
Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Protein Target and Ligands
2.2. Virtual Molecular Docking of Coumarin Derivatives and Its Validation with Molecular Dynamics Simulations
2.3. Pharmacophore Studies
2.4. Molecular Docking and Dynamics Simulation of Selected Compounds
2.5. Binding Free Energy Calculations
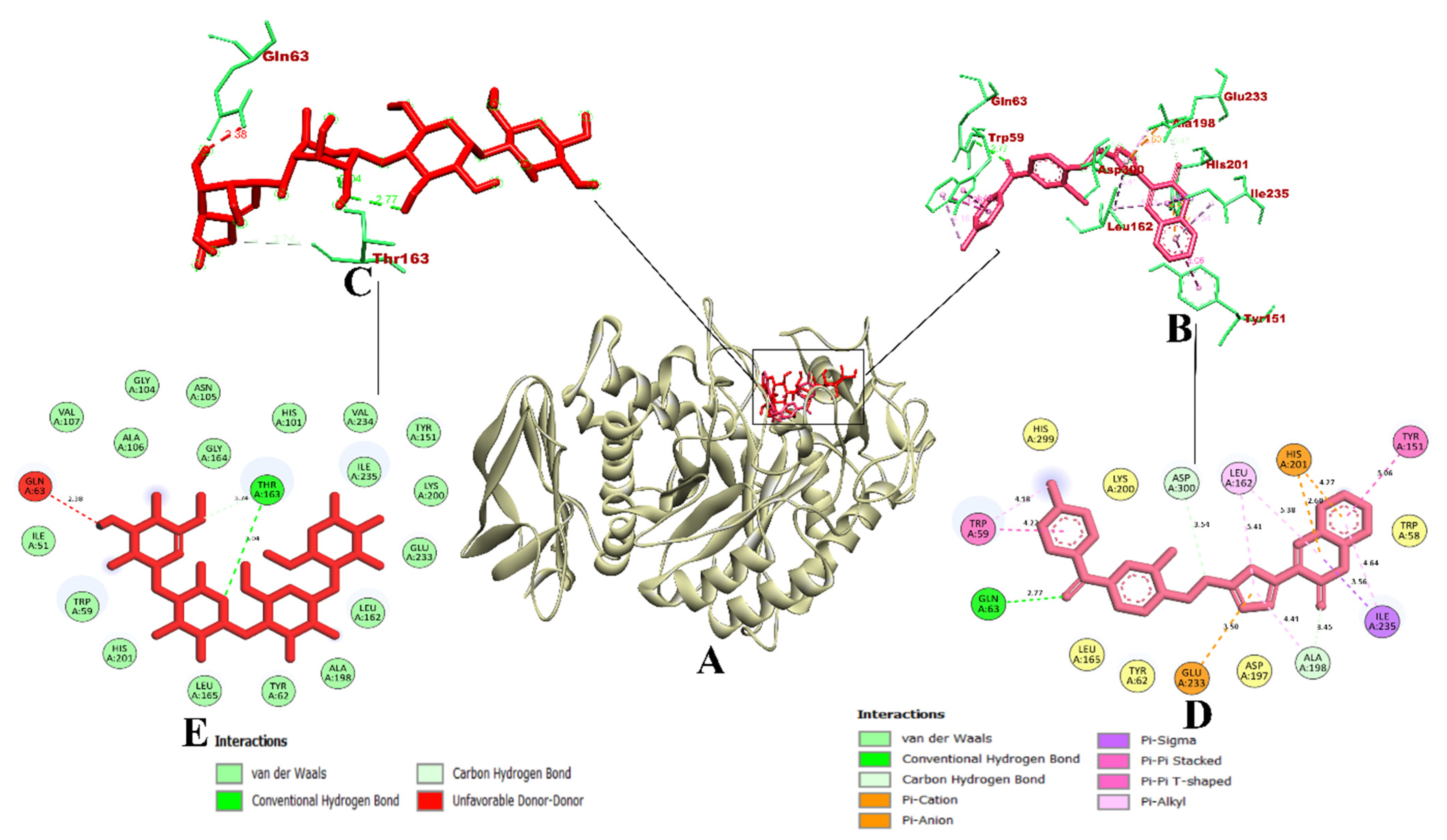
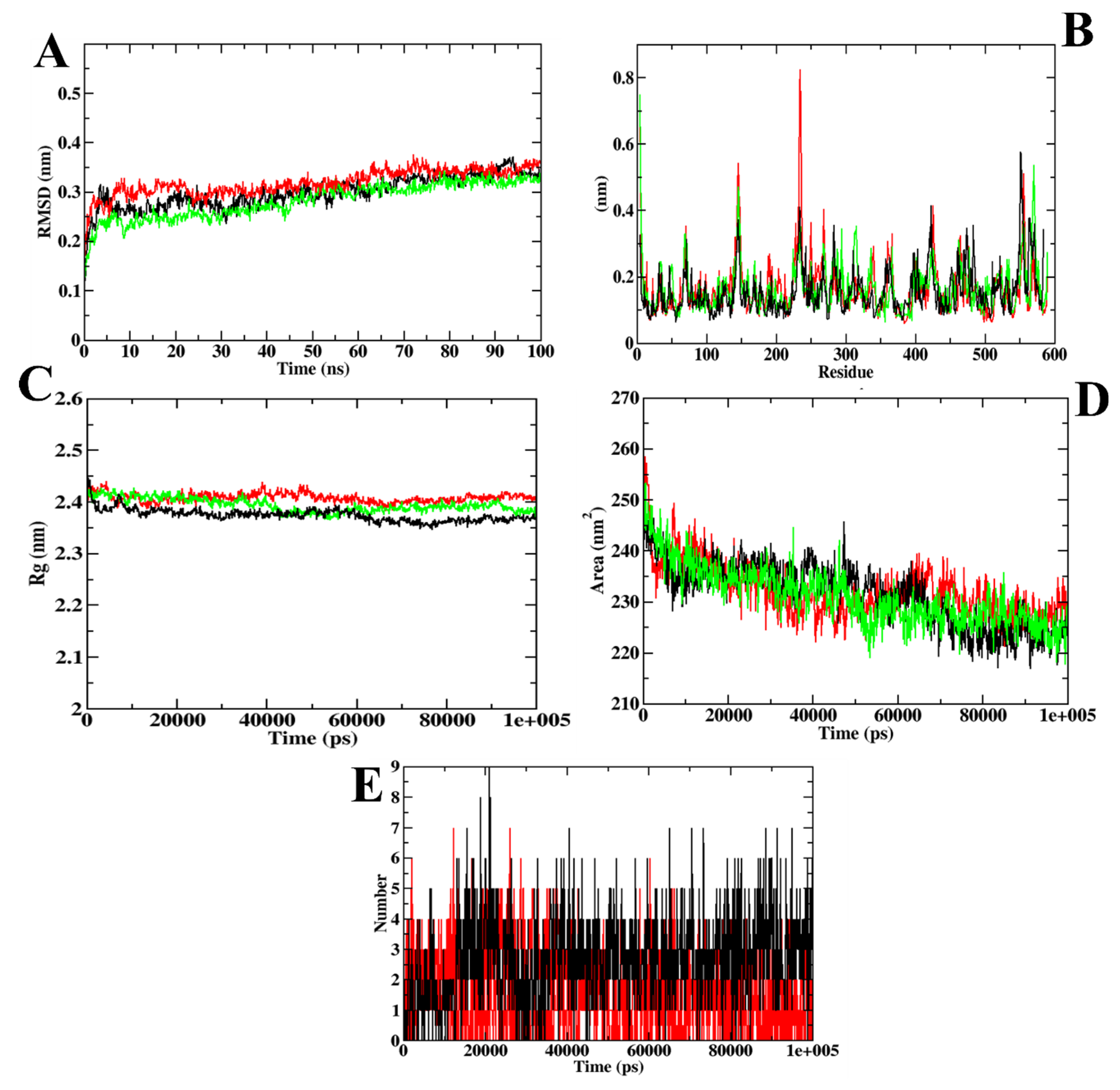
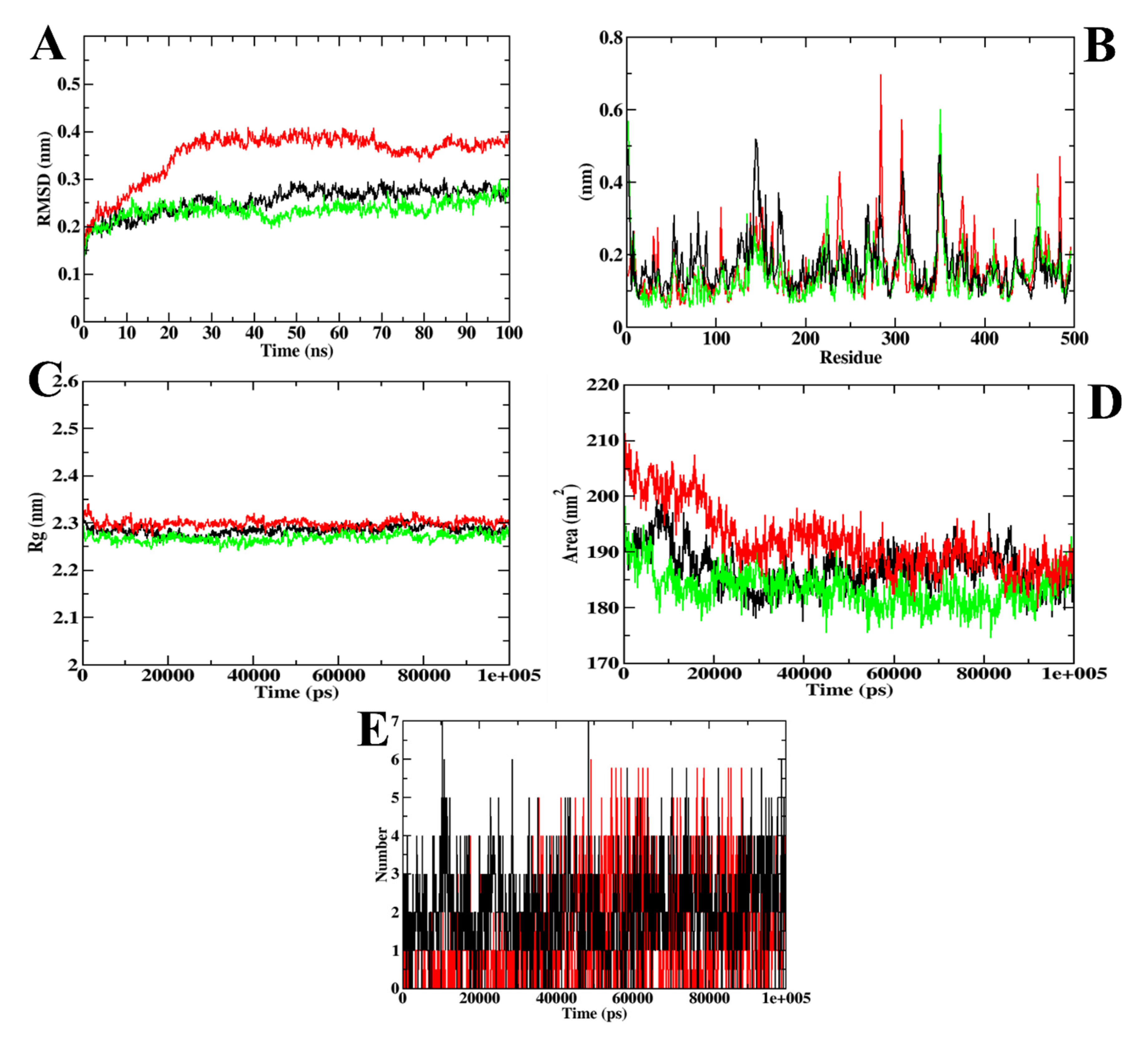
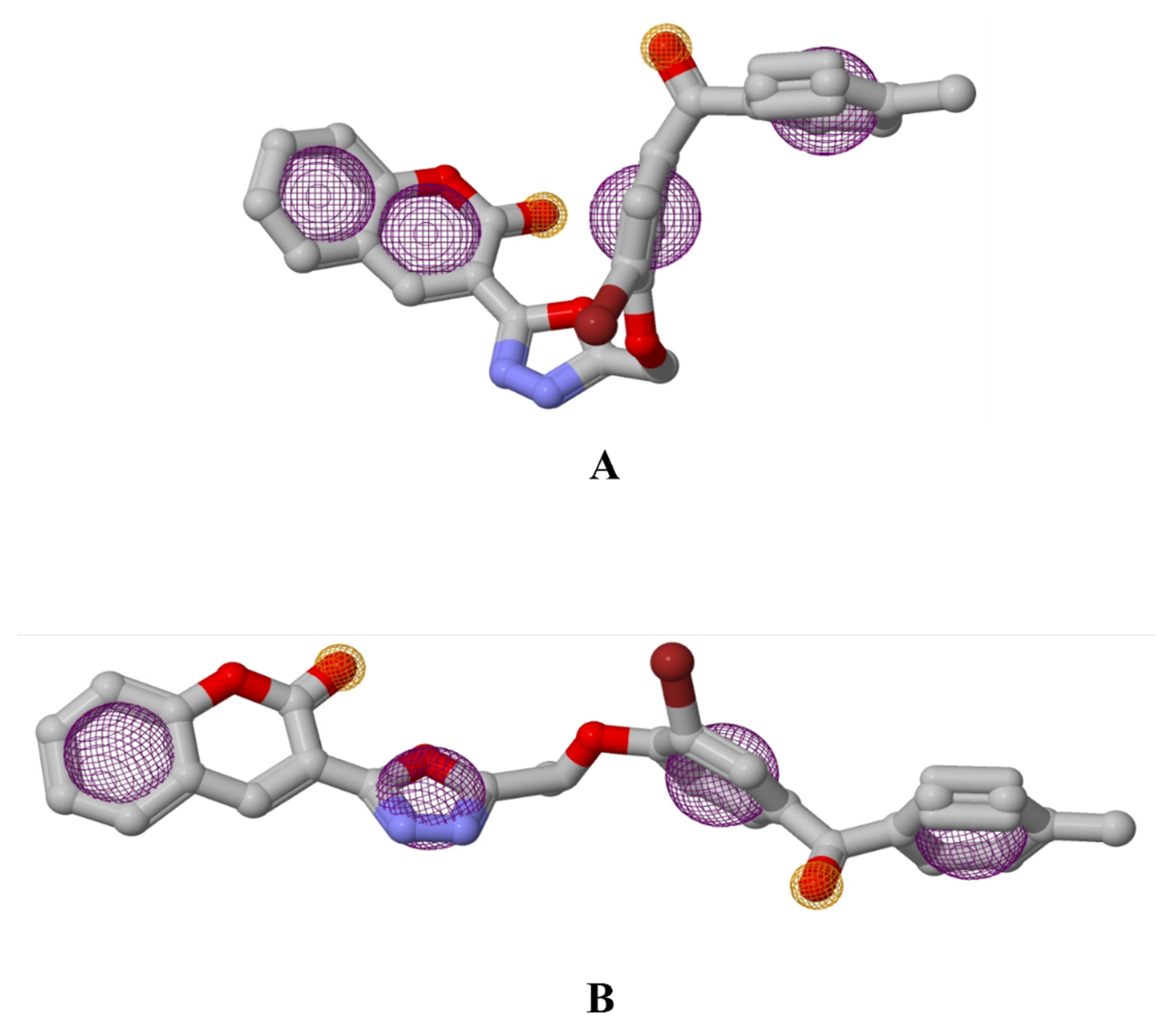
3. Results and Discussion
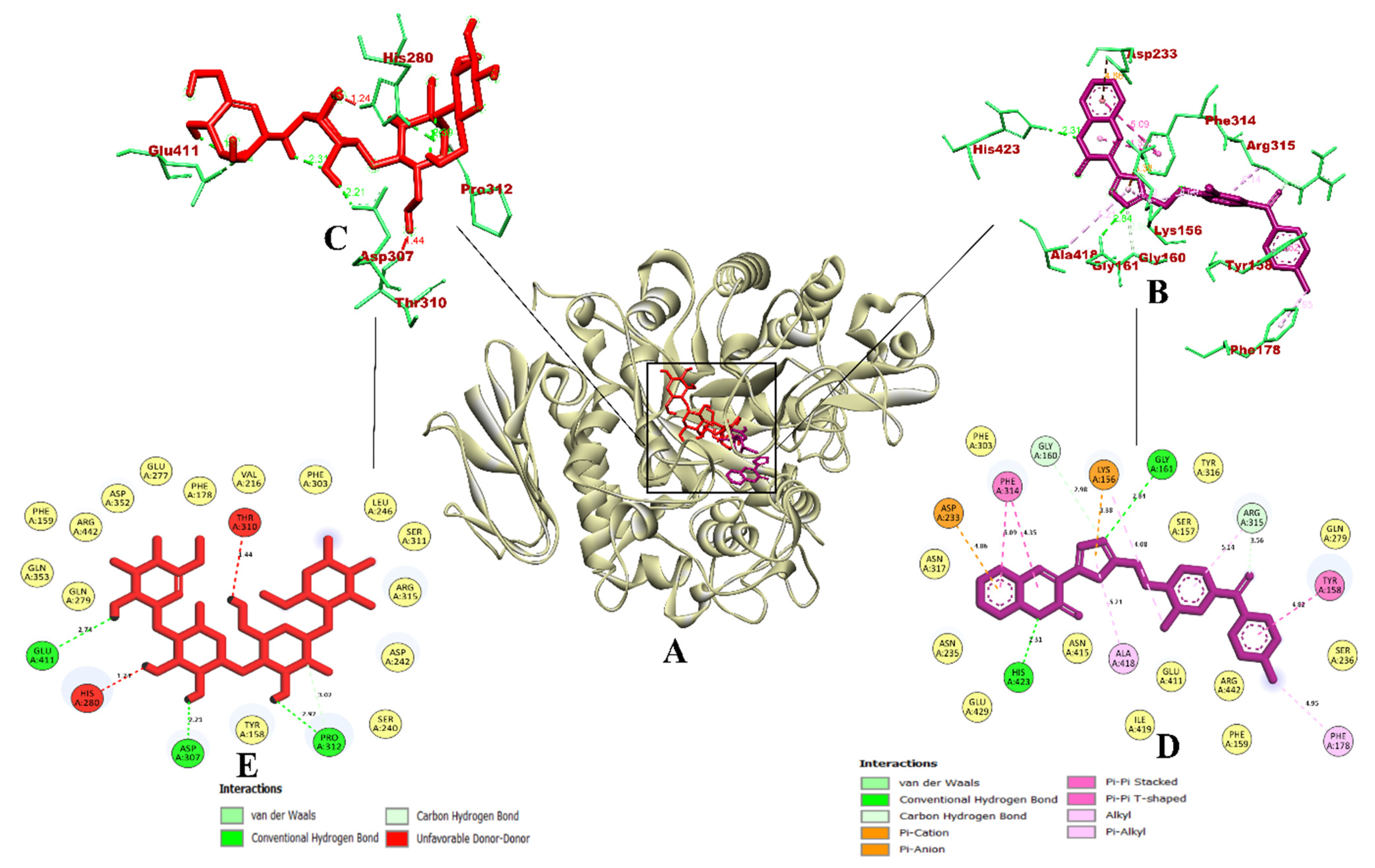
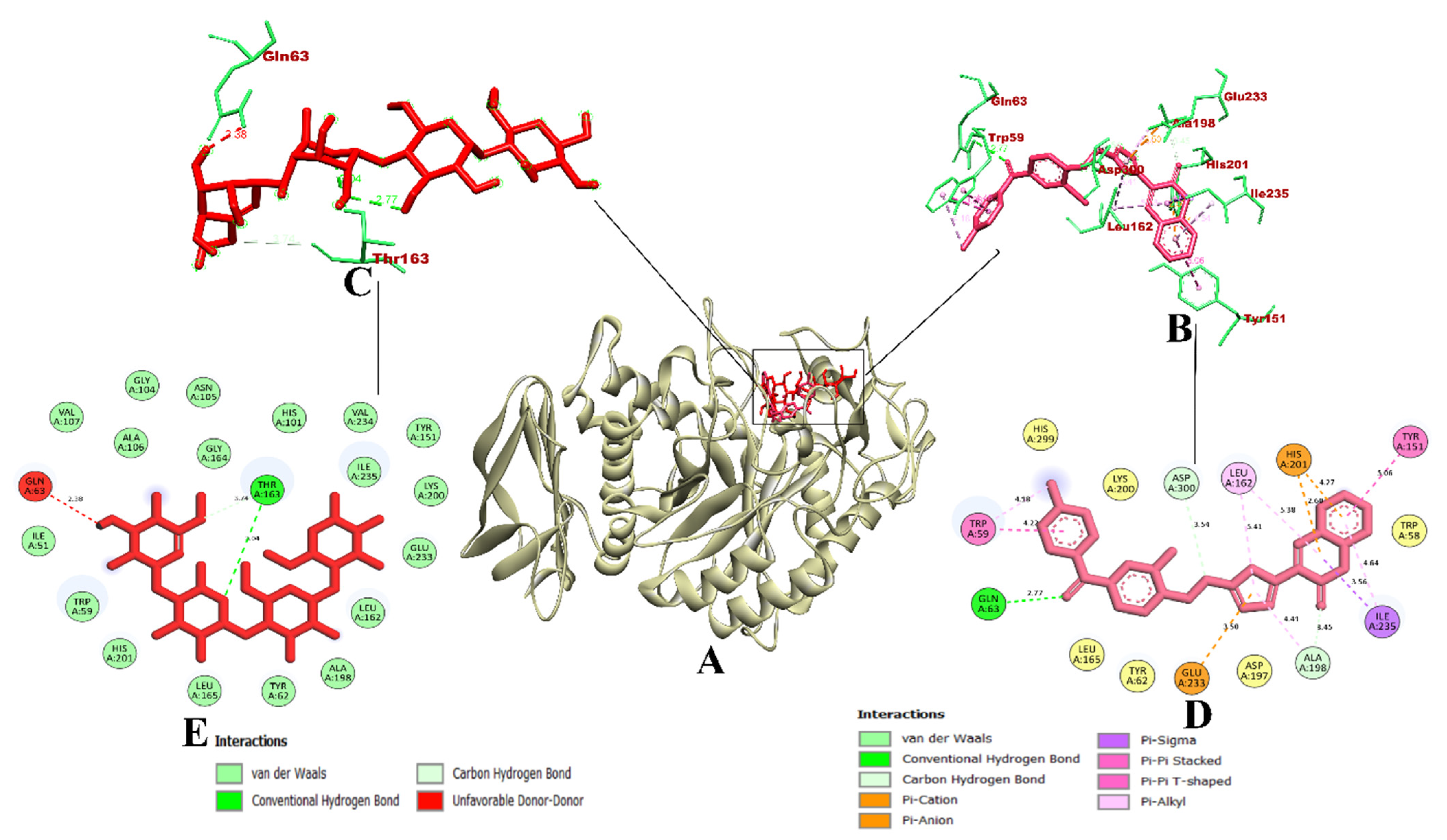
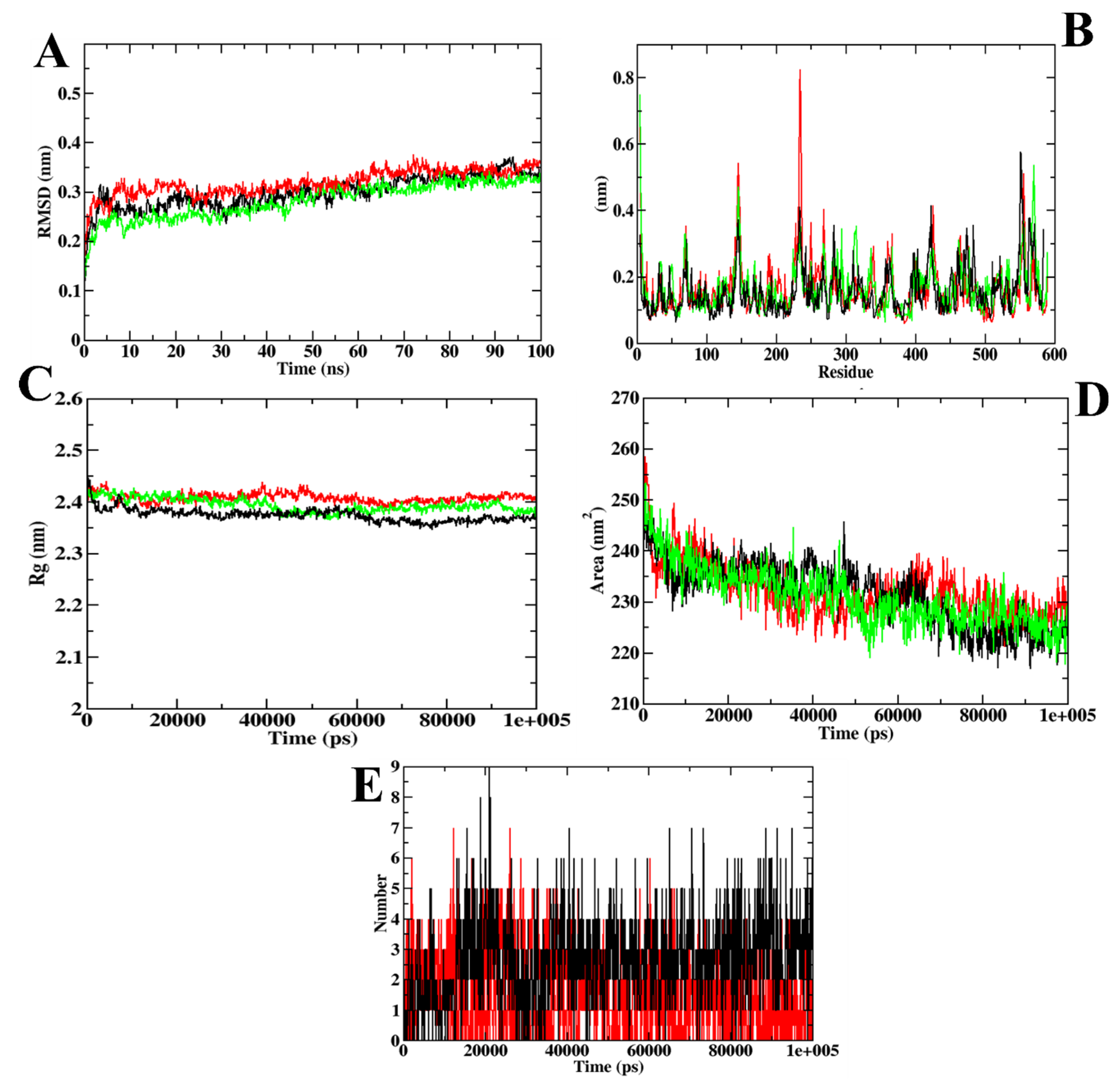
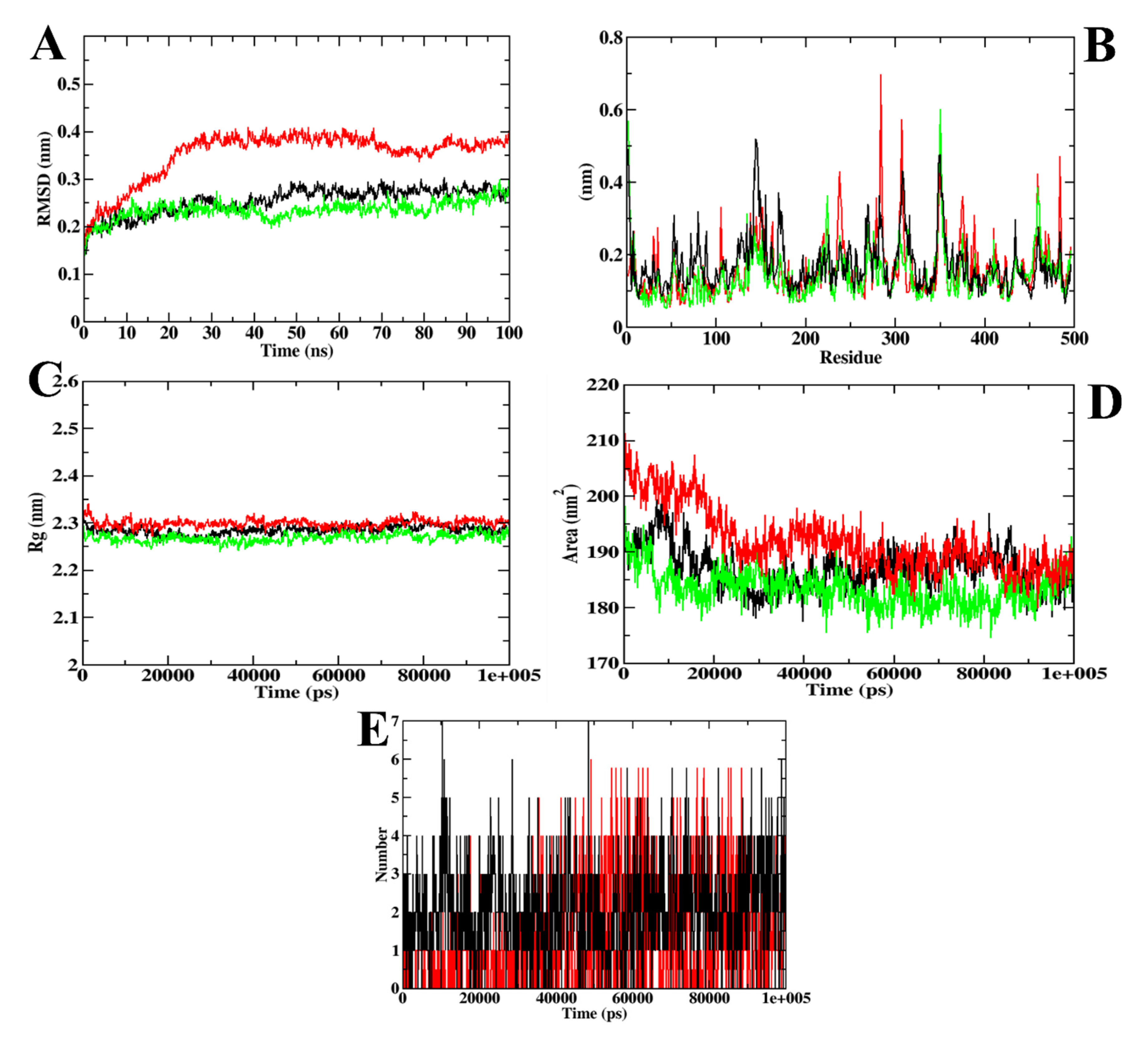
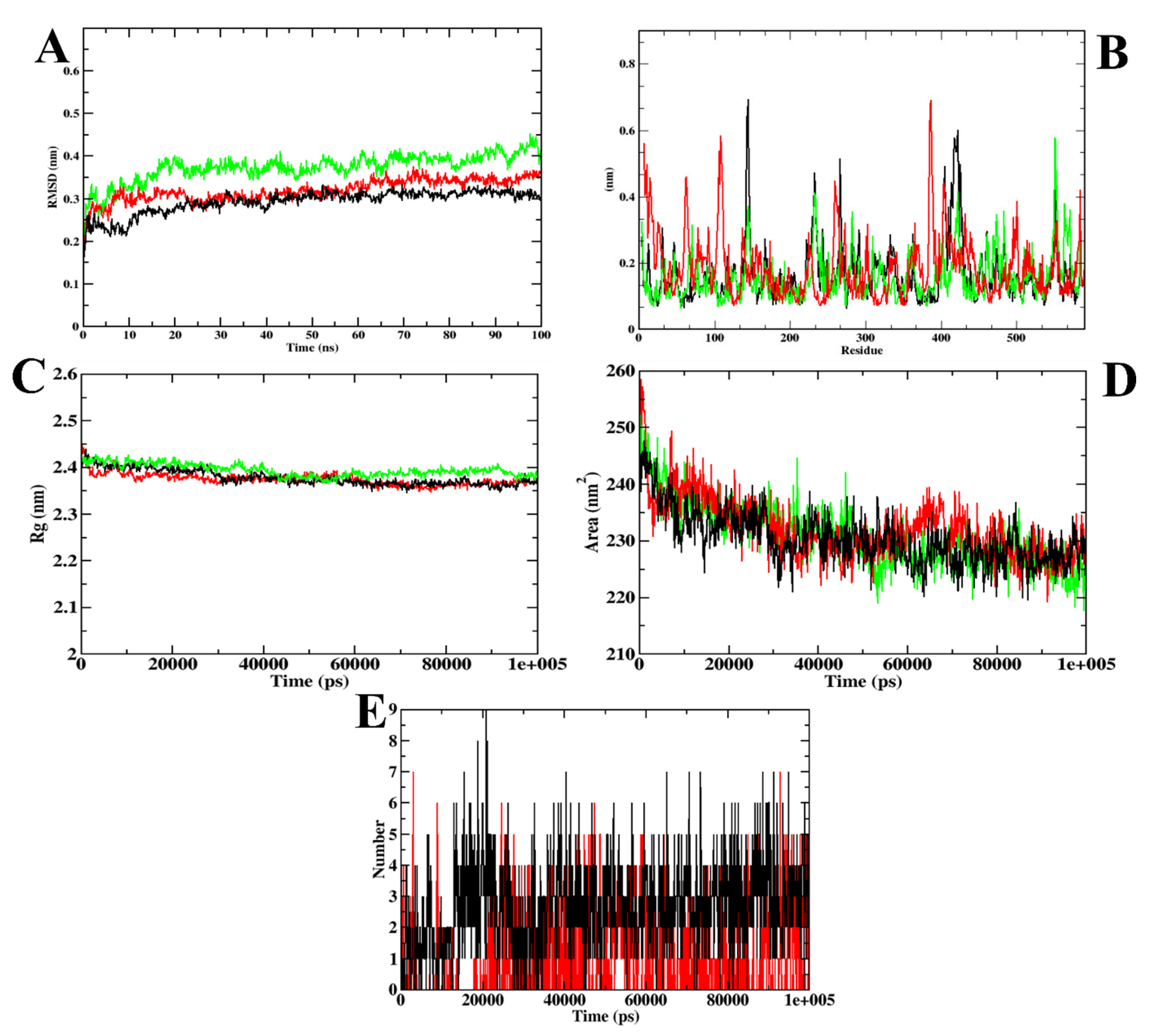
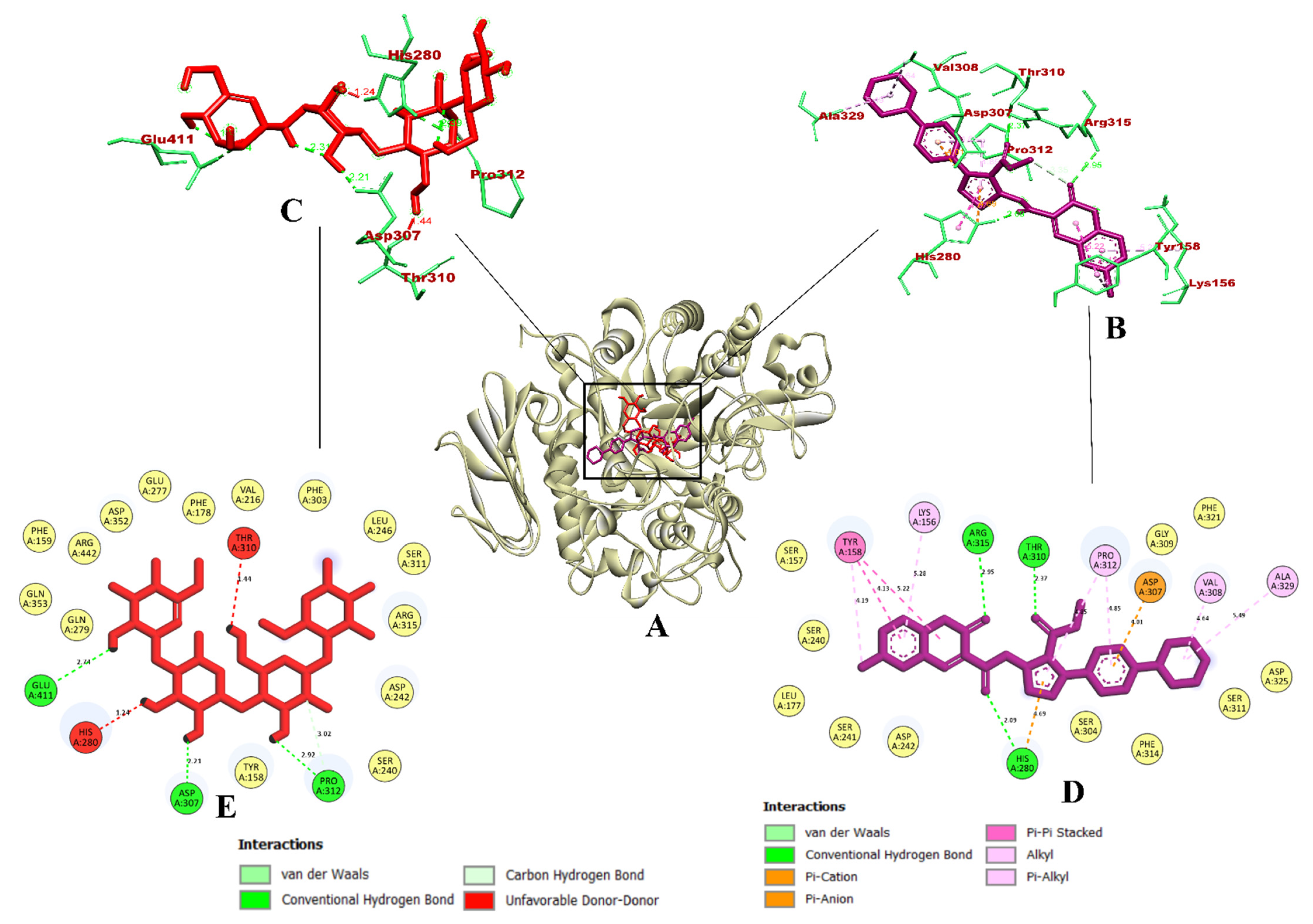
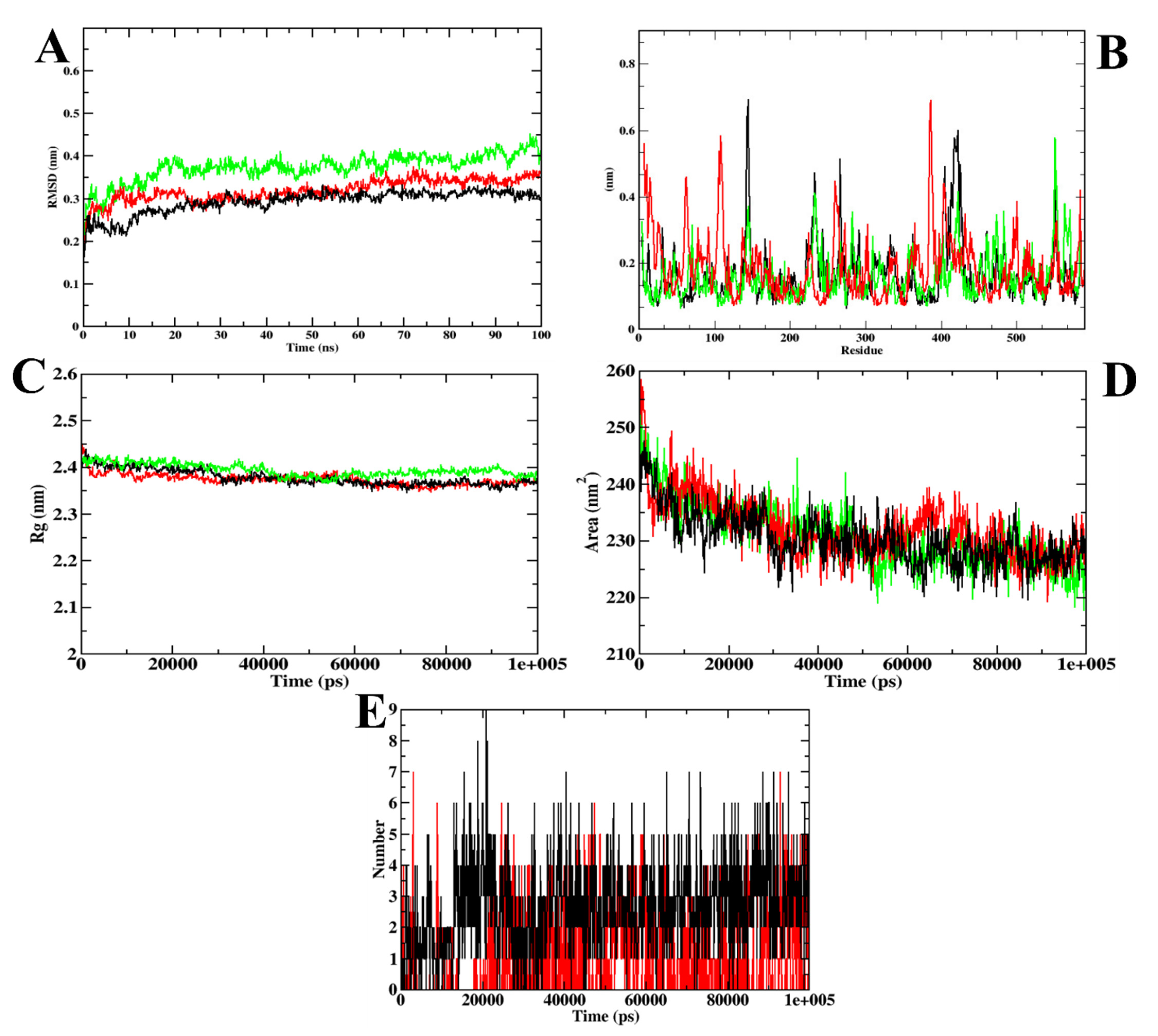
3.1. Virtual Screening of Ligands by Docking and Dynamics Simulation
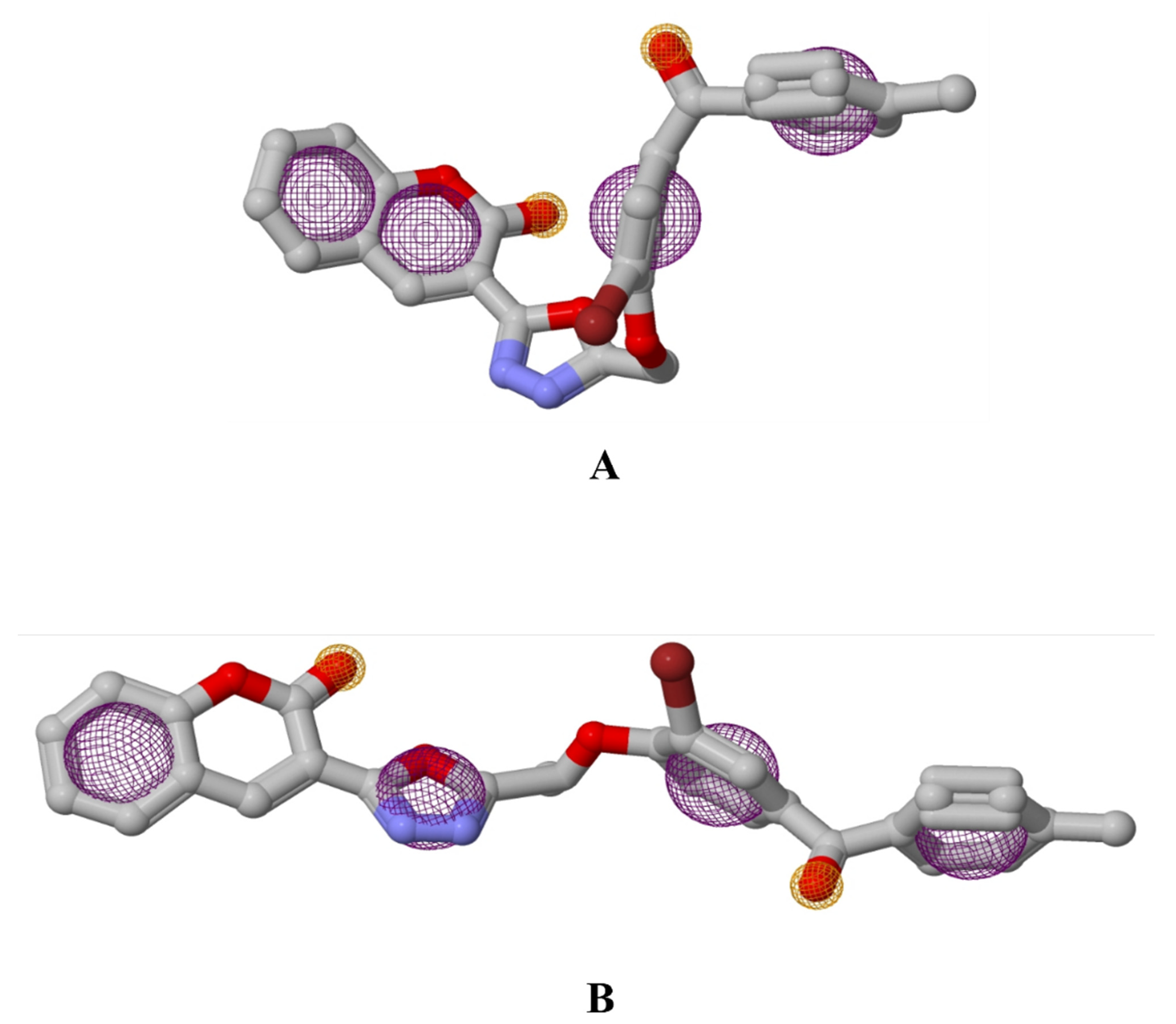
3.2. Pharmacophore Studies
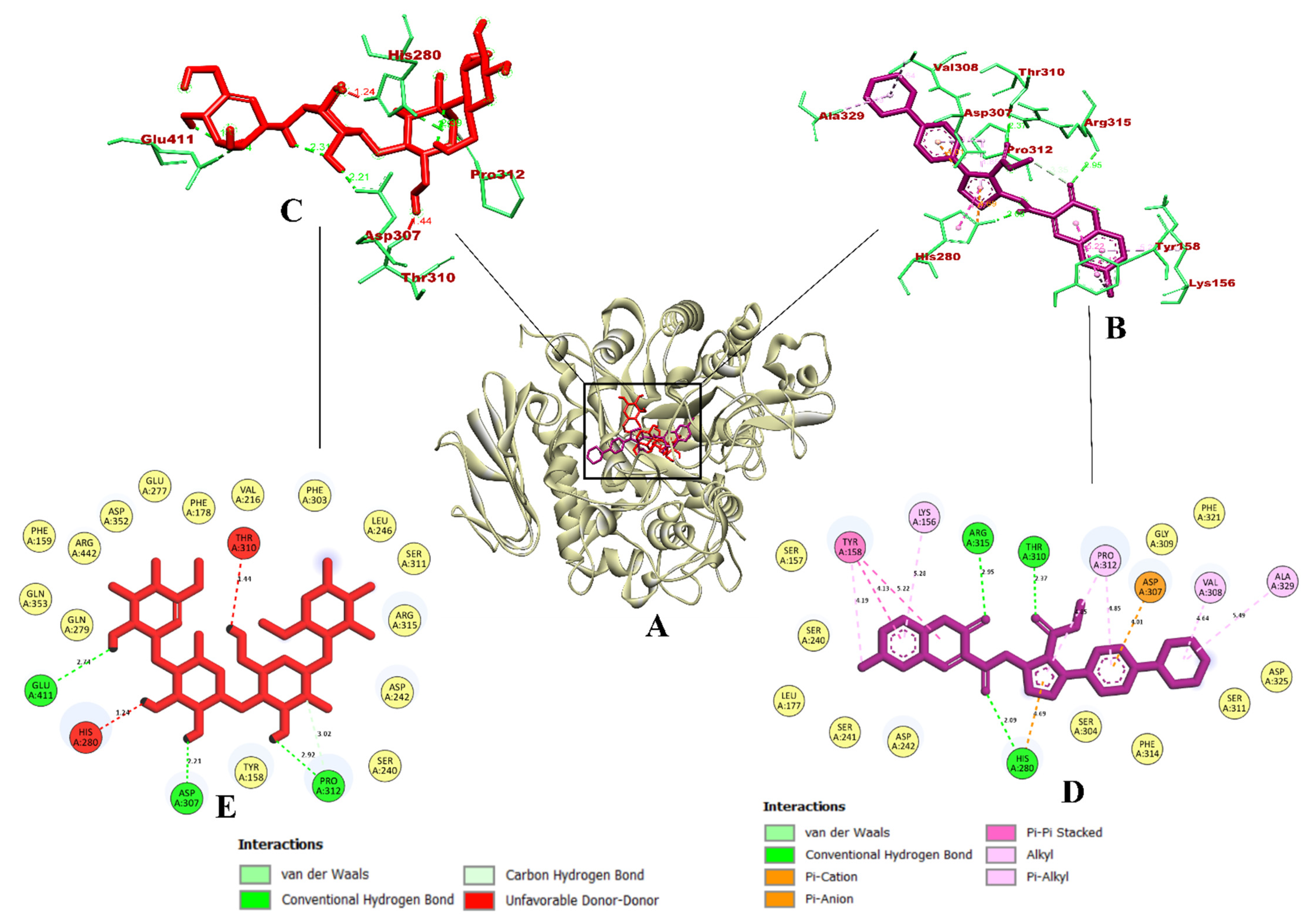
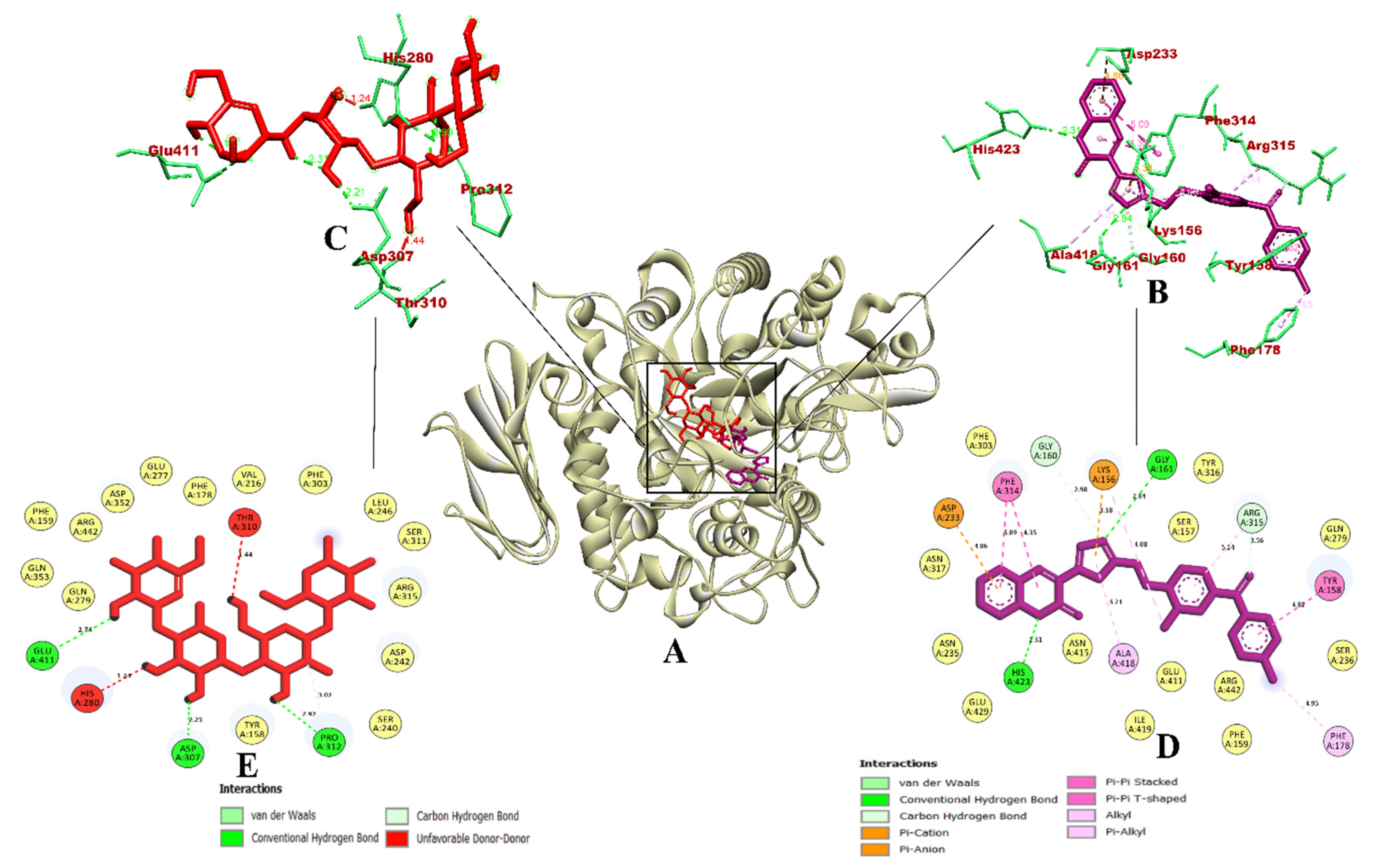
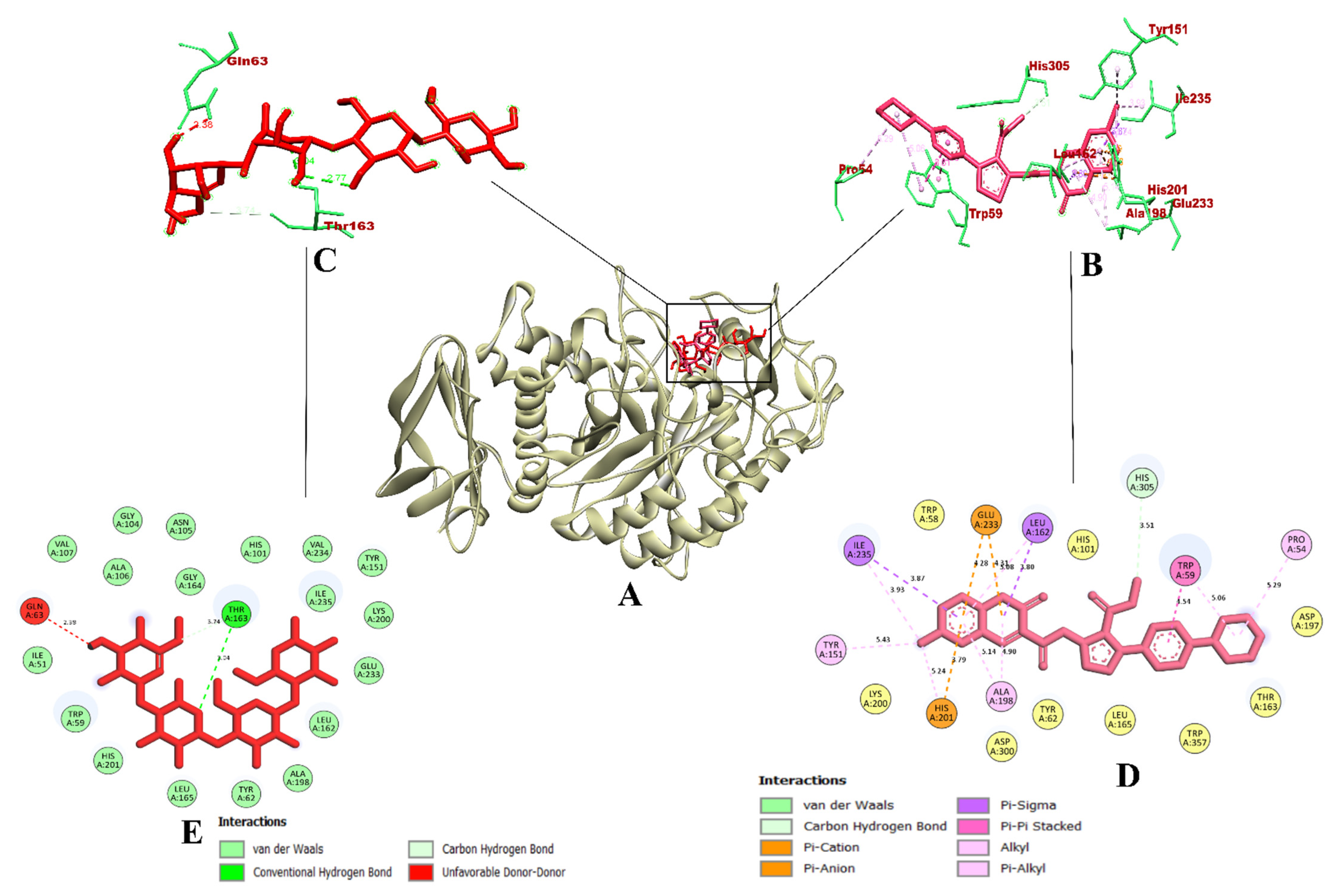
3.3. Molecular Docking and Dynamics Simulation of Selected Coumarin Derivatives
3.4. Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Patil, S.M.; Shirahatti, P.S.; Ramu, R. Azadirachta indica A. Juss (neem) against diabetes mellitus: A critical review on its phytochemistry, pharmacology, and toxicology. J. Pharm. Pharmacol. 2021, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Rachdaoui, N. Insulin: The friend and the foe in the development of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramu, R.; Shirahatti, P.S.; Deepika, T.H.; Bajpe, S.N.; Sreepathi, N.; Patil, S.M.; Prasad, N. Investigating Musa paradisiaca (Var. Nanjangud rasa bale) pseudostem in preventing hyperglycemia along with improvement of diabetic complications. J. Appl. Biol. Biotechnol. 2022, 10, 56–65. [Google Scholar] [CrossRef]
- Rizza, R.A.; Vella, A. Diabetes mellitus. In Pharmacology and Therapeutics: Principles to Practice, 1st ed.; Waldman, S.A., Terzic, A., Eds.; Elsevier: London, UK, 2009; Volume 37, pp. 557–570. [Google Scholar]
- Date, K. Regulatory Functions of α-Amylase in the Small Intestine Other than Starch Digestion: α-Glucosidase Activity, Glucose Absorption, Cell Proliferation, and Differentiation. In New Insights into Metabolic Syndrome; Takada, A., Ed.; IntechOpen: London, UK, 2020; Volume 5, pp. 1–37. [Google Scholar]
