl]prop-2-en-1-one (4k)
Yellow solid; yield 85% (376 mg); m.p. 179–180 °C; Rf = 0.15 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3102, 2918, 1564 (C=O), 1434, 1404, 1358, 1166, 1028, 977, 809, 742. 1H NMR (700 MHz, CDCl3): δH ppm 3.06 (s, 6H, CH3), 5.50 (s, 2H, CH2), 6.64–6.66 (m, 2H, (CH3)2NPh 3,5-H), 7.24–7.26 (m, 1H, NNPh 4-H), 7.38–7.40 (m, 1H, CH2Ph 4-H), 7.43–7.46 (m, 4H, CH2Ph 3,5-H, NNPh 3,5-H), 7.60–7.61 (m, 2H, CH2Ph 2,6-H), 7.64–7.65 (m, 2H, NNPh 2,6-H), 7.72 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.78 (d, J = 15.4 Hz, 1H, CHCHC(O)Ph), 7.94–7.96 (m, 3H, (CH3)2NPh 2,6-H, Pz 5-H). 13C NMR (176 MHz, CDCl3): δC ppm 40.2 (CH3), 71.0 (CH2), 107.7 (Pz C-4), 110.9 ((CH3)2NPh C-3,5), 118.1 (NNPh C-2,6), 121.1 (CHCHC(O)Ph), 126.1 (NNPh C-4), 126.4 ((CH3)2NPh C-1), 127.9 (CH2Ph C-2,6), 128.1 (CH2Ph C-4), 128.5 (Pz C-5), 128.6 (CH2Ph C-3,5), 129.6 (NNPh C-4), 130.8 ((CH3)2NPh C-2,6), 131.2 (CHCHC(O)Ph), 137.1 (CH2Ph C-1), 139.6 (NNPh C-1), 153.3 ((CH3)2NPh C-4), 162.6 (Pz C-3), 188.0 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −325.44 (N(CH3)2), −185.23 (Pz N-1), −118.32 (Pz N-2). HRMS (ESI+) for C27H25N3NaO2 ([M + Na]+) calcd 446.1839, found 446.1842.
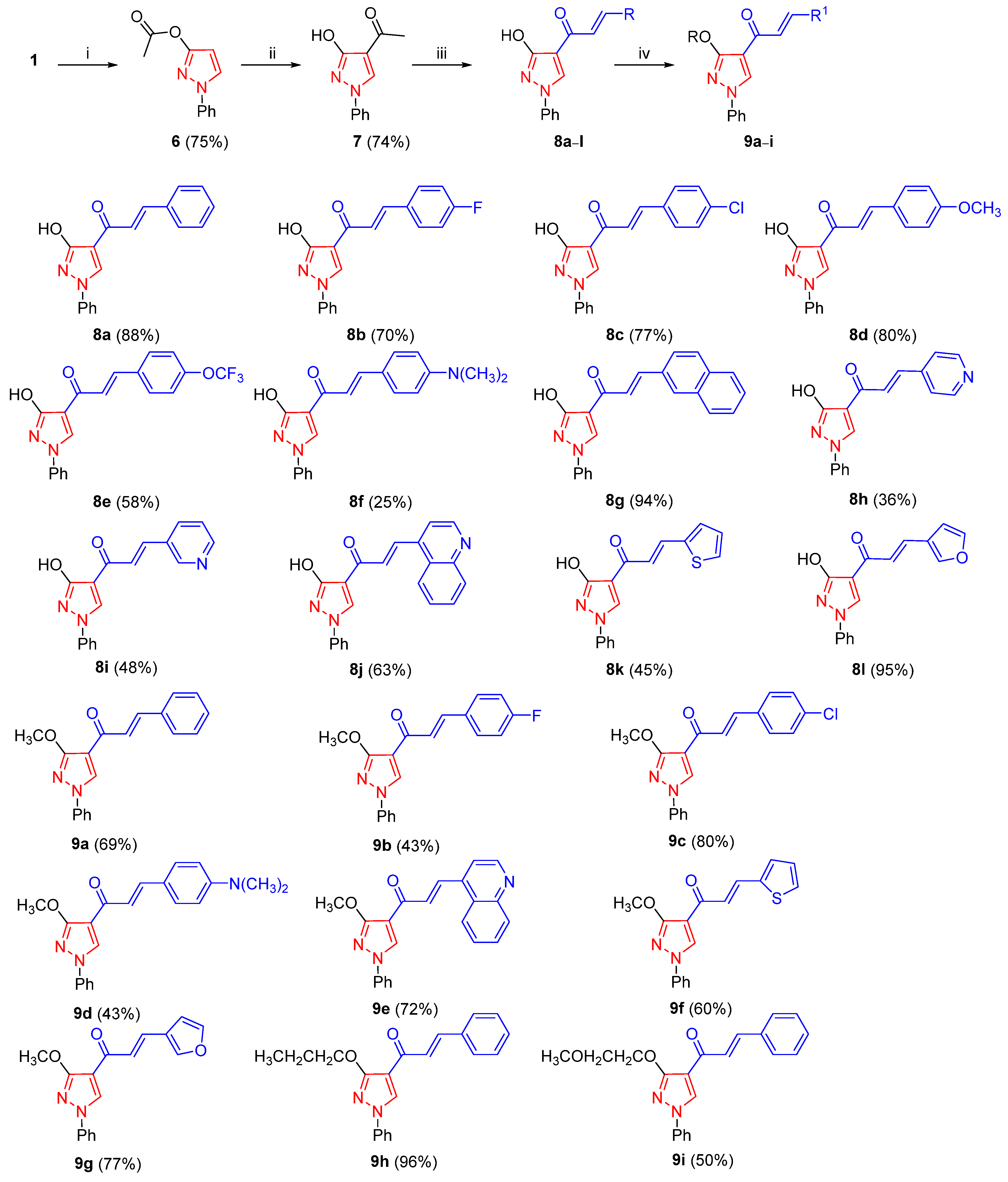
3.2.5. General Procedure for the Synthesis of 8a–l
To a solution of 1-(3-hydroxy-1-phenyl-1H-pyrazol-4-yl)ethan-1-one (7) (2.02 g, 10 mmol) in EtOH (96%, 80 mL), NaOH (2 g, 50 mmol) and appropriate aldehyde (20 mmol) were added. The reaction mixture was stirred at 55 °C for 3–5 h, cooled to room temperature and neutralized to pH = 7 using 6N HCl. The precipitate was filtered off, washed with water and cold ether and recrystallized from ACN to produce pure 8a–l.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-phenylprop-2-en-1-one (8a)
The reaction mixture was stirred for 3 h. White solid; yield 88% (2.56 g); m.p. 222–223 °C; Rf=0.24 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3075, 3058, 3026, 1654 (C=O), 1584, 1511, 1448, 1217, 1062, 746, 735, 693, 678. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.32–7.34 (m, 1H, NPh 4-H), 7.44–7.45 (m, 1H, CPh 4-H), 7.46–7.48 (m, 2H, CPh 3,5-H), 7.51–7.53 (m, 2H, NPh 3,5-H), 7.69 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 7.75 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.76–7.78 (m, 2H, CPh 2,6-H), 7.84–7.85 (m, 2H, NPh 2,6-H), 9.16 (s, 1H, Pz- 5-H), 11.22 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.1 (Pz- C-4), 118.0 (NPh C-2,6), 124.3 (C(O)CHCH), 126.5 (NPh C-4), 128.4 (CPh C-2,6), 129.0 (CPh C-3,5), 129.6 (NPh C-3,5), 130.4 (CPh C-4), 131.7 (Pz C-5), 134.7 (CPh C-1), 138.8 (NPh C-1), 141.4 (C(O)CHCH), 162.0 (Pz C-3), 182.6 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.9 (Pz N-1), −117.0 (Pz N-2). HRMS (ESI+) for C18H14N2O2 ([M + Na]+) calcd 313.0948, found 313.0947.
(2E)-3-(4-Fluorophenyl)-1-(3-hydroxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (8b)
The reaction mixture was stirred for 4.5 h. Orange solid; yield 70% (2.16 g); m.p. 239–240 °C; Rf = 0.46 (DCM/MeOH 100/1, v/v). IR (νmax, cm−1): 3360, 3111, 2978, 1646 (C=O), 1583, 1505, 1359, 1326, 821, 747, 673, 505. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.31–7.35 (m, 3H, NPh 4-H, CPh 3,5-H), 7.51–7.53 (m, 2H, NPh 3,5-H), 7.68 (s, 2H, C(O)CHCH), 7.83–7.86 (m, 4H, NPh 2,6-H, CPh 2,6-H), 9.14 (s, 1H, Pz 5-H), 11.20 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.1 (Pz C-4), 116.0 (d, 2J = 21.8 Hz, CPh C-3,5), 118.0 (NPh C-2,6), 124.2 (C(O)CHCH), 126.6 (NPh C-4), 129.6 (NPh C-3,5), 130.7 (d, 3J = 8.6 Hz, CPh C-2,6), 131.4 (d, 4J = 2.9 Hz, CPh C-1), 131.7 (Pz C-5), 138.8 (NPh C-1), 140.1 (C(O)CHCH), 161.9 (Pz C-3), 163.2 (d, 1J = 248.6 Hz, CPh C-4), 182.5 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −182.1 (Pz N-1), −119.5 (Pz N-2). HRMS (ESI+) for C18H14N2O2 ([M + H]+) calcd 331.0853, found 331.0853.