- Cheng, H.M.; Mah, K.K.; Seluakumaran, K. Carbohydrate Digestion: Small Intestine as the Site of Digestion and Absorption for Dietary Carbohydrate. In Defining Physiology: Principles, Themes, Concepts; Springer: Cham, Switzerland, 2020; Volume 2. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Kumar, B.P.; Kumar, N. Evaluation of flavonoids from banana pseudostem and flower (quercetin and catechin) as potent inhibitors of α-glucosidase: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lončar, M.; Jakovljević, M.; Šubarić, D.; Pavlić, M.; Buzjak Služek, V.; Cindrić, I.; Molnar, M. Coumarins in food and methods of their determination. Foods 2020, 9, 645. [Google Scholar] [CrossRef]
- Aoyama, Y.; Katayama, T.; Yamamoto, M.; Tanaka, H.; Kon, K. A new antitumor antibiotic product, demethylchartreusin isolation and biological activities. J. Antibiot. 1992, 45, 875–878. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.S.; Gupta, J.; Sharma, S.; Sahu, D. An insight into the therapeutic applications of coumarin compounds and their mechanisms of action. Eur. J. Pharm. Sci. 2020, 152, 105424. [Google Scholar] [CrossRef]
- Iranshahi, M.E.; Askari, M.; Sahebkar, A.; Hadjipavlou, L.D. Evaluation of antioxidant, anti-inflammatory and lipoxygenase inhibitory activities of the prenylated coumarin umbelliprenin. DARU 2009, 17, 99–103. [Google Scholar]
- Evans, W.C. Trease and Evans’ Pharmacognosy, 16th ed.; Elsevier Health Sciences: London, UK, 2009; pp. 571–603. [Google Scholar]
- Mead, J.A.; Smith, J.N.; Williams, R.T. Studies in detoxication. 72. The metabolism of coumarin and of o-coumaric acid. Biochem. J. 1958, 68, 67. [Google Scholar] [CrossRef] [Green Version]
- Hestrin, S.; Feingold, D.S.; Avigad, G. Synthesis of sucrose and other β-D-fructofuranosyl aldosides by levansucrase. J. Am. Chem. Soc. 1955, 77, 6710. [Google Scholar] [CrossRef]
- Abebe, W. Herbal medication: Potential for adverse interactions with analgesic drugs. J. Clin. Pharm. Ther. 2002, 27, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Ranganatha, L.V.; Prasad, M.N. Inhibitory effect of banana (Musa sp. var. Nanjangud rasa bale) flower extract and its constituents Umbelliferone and Lupeol on α-glucosidase, aldose reductase and glycation at multiple stages. S. Afr. J. Bot. 2014, 95, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Lakkappa Dhananjaya, B.; Prasad, M.N. Assessment of in vivo antidiabetic properties of umbelliferone and lupeol constituents of banana (Musa sp. var. Nanjangud Rasa Bale) flower in hyperglycaemic rodent model. PLoS ONE 2016, 11, e0151135. [Google Scholar]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Maurus, R.; Begum, A.; Williams, L.K.; Fredriksen, J.R.; Zhang, R.; Withers, S.G.; Brayer, G.D. Alternative catalytic anions differentially modulate human α-amylase activity and specificity. Biochemistry 2008, 47, 3332–3344. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Chandra, J.S.; Ranganatha, L.V. In silico identification of novel benzophenone-coumarin derivatives as SARS-CoV-2 RNAdependent RNA polymerase (RdRp) inhibitors. J. Biomol. Struct. Dyn. 2021, 11, 1–17. [Google Scholar]
- Maradesha, T.; Patil, S.M.; Al-Mutairi, K.A.; Ramu, R.; Madhunapantula, S.V.; Alqadi, T. Inhibitory effect of polyphenols from the whole green jackfruit flour against α-glucosidase, α-amylase, aldose reductase and glycation at multiple stages and their interaction: Inhibition kinetics and molecular simulations. Molecules 2022, 27, 1888. [Google Scholar] [CrossRef]
- Kumar, V.; Ramu, R.; Shirahatti, P.S.; Kumari, V.C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-Glucosidase; α-Amylase Inhibition; Kinetics and Docking Studies of Novel (2-Chloro-6-(trifluoromethyl) benzyloxy) arylidene) Based Rhodanine and Rhodanine Acetic Acid Derivatives. ChemistrySelect 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Patil, S.M.; Shirahatti, P.S.; Kumari, V.B.C.; Ramu, R.; Prasad, N. Azadirachta indica A. Juss (neem) as a contraceptive: An evidence-based review on its pharmacological efficiency. Phytomedicine 2021, 88, 153596. [Google Scholar] [CrossRef]
- Raphael, V.P.; Shanmughan, S.K. Computational evaluation of the inhibition efficacies of HIV antivirals on SARS-CoV-2 (COVID-19) protease and identification of 3D pharmacophore and hit compounds. Adv. Pharmacol. Sci. 2020, 2020, 8818008. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Maruthi, K.R.; Bajpe, N.S.; Vyshali, V.M.; Sushmitha, S.; Chagalamari, A.; Ramith, R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation 2021, 7, 932–939. [Google Scholar]
- Martiz, R.M.; Patil, S.M.; Ramu, R.; Jayanthi, M.K.; Ranganatha, L.V.; Khanum, S.A.; Silina, E.; Stupin, V.; Achar, R.R. Discovery of novel benzophenone integrated derivatives as anti-Alzheimer’s agents targeting presenilin-1 and presenilin-2 inhibition: A computational approach. PLoS ONE 2022, 17, e0265022. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Abdulaziz, M.; Babalghith, A.; Al-Areefi, M.; Al-Ghorbani, M.; Mallappa Kumar, J.; Prasad, A.; Mysore Nagalingaswamy, N.P.; Ramu, R.; et al. Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods. Molecules 2022, 27, 2398. [Google Scholar] [CrossRef] [PubMed]