(2E)-3-(4-Chlorophenyl)-1-(3-hydroxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (8c)
The reaction mixture was stirred for 4 h. Yellow solid; yield 77% (2505 mg); m.p. 354–355 °C; Rf = 0.17 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3110, 3071, 1654 (C=O), 1586, 1511, 1456, 1325, 1218, 1094, 1062, 815, 745, 709, 681, 497. 1H NMR (700 MHz, DMSO-d6) δH ppm 7.33–7.35 (m, 1H, NPh 4-H), 7.51–7.55 (m, 4H, NPh 3,5-H, CPh 3,5-H), 7.66 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 7.73 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 7.80–7.81 (m, 2H, CPh 2,6-H), 7.83–7.84 (m, 2H, NPh 2,6-H), 9.15 (s, 1H, Pz- 5-H), 11.22 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6) δC ppm 111.5 (Pz C-4), 118.5 (NPh C-2,6), 125.4 (C(O)CHCHPh), 127.1 (NPh C-4), 129.5 (CPh C-3,5), 130.0 (NPh C-3,5), 130.6 (CPh C-2,6), 132.3 (Pz 5-H), 134.2, 135.3, 139.3 (NPh C-1), 140.4 (C(O)CHCHPh), 162.3 (Pz C-3), 182.8 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.7 (Pz N-1), −118.1 (Pz N-2). HRMS (ESI+) for C18H13ClN2O2 ([M + Na]+) calcd 347.0558, found 347.0558.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-(4-methoxyphenyl)prop-2-en-1-one (8d)
The reaction mixture was stirred for 3.5 h. Brown solid; yield 80% (2.57 g); m.p. 200–201 °C; Rf = 0.14 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3110, 3071, 1653 (C=O), 1586, 1509, 1457, 1219, 1172, 1049, 818, 769, 743, 679. 1H NMR (700 MHz, DMSO-d6): δH ppm 3.82 (s, 3H, CH3), 7.03–7.04 (m, 2H, CPh 3,5-H), 7.32–7.34 (m, 1H, NPh 4-H), 7.51–7.53 (m, 2H, NPh 3,5-H), 7.60 (d, J = 15.6 Hz, 1H, C(O)CHCH), 7.66 (d, J = 15.6 Hz, 1H, C(O)CHCH), 7.73–7.74 (m, 2H, CPh 2,6-H), 7.84–7.85 (m, 2H, NPh 2,6-H), 9.14 (s, 1H, Pz 5-H), 11.11 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 55.4 (CH3), 111.0 (Pz C-4), 114.5 (CPh C-3,5), 118.0 (NPh C-2,6), 121.7 (C(O)CHCH), 126.5 (NPh C-4), 127.3 (CPh C-1), 129.6 (NPh C-3,5), 130.3 (CPh C-2,6), 131.5 (Pz C-5), 138.8 (NPh C-1), 141.4 (C(O)CHCH), 161.2 (Pz C-3), 161.9 (CPh C-4), 182.8 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −182.2 (Pz N-1). HRMS (ESI+) for C19H16N2O ([M + Na]+) calcd 343.1053, found 343.1053.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-[4-(trifluoromethoxy)phenyl]prop-2-en-1-one (8e)
The reaction mixture was stirred for 5 h. Yellow solid; yield 58% (2.21 g); m.p. 148–149 °C; Rf = 0.49 (DCM/MeOH 100/1, v/v). IR (νmax, cm−1): 3110, 3071, 1657 (C=O), 1599, 1583, 1525, 1506, 1214, 1146, 977, 925, 825, 745. 1H NMR (400 MHz, DMSO-d6): δH ppm 7.32–7.36 (m, 1H, NPh 4-H), 7.45–7.47 (m, 2H, CPh 2,6-H), 7.50–7.54 (m, 2H, NPh 3,5-H), 7.69 (d, J = 15.8 Hz, 1H, C(O)CHCH), 7.74 (d, J = 15.8 Hz, 1H, C(O)CHCH), 7.83–7.85 (m, 2H, CPh 3,5-H), 7.90–7.92 (m, 2H, NPh 2,6-H), 9.15 (s, 1H, Pz 5-H), 11.24 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δC ppm 111.0 (Pz C-4), 118.1 (NPh C-2,6), 120.0 (q, 1J = 256.8 Hz, OCF3), 121.4, 125.4 (C(O)CHCHPh), 126.6, 129.6, 130.3, 131.8, 134.1 (Pz C-5), 138.8 (NPh C-1), 139.6 (C(O)CH), 149.4 (CPh C-4), 161.9 (Pz C-3), 182.4 (C=O). HRMS (ESI+) for C19H13F3N2O3 ([M + Na]+) calcd 397.0770, found 397.0770.
(2E)-3-[4-(Dimethylamino)phenyl]-1-(3-hydroxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (8f)
The reaction mixture was stirred for 5 h. Dark red solid; yield 25% (835 mg); m.p. 203–204 °C; Rf = 0.09 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3111, 1635 (C=O), 1586, 1505, 1426, 1354, 1160, 1034, 808, 750, 687, 668. 1H NMR (700 MHz, DMSO-d6): δH ppm 2.99 (s, 6H, CH3), 6.75–6.76 (m, 2H, CPh 3,5-H), 7.31–7.33 (m, 1H, NPh 4-H), 7.47 (d, d, J = 15.5 Hz, 1H, C(O)CH), 7.50–7.52 (m, 2H, NPh 3,5-H), 7.59–7.61 (m, 2H, CPh 3,5-H), 7.63 (d, d, J = 15.5 Hz, 1H, C(O)CHCH), 7.83–7.84 (m, 2H, NPh 2,6-H), 9.10 (s, 1H, Pz 5-H), 11.02 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 39.7 (CH3), 111.0 (Pz C-4), 111.8 (CPh 3,5-C), 118.0 (NPh C-2,6), 118.3 (C(O)CHCH), 121.9 (CPh C-1), 126.4 (NPh C-4), 129.6 (CPh C-2,6), 130.3 (NPh C-3,5), 131.0 (Pz C-5), 138.9 (NPh C-1), 142.7 (C(O)CHCH), 151.9 (CPh C-4), 162.1 (Pz C-3), 183.1 (C=O). HRMS (ESI+) for C20H19N3O2 ([M + Na]+) calcd 356.1369, found 356.1369.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-(naphthalen-2-yl)prop-2-en-1-one (8g)
The reaction mixture was stirred for 3.5 h. Orange solid; yield 94% (3.20 g); m.p. 257–258 °C; Rf = 0.43 (DCM/MeOH 100/1, v/v). IR (νmax, cm−1): 3117, 3056, 1650 (C=O), 1586, 1509, 460, 1322, 1216, 1062, 847, 806, 754, 732, 680. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.34–7.36 (m, 1H, NPh 4-H), 7.52–7.55 (m, 2H, NPh 3,5-H), 7.57–7.60 (m, 2H, Naph 4,8-H), 7.85–7.87 (m, 4H, NPh 2,6-H, C(O)CHCH), C(O)CHCH), 7.96–8.02 (m, 4H, Naph 3,5,6,7-H), 8.26 (s, 1H, Naph 1-H), 9.21 (s, 1H, Pz 5-H), 11.19 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.1 (Pz C-4), 118.1 (NPh C-2,6), 123.8 (Naph C-6), 124.5 (C(O)CHCH), 126.6 (NPh C-4), 126.9 (Naph C-4), 127.4 (Naph C-8), 127.8 (Naph C-3), 128.5 (Naph C-5), 128.6 (Naph C-7), 129.6 (NPh C-3,5), 130.4 (Naph C-1), 131.8 (Pz C-5), 132.3 (Naph C-4a), 133.0 (Naph C-2), 133.8 (Naph C-8a), 138.8 (NPh C-1), 141.4 (C(O)CHCH), 162.0 (Pz C-3), 182.6 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.9 (Pz N-1). HRMS (ESI+) for C22H16N2O2 ([M + Na]+) calcd 363.1104, found 363.1104.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-(pyridin-4-yl)prop-2-en-1-one (8h)
The reaction mixture was stirred for 4 h. Yellow solid; yield 36% (1.05 g); m.p. 230–231 °C; Rf = 0.19 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3397, 3115, 3068, 1658 (C=O), 1585, 1509, 1453, 1318, 1216, 808, 748, 720, 675. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.33–7.36 (m, 1H, Ph 4-H), 7.52–7.54 (m, 2H, Ph 3,5-H), 7.62 (d, J = 15.8 Hz, 1H, C(O)CHCH), 7.73–7.74 (m, 2H, Pyr 3,5-H), 7.84–7.85 (m, 2H, Ph 2,6-H), 7.90 (d, J = 15.8 Hz, 1H, C(O)CHCH), 8.68–8.69 (m, 2H, Ph 2,6-H), 9.19 (s, 1H, Pz 5-H), 11.33 (s, 1H OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.0 (Pz C-4), 118.1 (Ph C-2,6), 122.3 (Pyr C-2,6), 126.7 (Ph C-4), 128.6 (C(O)CHCH), 129.6 (Ph C-3,5), 132.1 (Pz C-5), 138.4 (Pyr C-4), 138.7 (Ph C-1), 142.2 (C(O)CHCH), 150.2 (Pyr C-3,5), 161.9 (Pz C-3), 182.0 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.8 (Pz N-1), −118.1 (Pz N-2), −63.9 (Pyr N). HRMS (ESI+) for C17H13N3O2 ([M + H]+) calcd 292.1081, found 292.1081.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-(pyridin-3-yl)prop-2-en-1-one (8i)
The reaction mixture was stirred for 3.5 h. Yellow solid; yield 48% (1.40 mg); m.p. 230–231 °C; Rf = 0.21 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3115, 3026, 1656 (C=O), 1583, 1510, 1455, 1320, 1216, 1061, 800, 748, 703, 678. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.33–7.35 (m, 1H, Ph 4-H), 7.50–7.54 (m, 3H, Ph 3,5-H, Pyr 5-H), 7.70 (d, J = 15.8 Hz, 1H, C(O)CH), 7.81–7.84 (m, 3H, Ph 2,6-H, C(O)CHCH), 8.19–8.21 (m, 1H, Pyr 4-H), 8.61 (dd, J = 4.7, 1.7 Hz, 1H, Pyr 6-H), 8.97 (d, J = 2.3 Hz, 1H, Pyr 2-H), 9.19 (s, 1H, Pz 5-H), 11.22 (s, 1H, OH).13C NMR (176 MHz, DMSO-d6): δC ppm 111.0 (Pz C-4), 118.1 (Ph C-2,6), 124.0 (Pyr C-5), 126.1 (C(O)CH), 126.6 (Ph C-4), 129.6 (Ph C-3,5), 130.6 (Pyr C-3), 131.9 (Pz C-5), 134.8 (Pyr C-4), 137.9 (C(O)CHCH), 138.8 (Ph C-1), 149.8 (Pyr C-2), 150.7 (Pyr C-6), 161.9 (Pz C-3), 182.1 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −182.1 (Pz N-1), −119.4 (Pz N-2), −65.0 (Pyr N). HRMS (ESI+) for C17H13N3O2 ([M + H]+) calcd 292.1080, found 292.1081.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-(quinolin-4-yl)prop-2-en-1-one (8j)
The reaction mixture was stirred for 4 h. Orange solid; yield 63% (2.15 mg); m.p. 240–241 °C; Rf = 0.24 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3111, 1654 (C=O), 1574, 1507, 1441, 1221, 1050, 746, 730, 684, 669. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.34–7.36 (m, 1H, NPh 4-H), 7.52–7.54 (m, 2H, NPh 3,5-H), 7.74–7.76 (m, 1H, Quin 6-H), 7.86–7.87 (m, 2H, NPh 2,6-H), 7.88–7.89 (m, 1H, Quin 7-H), 7.95–7.98 (m, 2H, C(O)CHCH, Quin 3-H), 8.12–8.13 (m, 1H, Quin 5-H), 8.35–8.36 (m, 1H, Quin 8-H), 8.39 (d, J = 14.0 Hz, 1H, C(O)CHCH), 9.04 (d, J = 4.5 Hz, 1H, Quin 2-H), 9.22 (s, 1H, Pz 5-H), 11.43 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.0 (Pz C-4), 118.1 (NPh C-2,6), 118.5 (Quin C-3), 123.8 (Quin C-8), 125.8 (Quin C-4), 126.7 (NPh C-4), 127.7 (Quin C-6), 129.1 (Quin C-5), 129.6 (NPh C-3,5), 130.2 (Quin C-7), 131.0 (C(O)CHCH), 132.2 (Pz C-4), 134.8 (C(O)CHCH), 138.7 (NPh C-1), 140.7 (Quin C-4a), 147.5 (Quin C-8a), 149.9 (Quin C-2), 161.9 (Pz C-3), 181.8 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.1 (Pz N-1). HRMS (ESI+) for C21H15N3O2 ([M + H]+) calcd 342.1237, found 342.1237.
(2E)-1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)-3-(thiophen-2-yl)prop-2-en-1-one (8k)
The reaction mixture was stirred for 4 h. Yellow solid; yield 45% (1.33 mg); m.p. 196–197 °C; Rf = 0.54 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3104, 3067, 1647 (C=O), 1582, 1509, 1457, 1322, 1217, 1062, 967, 825, 699, 685. 1H NMR (700 MHz, DMSO-d6): δH ppm 7.17–7.18 (m, 1H, Th 5-H), 7.31–7.34 (m, 1H, Ph 4-H), 7.46 (d, J = 15.4 Hz, 1H, C(O)CHCH), 7.50–7.52 (m, 2H, Ph 3,5-H), 7.58–7.59 (m, 1H, Th 3-H), 7.74–7.75 (m, 1H, Th 4-H), 7.84–7.88 (m, 3H, Ph 2,6-H, C(O)CHCH), 9.08 (s, 1H, Pz 5-H), 11.31 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.0 (Pz C-4), 118.1 (Ph C-2,6), 122.7 (C(O)CHCH), 126.5 (Ph C-4), 128.7 (Th C-5), 129.5 (Ph C-3,5), 129.8 (Th C-4), 131.6 (Pz C-5), 132.9 (Th C-3), 134.5 (C(O)CHCH), 138.8 (Ph C-1), 139.9 (Th C-2), 161.7 (Pz C-3), 182.2 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.6 (Pz N-1), −118.4 (Pz N-2). HRMS (ESI+) for C16H12N2O2S ([M + Na]+) calcd 319.0512, found 319.0512.
(2E)-3-(Furan-3-yl)-1-(3-hydroxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (8l)
The reaction mixture was stirred for 5 h. White solid; yield 95% (2.67 mg); m.p. 195–196 °C; Rf = 0.62 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3111, 3071, 1653 (C=O), 1587, 1511, 1458, 1322, 1219, 1063, 1156, 727, 680. 1H NMR (700 MHz, DMSO-d6): δH ppm 6.67–6.68 (m, 1H, Furanyl 5-H), 7.00 (m, 1H, Furanyl 4-H), 7.31–7.33 (m, 1H, Ph 4-H), 7.49–7.53 (m, 4H, Ph 3,5-H, C(O)CHCH), 7.85–7.86 (m, 2H, Ph 2,6-H), 7.89 (m, 1H, Furanyl 2-H), 9.04 (s, 1H, Pz 5-H), 11.35 (s, 1H, OH). 13C NMR (176 MHz, DMSO-d6): δC ppm 111.1 (Pz C-4), 113.0 (Furanyl C-5), 116.8 (Furanyl C-4), 118.1 (Ph C-2,6), 121.1 (C(O)CHCH), 126.5 (Ph C-4), 128.1 (C(O)CHCH), 129.6 (Ph C-3,5), 131.5 (Pz C-5), 138.8 (Ph C-1), 145.9 (Furanyl C-2), 151.2 (Furanyl C-3), 161.6 (Pz C-3), 182.2 (C=O). 15N NMR (71 MHz, DMSO-d6): δN −181.6 (Pz N-1). HRMS (ESI+) for C16H12N2O3 ([M + Na]+) calcd 303.0740, found 303.0740.
3.2.6. General Procedure for the Synthesis of 9a–i
To a solution of an appropriate compound
8 (1 mmol) in abs. DMF (3 mL), NaH (60% suspension in mineral oil, 0.04 g, 1 mmol) and an appropriate alkylhalide (1.1 mmol) were added [
76]. The reaction mixture was stirred at room temperature for 1–2 h, diluted with KHSO
4 aq. (10 mL) and extracted with EtOAc (2 × 10 mL). The organic layers were combined, washed with H
2O (4 × 20 mL), dried with sodium sulphate, filtrated off and concentrated. The residue was purified by column chromatography (SiO
2, eliuent: hexane/ethyl acetate 9/1) to produce pure
9a–
i.