- Peytam, F.; Takalloobanafshi, G.; Saadattalab, T.; Norouzbahari, M.; Emamgholipour, Z.; Moghimi, S.; Firoozpour, L.; Bijanzadeh, H.R.; Faramarzi, M.A.; Mojtabavi, S.; et al. Design, synthesis, molecular docking, and in vitro α-glucosidase inhibitory activities of novel 3-amino-2,4-diarylbenzo [4,5] imidazo [1,2-a] pyrimidines against yeast and rat α-glucosidase. Sci. Rep. 2021, 11, 11911. [Google Scholar] [CrossRef]
- Shukla, A.K.; Shrivash, M.K.; Pandey, A.; Pandey, J. Synthesis, in vitro and computational studies of novel glycosyl-1,2,3-1H-triazolyl methyl benzamide derivatives as potential α-glucosidase inhibitory activity. Bioorg. Chem. 2021, 109, 104687. [Google Scholar] [CrossRef]
- Swilam, N.; Nawwar, M.A.; Radwan, R.A.; Mostafa, E.S. Antidiabetic Activity and In Silico Molecular Docking of Polyphenols from Ammannia baccifera L. subsp. Aegyptiaca (Willd.) Koehne Waste: Structure Elucidation of Undescribed Acylated Flavonol Diglucoside. Plants 2022, 11, 452. [Google Scholar] [CrossRef]
- Mor, S.; Sindhu, S. Synthesis, Type II diabetes inhibitory activity, antimicrobial evaluation and docking studies of indeno [1,2-c]pyrazol-4(1H)-ones. Med. Chem. Res. 2020, 1, 46–62. [Google Scholar] [CrossRef]
- Hajlaoui, A.; Laajimi, M.; Znati, M.; Jannet, H.B.; Romdhane, A. Novel pyrano-triazolo-pyrimidine derivatives as anti-α-amylase agents: Synthesis, molecular docking investigations and computational analysis. J. Mol. Struct. 2021, 1237, 130346. [Google Scholar] [CrossRef]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating Molecular Docking and Molecular Dynamics Simulations. Methods Mol. Biol. 2019, 2053, 13–34. [Google Scholar]
- Lee, Y.; Kim, S.; Kim, J.Y.; Arooj, M.; Kim, S.; Hwang, S.; Kim, B.W.; Park, K.H.; Lee, K.W. Binding mode analyses and pharmacophore model development for stilbene derivatives as a novel and competitive class of α-glucosidase inhibitors. PLoS ONE 2014, 9, e85827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhan, L.; Xu, C.; Jiang, H.; Zhu, C.; Sun, L.; Sun, C.; Li, X. α-Glucosidase inhibitors from Chinese bayberry (Morella rubra Sieb. et Zucc.) fruit: Molecular docking and interaction mechanism of flavonols with different B-ring hydroxylations. RSC Adv. 2020, 10, 29347–29361. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Shetty, P.; Arunodaya, H.S.; Chandra, K.S.; Ramu, R.; Patil, S.M.; Baliga, A.; Rai, V.M.; Udupi, V.; Poojary, V.; et al. Potential Fluorinated Anti-MRSA Thiazolidinone Derivatives with Antibacterial, Antitubercular Activity and Molecular Docking Studies. Chem. Biodivers. 2022, 19, e202100532. [Google Scholar] [CrossRef]