(2E)-1-(3-Methoxy-1-phenyl-1H-pyrazol-4-yl)-3-phenylprop-2-en-1-one (9a)
The reaction mixture was stirred for 1 h. Yellow solid; yield 69% (210 mg); m.p. 163–164 °C; Rf = 0.78 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3118, 3089, 1652 (C=O), 1550, 1498, 1408, 1308, 1217, 756, 731, 685, 672. 1H NMR (700 MHz, CDCl3): δH ppm 4.16 (s, 3H, CH3), 7.30–7.32 (m, 1H, NPh 4-H), 7.39–7.43 (m, 3H, CPh 3,4,5-H), 7.45–7.48 (m, 2H, NPh 3,5-H), 7.63 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 7.64–7.66 (m, 2H, CPh 2,6-H), 7.68–7.69 (m, 2H, NPh 2,6-H), 7.82 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 8.42 (s, 1H, Pz 5-H). 13C NMR (176 MHz, CDCl3): δC ppm 56.9 (CH3), 112.3 (Pz C-4), 118.6 (NPh C-2,6), 124.3 (C(O)CHCHPh), 126.9 (NPh C-4), 128.5 (CPh C-2,6), 128.8 (CPh C-3,5), 129.5 (NPh C-3,5), 130.2 (CPh C-4), 131.3 (Pz C-5), 135.2 (CPh C-1), 139.2 (NPh C-1), 142.6 (C(O)CHCHPh), 162.5 (Pz C-3), 183.3 (C=O). 15N NMR (71 MHz, CDCl3): δN ppm −181.4 (Pz N-1), −118.5 (Pz N-2). HRMS (ESI+) for C19H16N2O2 ([M + Na]+) calcd 327.1104, found 327.1104.
(2E)-3-(4-Fluorophenyl)-1-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (9b)
The reaction mixture was stirred for 2 h. Yellow solid; yield 43% (139 mg); m.p. 155–156 °C; Rf = 0.53 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3116, 3049, 1656 (C=O), 1560, 1500, 1408, 1327, 1221, 746, 706, 684, 641. 1H NMR (700 MHz, DMSO-d6): δH ppm 4.04 (s, 3H, CH3), 7.30–7.33 (m, 2H, CPh 3,5-H), 7.35–7.37 (m, 1H, NPh 4-H), 7.53–7.55 (m, 2H), 7.61 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 7.66 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.86–7.89 (m, 4H, NPh 2,6-H, CPh 2,6-H), 9.27 (s, 1H, Pz 5-H). 13C NMR (176 MHz, DMSO-d6): δC ppm 54.9 (CH3), 109.6 (Pz C-4), 114.3 (d, 2J= 21.7 Hz, CPh C-3,5), 116.5 (NPh C-2,6), 122.6 (C(O)CH), 125.1 (NPh C-4), 127.9 (NPh C-3,6), 129.1 (d, 3J= 8.5 Hz, CPh C-2,6), 129.7 (d, 4J = 3.1 Hz, CPh C-1), 131.4 (Pz C-5), 137.1 (NPh C-1), 138.5 (C(O)CHCH), 160.9 (Pz C-3), 161.6 (d, 1J = 248.3 Hz, CPh C-4), 179.6 (C=O). HRMS (ESI+) for C19H15FN2O2 ([M + H]+) calcd 323.1190, found 323.1191.
(2E)-3-(4-Chlorophenyl)-1-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (9c)
The reaction mixture was stirred for 2 h. Yellow solid; yield 80% (272 mg); m.p. 173–174 °C; Rf = 0.69 (DCM/MeOH 100/1, v/v). IR (νmax, cm−1): 3122, 3069, 1659 (C=O), 1597, 1560, 1490, 1403, 1329, 1222, 813, 744, 683, 638 497. 1H NMR (700 MHz, DMSO-d6): δH ppm 4.04 (s, 3H, CH3), 7.34–7.36 (m, 1H, NPh 4-H), 7.52–7.55 (m, 4H, NPh 3,5-H, CPh 2,6-H), 7.63 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.66 (d, J = 15.7 Hz, 1H, C(O)CHCHPh), 7.82–7.83 (m, 2H, CPh 3,5-H), 7.88–7.89 (m, 2H, NPh 2,6-H), 9.27 (s, 1H, Pz 5-H). 13C NMR (176 MHz, DMSO-d6): δC ppm 56.5 (CH3), 111.2 (Pz C-4), 118.1 (NPh C-2,6), 125.1 (C(O)CHCHPh), 126.7 (NPh C-4), 129.0 (CPh C-2,6), 129.6 (NPh C-3,5), 130.2 (CPh C-3,5), 133.1(Pz C-5), 133.7 (CPh C-1), 134.8 (CPh C-4), 138.7 (NPh C-4), 140.0 (C(O)CHCHPh), 162.6 (Pz C-3), 181.2 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −182.2 (Pz N-1), −119.1 (Pz N-2). HRMS (ESI+) for C19H15ClN2O2 ([M + Na]+) calcd 361.0714, found 361.0714.
(2E)-3-[4-(Dimethylamino)phenyl]-1-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (9d)
The reaction mixture was stirred for 1 h. Yellow solid; yield 43% (277 mg); m.p. 186–187 °C; Rf=0.15 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3111, 1634 (C=O), 1580, 1503, 1421, 1358, 1157, 1032, 810, 748, 688, 667. 1H NMR (700 MHz, DMSO-d6): δH ppm 3.00 (s, 6H, N(CH3)2), 4.04 (s, 3H, OCH3), 6.75–6.76 (m, 2H, CPh 3,5-H), 7.33–7.35 (m, 1H, NPh 4-H), 7.41 (d, J = 15.4 Hz 1H C(O)CHCHPh), 7.51–7.54 (m, 2H), 7.57–7.61 (m, 3H, CPh 2,6-H, C(O)CHCHPh), 7.88–7.90 (m, 2H, NPh 2,6-H), 9.16 (s, 1H, Pz 5-H). 13C NMR (176 MHz, DMSO-d6): δC ppm 39.7 (N(CH3)2), 56.5 (OCH3), 111.7 (Pz C-4), 111.8 (CPh 3,5-C), 118.0 (NPh C-2,6), 118.8 (C(O)CHCHPh), 122.0 (CPh C-1), 126.5 (NPh C-4), 129.5 (CPh C-2,6), 130.2 (NPh C-3,5), 132.4 (Pz C-5), 138.8 (NPh C-1), 142.5 (C(O)CHCHPh), 151.8 (CPh C-4), 162.3 (Pz C-3), 181.3 (C=O). HRMS (ESI+) for C21H21N3NaO2 ([M + Na]+) calcd 370.1526, found 370.1526.
(2E)-1-(3-Methoxy-1-phenyl-1H-pyrazol-4-yl)-3-(quinolin-4-yl)prop-2-en-1-one (9e)
The reaction mixture was stirred for 1 h. Yellow solid; yield 72% (256 mg); m.p. 182–183 °C, Rf = 0.6 (DCM/MeOH 100/3, v/v). IR (νmax, cm−1): 3120, 3091, 1654 (C=O), 1596, 1556, 1500, 1414, 1309, 1218, 834, 749, 726, 682. 1H NMR (700 MHz, DMSO-d6): δH ppm 4.06 (s, 3H, CH3), 7.35–7.38 (m, 1H, NPh 4-H), 7.53–7.56 (m, 2H, NPh 3,5-H), 7.71–7.73 (m, 1H, Quin 7-H), 7.83–7.85 (m, 1H, Quin 6-H), 7.87–7.91 (m, 3H, C(O)CHCH, NPh 2,6-H), 7.95–7.96 (m, 1H, Quin 4-H), 8.10–8.11 (m, 1H, Quin 5-H), 8.31–8.32 (m, 1H, Quin 8-H), 8.36 (d, J = 15.5 Hz, 1H, C(O)CHCH), 9.01 (m, 1H, Quin 2-H), 9.34 (s, 1H, Pz 5-H). 13C NMR (176 MHz, DMSO-d6): δC ppm 56.6 (CH3), 111.1 (Pz C-4), 118.2 (NPh C-2,6), 118.5 (Quin C-3), 123.6, 125.7, 126.9, 127.5, 129.6 (NPh C-3,5), 129.7, 129.8, 130.7, 133.4 (Pz C-5), 135.1 (C(O)CHCH), 138.7 (NPh C-1), 139.8 (Quin C-4a), 148.3 (Quin C-8a), 150.3 (Quin C-2), 162.7 (Pz C-3), 181.0 (C=O). HRMS (ESI+) for C22H17N3O2 ([M + H]+) calcd 356.1394, found 356.1394.
(2E)-1-(3-Methoxy-1-phenyl-1H-pyrazol-4-yl)-3-(thiophen-2-yl)prop-2-en-1-one (9f)
The reaction mixture was stirred for 1 h. Yellow solid; yield 60% (186 mg); m.p. 131–132 °C; Rf = 0.37 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3127, 3096, 1664 (C=O), 1560, 1502, 1410, 1399, 1225, 1014, 943, 750, 685, 669, 505. 1H NMR (700 MHz, DMSO-d6): δH ppm 4.05 (s, 3H, CH3), 7.18–7.19 (m, 1H, Th 5-H), 7.34–7.38 (m, 2H, Ph 4-H, C(O)CHCH), 7.51–7.54 (m, 2H, Ph 3,5-H), 7.60 (m, 1H, Th 3-H), 7.75–7.76 (m, 1H, Th 4-H), 7.84 (d, J = 15.3 Hz, 1H, C(O)CHCH), 7.88–7.91 (m, 2H, Ph 2,6-H), 9.21 (s, 1H, Pz 5-H). 13C NMR (176 MHz, DMSO-d6): δH ppm 57.1 (CH3), 111.6 (Pz C-4), 118.6 (Ph C-2,6), 123.3 (C(O)CHCH), 127.1 (Ph C-4), 129.1 (Th C-5), 130.0 (Ph C-3,5), 130.2 (Th C-4), 133.2 (Pz C-5), 133.3 (Th C-3), 135.0 (C(O)CHCH), 139.2 (Ph C-1), 140.3 (Th C-2), 162.8 (Pz C-3), 181.4 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.9 (Pz N-1), −119.4 (Pz N-2). HRMS (ESI+) for C17H14N2O2S ([M + Na]+) calcd 333.0668, found 333.0668.
(2E)-3-(Furan-3-yl)-1-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-one (9g)
The reaction mixture was stirred for 1 h. Yellow solid; yield 77% (227 mg); m.p. 136–137 °C; Rf = 0.34 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3122, 3063, 1658 (C=O), 1558, 1501, 1461, 1404, 1220, 1014, 742, 681, 631, 595. 1H NMR (700 MHz, DMSO-d6): δH ppm 4.05 (s, 3H, CH3), 6.67–6.68 (m, 1H, Furanyl 4-H), 7.01 (m, 1H, Furanyl 5-H), 7.33–7.35 (m, 1H, Ph 4-H), 7.40 (d, J = 14.0 Hz, 1H, C(O)CHCH), 7.48–7.53 (m, 3H, Ph 3,5-H, C(O)CHCH), 7.90–7.91 (m, 3H, Ph 2,6-H, Furanyl 2-H), 9.16 (s, 1H, Pz 5-H). 13C NMR (176 MHz, DMSO-d6): δC ppm 56.6 (CH3), 111.3 (Pz C-4), 113.0 (Furanyl C-4), 116.8 (Furanyl C-5), 118.1 (Ph C-2,6), 121.2 (C(O)CHCH), 126.7 (Ph C-4), 128.3 (C(O)CHCH), 129.5 (Ph C-3,5), 132.7 (Pz C-5), 138.7 (Ph C-1), 145.9 (Furanyl C-2), 151.1 (Furanyl C-3), 162.2 (Pz C-3), 181.1 (C=O). 15N NMR (71 MHz, DMSO-d6): δN ppm −181.6 (Pz N-1), −119.0 (Pz N-2). HRMS (ESI+) for C17H14N2O3 ([M + Na]+) calcd 317.0897, found 317.0897.
(2E)-3-Phenyl-1-(1-phenyl-3-propoxy-1H-pyrazol-4-yl)prop-2-en-1-one (9h)
The reaction mixture was stirred for 1 h. Yellow solid; yield 96% (320 mg); m.p. 131–132 °C; Rf = 0.77 (DCM/MeOH 100/1, v/v). IR (νmax, cm−1): 3118, 3082, 1657 (C=O), 1597, 1561, 1491, 1447, 1349, 1330, 1222, 761, 746, 681, 638. 1H NMR (400 MHz, DMSO-d6): δH ppm 1.05 (t, J = 7.4 Hz, 3H, CH3), 1.85 (sext, J = 7.1 Hz, 2H, CH3CH2CH2), 4.33 (t, J = 6.5 Hz, 2H, CH3CH2CH2), 7.32–7.36 (m, 1H, NPh 4-H), 7.44–7.54 (m, 5H, NPh 3,5-H, CPh 3,4,5-H), 7.64–7.72 (m, 2H, C(O)CHCHPh), 7.74–7.78 (m, 2H, CPh 2,6-H), 7.86–7.90 (m, 2H, NPh 2,6-H), 9.20 (s, 1H, Pz 5-H). 13C NMR (101 MHz, DMSO-d6): δC ppm 10.4 (CH3), 22.0 (CH3CH2CH2), 70.6 (CH3CH2CH2), 111.5 (Pz C-4), 118.1 (NPh C-2,6), 124.5 (C(O)CHCHPh), 126.7 (NPh C-4), 128.3 (CPh C-2,6), 129.0 (NPh C-3,5), 129.5 (CPh C-3,5), 130.4 (CPh C-4), 132.6 (Pz C-5), 134.7 (CPh C-1), 138.7 (NPh C-1), 141.3 (C(O)CHCHPh), 161.9 (Pz C-3), 181.5 (C=O). HRMS (ESI+) for C21H20N2O2 ([M + Na]+) calcd 355.1417, found 355.1417.
(2E)-1-[3-(2-Methoxyethoxy)-1-phenyl-1H-pyrazol-4-yl]-3-phenylprop-2-en-1-one (9i)
The reaction mixture was stirred for 1 h. Yellow solid; yield 50% (175 mg); m.p. 133–134 °C; Rf = 0.43 (DCM/MeOH 100/1, v/v). IR (νmax, cm−1): 3114, 3089, 1656 (C=O), 1596, 1556, 1493, 1468, 1371, 1220, 766, 750, 686, 677. 1H NMR (400 MHz, DMSO-d6): δH ppm 3.39 (s, 3H, CH3), 3.78–3.80 (m, 2H, CH3OCH2CH2), 4.49–4.51 (m, 2H, CH3OCH2CH2), 7.33–7.37 (m, 1H, NPh 4-H), 7.46–7.55 (m, 5H, NPh 3,5-H, CPh 3,4,5-H), 7.66 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.75–7.79 (m, 3H, C(O)CHCHPh, CPh 2,6-H), 7.88–7.90 (m, 2H, NPh 2,6-H), 9.19 (s, 1H, Pz 5-H). 13C NMR (101 MHz, DMSO-d6): δC ppm 58.3 (CH3), 68.5 (CH3OCH2CH2), 70.2 (CH3OCH2CH2), 111.5 (Pz C-4), 118.2 (NPh C-2,6), 124.5 (C(O)CHCHPh), 126.7 (NPh C-4), 128.4 (CPh C-2,6), 129.0 (CPh C-3,5), 129.5 (NPh C-3,5), 130.4 (CPh C-4), 132.7 (Pz C-5), 134.7 (CPh C-1), 138.7 (NPh C-1), 141.3 (C(O)CHCHPh), 161.7 (Pz C-3), 181.5 (C=O). HRMS (ESI+) for C21H20N2O3 ([M + Na]+) calcd 371.1366, found 371.1366.
3.2.8. General Procedure for the Synthesis of 12a–j
To a solution of an appropriate pyrazole ethanone (11a,b) (50 mg, 0.19 mmol) in EtOH (96%, 2 mL), NaOH (75 mg, 1.9 mmol) and appropriate benzaldehyde (0.47 mmol) were added. The reaction mixture was heated at 55 °C for 10 min. After the completion of the reaction as monitored by TLC, EtOH was evaporated, the mixture was diluted with water (10 mL) and extracted with ethyl acetate (3 × 10 mL). The organic layers were combined, washed with brine, dried over sodium sulphate, filtrated off, and the solvent was evaporated. The residue was purified by flash column chromatography (SiO2, eluent: ethyl acetate/n-hexane, 1:2, v/v) to provide the desired compounds 12a–j.