- Lolok, N.; Sumiwi, S.A.; Muhtadi, A.; Susilawati, Y.; Hendriani, R.; Ramadhan, D.S.; Levita, J.; Sahidin, I. Molecular docking and molecular dynamics studies of bioactive compounds contained in noni fruit (Morinda citrifolia L.) against human pancreatic α-amylase. J. Biomol. Struct. Dyn. 2021, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Sathyanarayana, R.; Poojary, B.; Holla, B.S.; Poojary, V.; et al. In vitro and in silico studies of fluorinated 2,3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2021, 12, e2100342. [Google Scholar] [CrossRef]
- Chenafa, H.; Mesli, F.; Daoud, I.; Achiri, R.; Ghalem, S.; Neghra, A. In silico design of enzyme α-amylase and α-glucosidase inhibitors using molecular docking, molecular dynamic, conceptual DFT investigation and pharmacophore modelling. J. Biomol. Struct. Dyn. 2021, 2, 1–22. [Google Scholar] [CrossRef]
- Gurupadaswamy, H.D.; Ranganatha, V.L.; Ramu, R.; Patil, S.M.; Khanum, S.A. Competent synthesis of biaryl analogs via asymmetric Suzuki–Miyaura cross-coupling for the development of anti-inflammatory and analgesic agents. J. Iran. Chem. Soc. 2022, 19, 2421–2436. [Google Scholar] [CrossRef]
- Nipun, T.S.; Khatib, A.; Ibrahim, Z.; Ahmed, Q.U.; Redzwan, I.E.; Saiman, M.Z.; Supandi, F.; Primaharinastiti, R.; El-Seedi, H.R. Characterization of α-glucosidase inhibitors from Psychotria malayana jack leaves extract using LC-MS-based multivariate data analysis and in-silico molecular docking. Molecules 2020, 25, 5885. [Google Scholar] [CrossRef]
- Murugesu, S.; Ibrahim, Z.; Ahmed, Q.U.; Uzir, B.F.; Yusoff, N.I.; Perumal, V.; Abas, F.; Shaari, K.; Khatib, A. Identification of α-glucosidase inhibitors from Clinacanthus nutans leaf extract using liquid chromatography-mass spectrometry-based metabolomics and protein-ligand interaction with molecular docking. J. Pharm. Anal. 2019, 9, 91–99. [Google Scholar] [CrossRef]
- Trimurtulu, G.; Raghavendra, N.M. Isolation, in vitro antidiabetic, antioxidant activity and molecular docking studies of pentacyclic triterpenoids from Syzygium alternifolium (wt.) Walp bark. IOSR J. Pharm. Biol. Sci. 2015, 10, 22–27. [Google Scholar]