(2E/Z)-3-Phenyl-1-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12a,13a)
A mixture of isomers was obtained in the ratio E-12a:Z-13a = 1:0.15. Yellow solid; yield 70% (55 mg); m.p. 169–170.8 °C; Rf = 0.67 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 6.45 (d, J = 12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 6.91 (d, J =12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 7.12 (d, J =15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.36–7.42 (m, 5H, NPh 4-H, CPh 3,4,5-H, Pyr 5-H), 7.49–7.50 (m, 2H, CPh 2,6-H), 7.52–7.55 (m, 2H, NPh 3,5-H), 7.64–7.66 (m, 2H, NPh 2,6-H of minor isomer), 7.76 (d, J = 15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.82–7.83 (m, 2H, NPh 2,6-H), 8.18 (dd, JPyr 4-H,5-H = 7.8 Hz, JPyr 4-H,6-H = 1.8 Hz, 1H, Pyr 4-H), 8.32 (s, 1H, Pz 5-H of minor isomer), 8.59 (s, 1H, Pz 5-H of major isomer), 8.65 (d, J = 4.8 Hz, 1H, Pyr 4-H of minor isomer), 8.68 (d, JPyr 5-H,6-H = 4.8 Hz, 1H, Pyr 6-H of major isomer), 9.04 (s, 1H, Pyr 2-H of minor isomer), 9.07 (s, 1H, Pyr 2-H of major isomer). 13C NMR (176 MHz, CDCl3): δC ppm 119.8 (NPh C-2,6), 123.0 (Pyr C-5), 123.5 (Pz C-4), 124.4 (C(O)CH=CHPh), 128.1 (NPh C-4), 128.5 (CPh C-2,6), 128.7 (Pyr C-3), 129.1 (CPh C-3,5), 129.9 (NPh C-3,5), 130.8 (CPh C-4), 131.5 (Pz C-5), 134.7 (CPh C-1), 137.1 (Pyr C-4), 139.2 (NPh C-1), 144.2 (C(O)CH=CHPh), 149.9 (Pyr C-6), 150.2 (Pyr C-2), 150.9 (Pz C-3), 184.4 (CO). 15N NMR (71 MHz, CDCl3): δN ppm −162.0 (Pz N-1), −71.4 (Pyr N). HRMS (ESI+) for C23H18N3O ([M + H]+) calcd 352.1444, found 352.1444.
(2E/Z)-3-(4-Methylphenyl)-1-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12b,13b)
A mixture of isomers was obtained in the ratio E-12b:Z-13b = 1:0.17. Yellow crystals; yield 61% (42 mg); m.p. 150.2–151.3 °C; Rf=0.61 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 1231, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 2.29 (s, 3H, CH3 of minor isomer), 2.38 (s, 3H, CH3 of major isomer), 6.40 (d, J =12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 6.86 (d, J =12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 7.07 (d, J =15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.19 (d, J = 7.8 Hz, 2H, CPh 3,5-H), 7.36–7.42 (m, 4H, NPh 4-H, CPh 2,6-H, Pyr 5-H), 7.47–7.49 (m, 2H, NPh 3,5-H of minor isomer), 7.51–7.54 (m, 2H, NPh 3,5-H of major isomer), 7.65–7.67 (m, 2H, NPh 2,6-H of minor isomer), 7.74 (d, J = 15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.81–7.83 (m, 2H, NPh 2,6-H), 8.17 (dt, JPyr 4-H,5-H = 7.9 Hz, J = 2.0 Hz, 1H, Pyr 4-H), 8.34 (s, 1H, Pz 5-H of minor isomer), 8.57 (s, 1H, Pz 5-H, of major isomer), 8.67 (dd, JPyr 5-H,6-H = 4.9 Hz, J = 1.7 Hz 1H, Pyr 4-H), 9.04 (d, J = 2.2 Hz, 1H, Pyr 2-H of minor isomer), 9.07 (d, J = 2.0 Hz, 1H, Pyr 2-H of major isomer). 13C NMR (176 MHz, CDCl3): δC ppm 21.7 (CH3), 119.8 (NPh C-2,6), 123.0 (Pyr C-5), 123.4 (Pz C-4), 123.6 (C(O)CHCHPh), 128.0 (NPh C-4), 128.6 (CPh C-2,6), 128.8 (Pyr C-3), 129.2 129.9 (CPh C-3,5, NPh C-3,5), 131.4 (Pz C-5), 131.9 (CPh C-1), 137.1 (Pyr C-4), 139.2 (NPh C-1), 141.4 (CPh C-4), 144.3 (C(O)CHCHPh), 149.9 (Pyr C-6), 150.2 (Pyr C-2), 150.8 (Pz C-3), 184.6 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.9 (Pz N-1), −70.8 (Pyr N). HRMS (ESI+) for C24H20N3O ([M + H]+) calcd 366.1601, found 366.1601.
(2E/Z)-1-[1-Phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]-3-[4-(trifluoromethoxy)phenyl]prop-2-en-1-one (12c,13c)
A mixture of isomers was obtained in the ratio E-12c:Z-13c = 1:0.4. Yellow crystals; yield 55% (46 mg); m.p. 183–184 °C; Rf = 0.57 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 1165, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 6.52 (d, J =12.8 Hz, 1H, C(O)CHCHPh of minor isomer), 6.84 (d, J =12.8 Hz, 1H, C(O)CHCHPh of minor isomer), 7.06 (d, J =15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.12–7.13 (m, 2H, CPh 3,5-H of minor isomer), 7.22–7.23 (m, 2H, CPh 3,5-H of major isomer), 7.37–7.43 (m, 2H, NPh 4-H, Pyr 5-H), 7.49–7.55 (m, 4H, CPh 2,6-H, NPh 3,5-H), 7.73 (d, J = 15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.81–7.83 (m, 2H, NPh 2,6-H), 8.17 (d, JPyr 4-H,5-H = 7.8 Hz, 1H, Pyr 4-H), 8.41 (s, 1H, Pz 5-H of minor isomer), 8.59 (s, 1H, Pz 5-H of major isomer), 8.66 (d, JPyr 5-H,6-H = 4.7 Hz, 1H, Pyr 6-H of major isomer), 8.69 (d, JPyr 5-H,6-H = 4.3 Hz, 1H, Pyr 6-H of minor isomer), 9.03 (s, 1H, Pyr 2-H of minor isomer), 9.06 (s, 1H, Pyr 2-H of major isomer). 13C NMR (176 MHz, CDCl3): δC ppm 119.8 (NPh C-2,6), 121.3 (CPh C-3,5), 123.1 (Pyr C-5), 123.4 (Pz C-4), 125.1 (C(O)CHCHPh), 127.9 (NPh C-4 of minor isomer), 128.2 (NPh C-4 of major isomer), 128.7 (Pyr C-3), 129.9 (CPh C-2,6, NPh C-3,5), 130.5 (q, 1J = 254.6 Hz, OCF3), 131.6 (Pz C-5 of major isomer), 133.3 (CPh C-1 of major isomer), 133.6 (CPh C-1 of minor isomer), 137.0 (Pyr C-4 of minor isomer), 137.1 (Pyr C-4 of major isomer), 138.6 (NPh C-1 of minor isomer), 139.1 (NPh C-1 of major isomer), 142.3 (C(O)CHCHPh), 150.0 (Pyr C-6), 150.2 (Pyr C-2), 150.7 (Pz C-3), 150.9 (CPh C-4), 184.0 (C(O)CHCHPh of major isomer), 186.8 (C(O)CHCHPh of minor isomer). 15N NMR (71 MHz, CDCl3): δN ppm −161.6 (Pz N-1), −70.9 (Pyr N). HRMS (ESI+) for C24H17N3O2F3 ([M + H]+) calcd 436.1267, found 436.1267.
(2E/Z)-3-Phenyl-1-[1-phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12d,13d)
A mixture of isomers was obtained in the ratio E-12d:Z-13d = 1:0.18. White crystals; yield 58% (39 mg); m.p. 162.8–165 °C; Rf = 0.29 (EtOAc/Hex 1/4, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 6.47 (d, J =12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 6.93 (d, J =12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 7.12 (d, J =15.7 Hz, 1H, C(O)CHCHPh of major isomer), 7.38–7.42 (m, 4H, NPh 4-H, CPh 3,4,5-H), 7.48–7.51 (m, 2H, CPh 2,6-H), 7.52–7.54 (m, 2H, NPh 3,5-H), 7.63–7.64 (m, 2H, NPh 2,6-H of minor isomer), 7.76 (d, J = 15.7 Hz, 1H, C(O)CHCHPh of major isomer), 7.79–7.83 (m, 4H, Pyr 3,5-H, NPh 2,6-H), 8.31 (s, 1H, Pz 5-H of minor isomer), 8.56 (s, 1H, Pz 5-H of major isomer), 8.68–8.71 (m, 2H, Pyr 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 119.8 (NPh C-2,6), 123.6 (Pz C-4), 123.8 (Pyr C-3,5), 124.5 (C(O)CHCHPh), 128.2 (NPh C-4), 128.6 (CPh C-2,6), 129.1 (CPh C-3,5), 129.9 (NPh C-3,5), 130.9 (CPh C-4), 131.7 (Pz C-5), 134.5 (CPh C-1), 139.1 (NPh C-1), 140.3 (Pyr C-4), 144.4 (C(O)CHCHPh), 149.9 (Pyr C-2,6), 151.1 (Pz C-3), 184.6 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.5 (Pz N-1), −70.6 (Pyr N). HRMS (ESI+) for C23H18N3O ([M + H]+) calcd 352.1444, found 352.1444.
(2E/Z)-3-(4-Methylphenyl)-1-[1-phenyl-3-(pyridine-4-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12e,13e)
A mixture of isomers was obtained in the ratio E-12e:Z-13e = 1:0.13. Yellow crystals; yield 58% (40 mg); m.p. 176–177 °C; Rf=0.35 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 2.29 (s, 3H, CH3 of minor isomer), 2.38 (s, 3H, CH3 of major isomer), 6.42 (d, J =12.7 Hz, 1H, C(O)CHCHPh of minor isomer), 6.89 (d, J =12.7 Hz, 1H, C(O)CH=CHPh of minor isomer), 7.07 (d, J =15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.19 (d, J = 7.8 Hz, 2H, CPh 3,5-H), 7.38–7.42 (m, 3H, NPh 4-H, CPh 2,6-H), 7.47–7.50 (m, 2H, NPh 3,5-H of minor isomer), 7.52–7.54 (m, 2H, NPh 3,5-H of major isomers), 7.64–7.66 (m, 2H, NPh 2,6-H of minor isomer), 7.74 (d, J = 15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.79–7.83 (m, 4H, Pyr 3,5-H, NPh 2,6-H), 8.32 (s, 1H, Pz 5-H of minor isomer), 8.55 (s, 1H, Pz 5-H of major isomer), 8.70 (d, 3J = 5.3 Hz, 1H, Pyr 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 21.7 (CH3), 119.9 (NPh C-2,6), 123.6 (C(O)CHCHPh), 123.7 (Pz C-4), 123.8 (Pyr C-3,5), 128.1 (NPh C-4), 128.6 (CPh C-2,6), 128.8 (Pyr C-1), 129.9 (CPh C-3,5, NPh C-3,5), 131.6 (Pz C-5), 131.8 (CPh C-1), 139.1 (NPh C-1), 141.5 (CPh C-4), 144.6 (C(O)CHCHPh), 149.9 (Pyr C-2,6), 151.0 (Pz C-3), 184.8 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.7 (Pz N-1), −70.2 (Pyr N). HRMS (ESI+) for C24H20N3O ([M + H]+) calcd 366.1601, found 366.1601.
(2E/Z)-1-[1-Phenyl-3-(pyridine-4-yl)-1H-pyrazol-4-yl]-3-[4-(trifluoromethoxy)phenyl]prop-2-en-1-one (12f,13f)
A mixture of isomers was obtained in ratio E-12f:Z-13f = 1:0.24. Yellow crystals; yield 51% (42 mg); m.p. 168–170 °C; Rf = 0.42 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 1165, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 6.54 (d, J =12.8 Hz, 1H, C(O)CHCHPh of minor isomer), 6.87 (d, J =12.8 Hz, 1H, C(O)CHCHPh of minor isomer), 7.07 (d, J =15.6 Hz, 1H, C(O)CHCHPh of major isomer), 7.12–7.13 (m, 2H, CPh 3,5-H of minor isomer), 7.20–7.24 (m, 2H, CPh 3,5-H of major isomer), 7.40–7.43 (m, 1H, NPh 4-H), 7.49–7.56 (m, 4H, CPh 2,6-H, NPh 3,5-H), 7.68–7.69 (m, 2H, NPh 2,6-H of minor isomer), 7.73 (d, J = 15.7 Hz, 1H, C(O)CHCHPh of major isomer), 7.79–7.82 (m, 4H, NPh 2,6-H, Pyr 3,5-H), 8.57 (s, 1H, Pz 5-H), 8.67–8.77 (m, 2H, Pyr 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 119.8 (NPh C-2,6 of minor isomer), 119.9 (NPh C-2,6 of major isomer), 120.7 (CPh C-3,5 of minor isomer), 121.4 (CPh C-3,5 of major isomers), 123.5 (Pyr C-3,5), 123.9 (Pz C-4), 125.2 (C(O)CH=CHPh), 127.9 (NPh C-4 of minor isomer), 128.3 (NPh C-4 of major isomer), 128.4 (Pyr C-4), 129.9 (NPh C-3,5 of major isomer), 130.0 (CPh C-2,6), 130.7 (q, 1J = 266.6 Hz, OCF3), 131.7 (Pz C-5), 133.1 (CPh C-1), 138.9 (NPh C-1 of minor isomer), 139.1 (NPh C-1 of major isomer), 140.3 (Pyr C-4 of major isomer), 140.7 (Pyr C-4 of minor isomer), 142.5 (C(O)CHCHPh), 149.8 (Pyr C-2,6 of minor isomer), 149.9 (Pyr C-2,6 of major isomer), 150.8 (CPh C-4 of major isomer), 151.0 (CPh C-4 of minor isomer), 151.0 (Pz C-3 of minor isomer), 151.1 (Pz C-3 of major isomer), 184.1 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.2 (Pz N-1). HRMS (ESI+) for C24H17N3O2F3 ([M + H]+) calcd 436.1267, found 436.1267.
(2E)-3-(4-Chlorophenyl)-1-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12g)
White crystals; yield 67% (49 mg); m.p. 169.7–172.3 °C; Rf = 0.50 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 987, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 6.99 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.26 (d, J = 8.5 Hz, 2H, CPh 3,5-H), 7.28–7.33 (m, 4H, NPh 4-H, CPh 2,6-H, Pyr 5-H), 7.42–7.45 (m, 2H, NPh 3,5-H), 7.60 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.72–7.74 (m, 2H, NPh 2,6-H), 8.08 (dt, JPyr 4-H,5-H = 7.9 Hz, J = 2.0 Hz, 1H, Pyr 4-H), 8.51 (s, 1H, Pz 5-H), 8.58 (dd, JPyr 5-H,6-H = 4.9 Hz, J = 1.6 Hz, 1H, Pyr 6-H), 8.98 (d, J = 2.2 Hz, 1H, Pyr 2-H). 13C NMR (176 MHz, CDCl3): δC ppm 118.6 (NPh C-2,6), 121.9 (Pyr C-5), 122.2 (Pz C-4), 123.6 (C(O)CHCHPh), 126.9 (NPh C-4), 127.5 (Pyr C-3), 128.2 (CPh C-3,5), 128.5 (CPh C-2,6), 128.7 (NPh C-3,5), 130.4 (Pz C-5), 132.0 (CPh C-1), 135.5 (CPh C-4), 135.9 (Pyr C-4), 138.0 (NPh C-1), 141.4 (C(O)CHCHPh), 148.8 (Pyr C-6), 149.0 (Pyr C-2), 149.7 (Pz C-3), 182.8 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.8 (Pz N-1), −78.2 (Pz N-1), −70.6 (Pyr N). HRMS (ESI+) for C23H17N3OCl ([M + H]+) calcd 386.1055, found 386.1055.
(2E)-3-(4-Methoxyphenyl)-1-[1-phenyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12h)
Yellow crystals; yield 54% (39 mg); m.p. 157–159 °C; Rf = 0.51 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1659 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 3.84 (s, 3H, OCH3), 6.89 (d, J = 8.6 Hz, 2H, CPh 3,5-H), 6.99 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.37–7.41 (m, 2H, NPh 4-H, Pyr 5-H), 7.43–7.45 (m, 2H, CPh 2,6-H), 7.51–7.54 (m, 2H, NPh 3,5-H), 7.72 (d, J = 15.6 Hz, 1H, C(O)CH=CHPh), 7.81–7.83 (m, 2H, NPh 2,6-H), 8.17 (d, JPyr 4-H,5-H = 7.9 Hz, 1H, Pyr 4-H), 8.56 (s, 1H, Pz 5-H), 8.67 (d, JPyr 5-H,6-H = 4.2 Hz, 1H, Pyr 6-H), 9.07 (s, 1H, Pyr 2-H). 13C NMR (176 MHz, CDCl3): δC ppm 55.6 (OCH3), 114.6 (CPh C-3,5), 119.8 (NPh C-2,6), 122.1 (C(O)CHCHPh), 123.0 (Pyr C-5), 123.7 (Pz C-4), 127.4 (CPh C-1), 128.0 (NPh C-4), 128.8 (Pyr C-3), 129.9 (NPh C-3,5), 130.3 (CPh C-2,6), 131.3 (Pz C-5), 137.1 (Pyr C-4), 139.2 (NPh C-1), 144.1 (C(O)CHCHPh), 149.8 (Pyr C-6), 150.2 (Pyr C-2), 150.8 (Pz C-3), 161.9 (CPh C-4), 184.6 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −162.2 (Pz N-1), −70.9 (Pyr N). HRMS (ESI+) for C24H20N3O2 ([M + H]+) calcd 382.1550, found 382.1550.