- Kumar, P.; Duhan, M.; Kadyan, K.; Sindhu, J.; Kumar, S.; Sharma, H. Synthesis of novel inhibitors of α-amylase based on the thiazolidine-4-one skeleton containing a pyrazole moiety and their configurational studies. MedChemComm 2017, 8, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Maliwal, D.; Pissurlenkar, R.R.; Telvekar, V. Identification of novel potential anti-diabetic candidates targeting human pancreatic α-amylase and human α-glycosidase: An exhaustive structure-based screening. Can. J. Chem. 2022, 100, 338–352. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
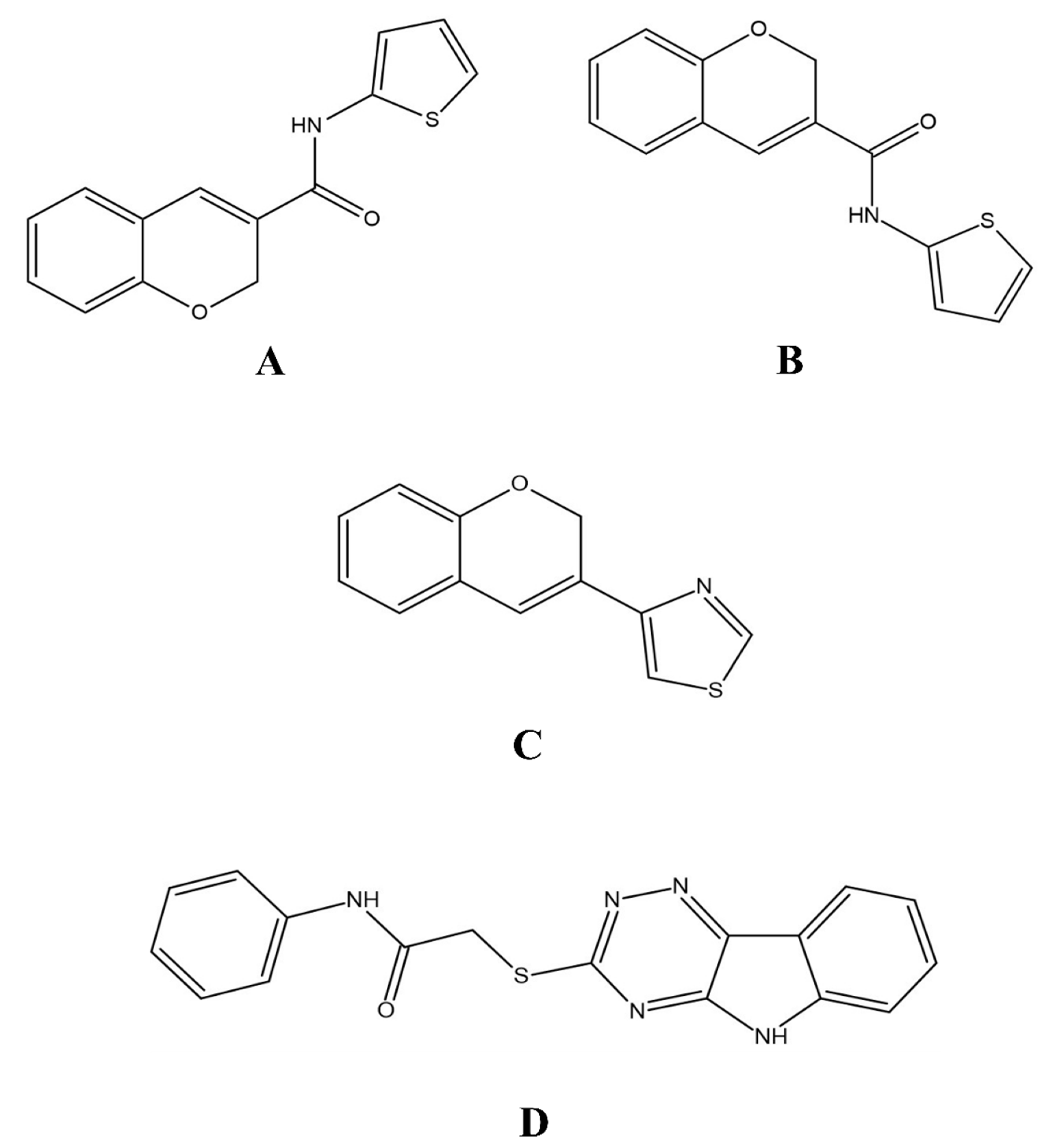{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No | Structure | Molecular Structural Differences |
|---|---|---|
| 1 | 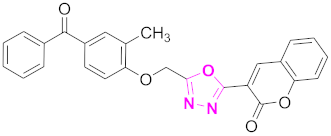 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(phenyl)methanone (CD-1) | Basic skeleton comprises coumarin, and benzophenone bridged via 1,3,4-oxadiazole, Additional one methyl substituent present at ortho position of phenyl ring of benzophenone, no substituents at benzoyl ring as well as coumarin |
| 2 | 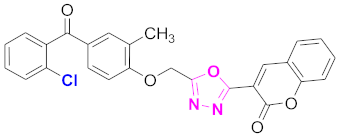 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-chlorophenyl)methanone (CD-2) | With basic skeleton, chloro group is present at ortho position of benzoyl ring of benzophenone |
| 3 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-bromophenyl)methanone (CD-3) | With basic skeleton, bromo group is present at ortho position of benzoyl ring of benzophenone |
| 4 | 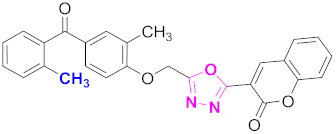 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-methylphenyl)methanone (CD-4) | With basic skeleton, methyl group is present at ortho position of benzoyl ring of benzophenone |
| 5 | 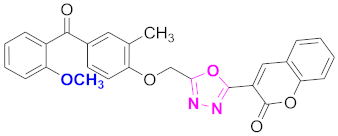 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-methoxyphenyl)methanone (CD-5) | With basic skeleton, methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 6 | 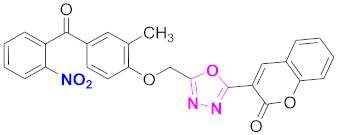 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-nitrophenyl)methanone (CD-6) | With basic skeleton, nitro group is present at ortho position of benzoyl ring of benzophenone |
| 7 | 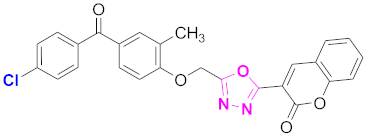 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-chlorophenyl)methanone (CD-7) | With basic skeleton, chloro group is present at para position of benzoyl ring of benzophenone |
| 8 | 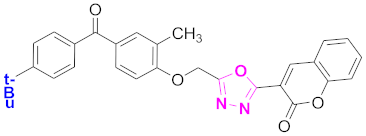 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-t-butylphenyl)methanone (CD-8) | With basic skeleton, tertiary butyl group is present at para position of benzoyl ring of benzophenone |
| 9 | 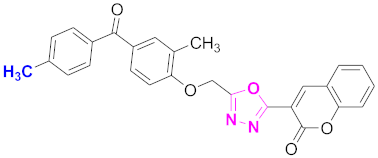 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-methylphenyl)methanone (CD-9) | With basic skeleton, methyl group is present at para position of benzoyl ring of benzophenone |
| 10 | 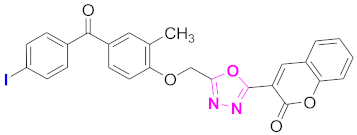 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-iodophenyl)methanone (CD-10) | With basic skeleton, iodo group is present at para position of benzoyl ring of benzophenone |
| 11 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(phenyl)methanone (CD-11) | With basic skeleton, 2-methyl groups present at ortho position of phenyl ring of benzophenone, no substitutions at benzoyl ring of benzophenone |
| 12 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-chlorophenyl)methanone (CD-12) | With basic skeleton, chloro group is present at ortho position of benzoyl ring of benzophenone |
| 13 | 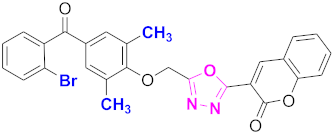 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-bromophenyl)methanone (CD-13) | With basic skeleton, bromo group is present at ortho position of benzoyl ring of benzophenone |
| 14 | 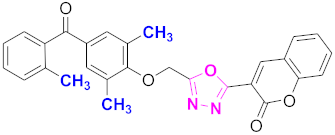 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-methylphenyl)methanone (CD-14) | With basic skeleton, methyl group is present at ortho position of benzoyl ring of benzophenone |
| 15 | 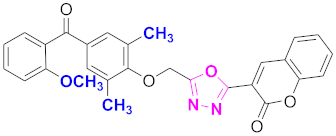 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-methoxyphenyl)methanone (CD-15) | With basic skeleton, methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 16 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-nitrophenyl)methanone (CD-16) | With basic skeleton, nitro group is present at ortho position of benzoyl ring of benzophenone |
| 17 | 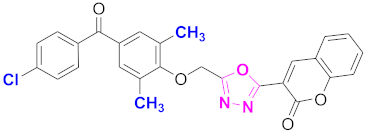 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-chlorophenyl)methanone (CD-17) | With basic skeleton, chloro group is present at para position of benzoyl ring of benzophenone |
| 18 | 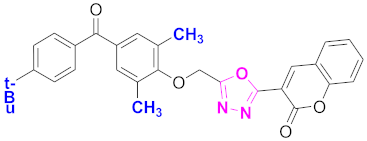 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-t-butylphenyl)methanone (CD-18) | With basic skeleton, t-butyl group is present at para position of benzoyl ring of benzophenone |