(2E)-3-(4-Chlorophenyl)-1-[1-phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12i)
White crystals; yield 55% (40 mg); m.p. 205–207 °C; Rf = 0.41 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1684 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 987, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 7.07 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.34–7.37 (m, 2H, CPh 3,5-H), 7.40–7.43 (m, 3H, NPh 4-H, CPh 2,6-H), 7.52–7.55 (m, 2H, NPh 3,5-H), 7.71 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.77–7.79 (m, 2H, Pyr 3,5-H), 7.80–7.81 (m, 2H, NPh 2,6-H), 8.56 (s, 1H, Pz 5-H), 8.69–8.72 (m, 2H, Pyr 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 119.9 (NPh C-2,6), 123.5 (Pz C-4), 123.8 (Pyr C-3,5), 124.9 (C(O)CHCHPh), 128.2 (NPh C-4), 129.5 (CPh C-3,5), 129.7 (CPh C-2,6), 129.9 (NPh C-3,5), 131.7 (Pz C-5), 133.0 (CPh C-1), 136.8 (CPh C-4), 139.1 (NPh C-1), 140.3 (Pyr C-4), 142.9 (C(O)CHCHPh), 150.0 (Pyr C-2,6), 151.1 (Pz C-3), 184.2 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.3 (Pz N-1), −70.3 (Pyr N). HRMS (ESI+) for C23H17N3OCl ([M + H]+) calcd 386.1055, found 386.1055.
(2E)-3-(4-Methoxyphenyl)-1-[1-phenyl-3-(pyridin-4-yl)-1H-pyrazol-4-yl]prop-2-en-1-one (12j)
Yellow crystals; yield 50% (36 mg); m.p. 167–169 °C; Rf = 0.36 (EtOAc/Hex 1/2, v/v). IR (νmax, cm−1): 3120, 3041, 1659 (C=O), 1599, 1521, 1448, 1362, 1260, 1241, 1221, 977, 940, 863, 751, 705, 683. 1H NMR (700 MHz, CDCl3): δH ppm 3.84 (s, 3H, OCH3), 6.89–6.90 (m, 2H, CPh 3,5-H), 6.98 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.39–7.41 (m, 1H, NPh 4-H), 7.44–7.46 (m, 2H, CPh 2,6-H), 7.51–7.54 (m, 2H, NPh 3,5-H), 7.73 (d, J = 15.6 Hz, 1H, C(O)CHCHPh), 7.78–7.81 (m, 4H, NPh 2,6-H, Pyr 3,5-H), 8.54 (s, 1H, Pz 5-H), 8.69–8.70 (m, 2H, Pyr 2,6-H). 13C NMR (176 MHz, CDCl3): δC ppm 55.6 (OCH3), 114.6 (CPh C-3,5), 119.8 (NPh C-2,6), 122.3 (C(O)CHCHPh), 123.78 (Pyr C-3,5), 123.83 (Pz C-4), 127.2 (CPh C-1), 128.1 (NPh C-4), 129.9 (NPh C-3,5), 130.4 (CPh C-2,6), 131.5 (Pz C-5), 139.2 (NPh C-1), 140.4 (Pyr C-4), 144.3 (C(O)CHCHPh), 149.9 (Pyr C-2,6), 150.9 (Pz C-3), 162.0 (CPh C-4), 184.7 (C(O)CHCHPh). 15N NMR (71 MHz, CDCl3): δN ppm −161.8 (Pz N-1), −70.4 (Pyr N). HRMS (ESI+) for C24H20N3O2 ([M + H]+) calcd 382.1550, found 382.1550.
3.2.10. Procedure for the Preparation of Mixture of 3-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)-5-phenyl-15N-1,2-oxazole (16) and 5-(3-methoxy-1-phenyl-1H-pyrazol-4-yl)-3-phenyl-15N-1,2-oxazole (17)
To a solution of 15N hydroxylamine hydrochloride (139 mg, 2 mmol) and potassium hydroxide (160 mg, 4 mmol) in EtOH (96%, 15 mL), chalcone 9a was added (304 mg, 1 mmol). The reaction mixture was stirred at 80 °C for 3 h, poured into water (30 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were washed with brine (30 mL) and dried over sodium sulphate, filtrated and concentrated. The residue was purified by column chromatografy (SiO2, eliuent: hexane/ethyl acetate, 3/1, v/v) to produce an inseparable mixture of compounds 16 and 17 with a 23% yield. The inseparable mixture of regioisomers 16 (major) and 17 (minor) was obtained in a ratio of about 8:1.
Yellow solid; yield 23% (73 mg mixture); 1H NMR (700 MHz, CDCl3): δH ppm 4.15 (s, 3H, CH3 of minor regioisomer), 4.16 (s, 3H, CH3 of major regioisomer), 6.77 (d, J = 1.23 Hz, 1H, Ox 4-H of major regioisomer), 6.93 (d, J = 1.31 Hz, 1H, Ox 4-H of minor regioisomer), 7.27–7.29 (m, 1H, NPh 4-H of both regioisomers), 7.45–7.49 (m, 5H, NPh 3,5-H, CPh 3,5-H, CPh 4-H of both regioisomers), 7.67–7.69 (m, 2H, NPh 2,6-H of both regioisomers), 7.84–7.86 (m, 1H, NPh 2,6-H of minor regioisomer), 7.87–7.89 (m, 2H, CPh 2,6-H of major regioisomer), 8.24 (s, 1H, Pz 5-H of major regioisomer), 8.33 (s, 1H, Pz 5-H of minor regioisomer). 13C NMR (176 MHz, CDCl3): δC ppm 56.62 (CH3 of minor regioisomer), 56.73 (CH3 of major regioisomer), 97.51 (d, 2JC,N = 1.23 Hz, Ox C-4 of major regioisomer), 98.45 (d, 2JC,N = 1.11 Hz, Ox C-4 of minor regioisomer), 98.82 (d, 2JC,N = 8.40 Hz, Pz C-4 of minor regioisomer), 98.96 (d, 3JC,N = 0.66 Hz, Pz C-4 of major regioisomer), 118.12 (NPh C-2,6 of minor regioisomer), 118.20 (NPh C-2,6 of major regioisomer), 125.53 (Pz C-5 of major regioisomer), 125.88 (CPh C-2,6 of minor regioisomer), 126.03 (d, 3JC,N = 1.47 Hz, Pz C-5), 126.06 (NPh C-4 of minor regioisomer), 126.27 (NPh C-4 of major regioisomer), 126.91 (d, 3JC,N = 2.28 Hz, CPh C-2,6 of major regioisomer), 127.59 (CPh C-1 of minor regioisomer), 128.85 (CPh C-3,5 of major regioisomer), 128.94 (NPh C-3,5 of minor regioisomer), 129.28 (d, 2JC,N = 7.11 Hz, CPh C-1 of major regioisomer), 129.50 (CPh C-3,5 of minor regioisomer), 129.54 (NPh C-3,5 of major regioisomer), 129.89 (CPh C-4 of major regioisomer), 130.06 (CPh C-4 of minor regioisomer), 139.51 (NPh C-1 of major regioisomer), 139.68 (NPh C-1 of minor regioisomer), 155.35 (d, 1JC,N = 2.25 Hz, Ox C-3 of minor regioisomer), 161.09 (Pz C-3 of major regioisomer), 162.27 (d, 3JC,N = 1.92 Hz, Pz C-3 of minor regioisomer), 162.77 (d, 1JC,N = 2.89 Hz, Ox C-3 of major regioisomer), 162.84 (d, 2JC,N = 1.39 Hz, Ox C-5 of major regioisomer), 169.61 (d, 2JC,N = 1.52 Hz, Ox C-5 of minor regioisomer). 15N NMR (71 MHz, CDCl3): δN ppm −184.8 (Pz N-1 of minor regioisomer), −184.5 (Pz N-1 of major regioisomer), −119.9 (Pz N-2 of minor regioisomer), −119.7 (Pz N-2 of major regioisomer), −19.5 (Ox N of minor regioisomer), −18.6 (Ox N of major regioisomer). HRMS (ESI+) for C19H1515NN2NaO2 ([M + Na]+) calcd 340.1056, found 340.1061.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}