| 19 | 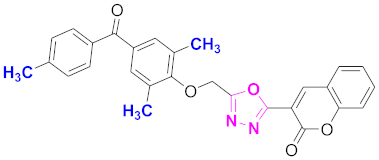 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-methylphenyl)methanone (CD-19) | With basic skeleton, methyl group is present at para position of benzoyl ring of benzophenone |
| 20 | 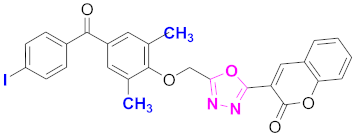 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-iodophenyl)methanone (CD-20) | With basic skeleton, iodo group is present at para position of benzoyl ring of benzophenone |
| 21 | 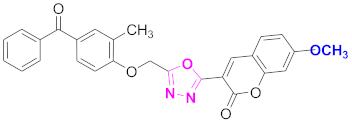 (4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(phenyl)methanone (CD-21) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone |
| 22 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-chlorophenyl)methanone (CD-22) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and chloro group is present at ortho position of benzoyl ring of benzophenone |
| 23 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-bromophenyl)methanone (CD-23) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and bromo group is present at ortho position of benzoyl ring of benzophenone |
| 24 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-methylphenyl)methanone (CD-24) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and methyl group is present at ortho position of benzoyl ring of benzophenone |
| 25 | 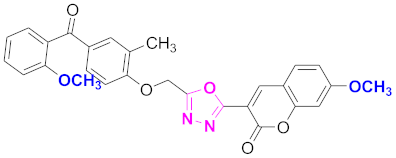 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-methoxyphenyl)methanone (CD-25) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 26 | 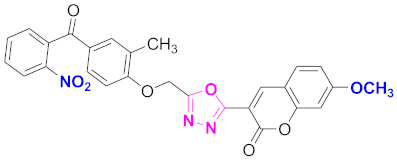 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(2-nitrophenyl)methanone (CD-26) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and nitro group is present at ortho position of benzoyl ring of benzophenone |
| 27 | 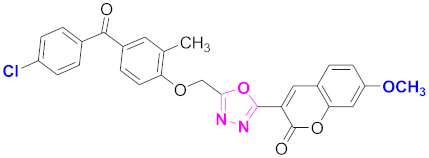 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-chlorophenyl)methanone (CD-27) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and chloro group is present at para position of benzoyl ring of benzophenone |
| 28 | 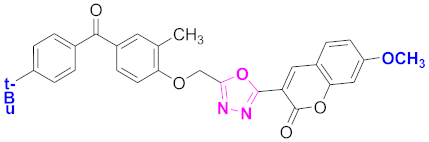 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-t-butylphenyl)methanone (CD-28) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and t-butyl group is present at para position of benzoyl ring of benzophenone |
| 29 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-methylphenyl)methanone (CD-29) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and methyl group is present at para position of benzoyl ring of benzophenone |
| 30 | 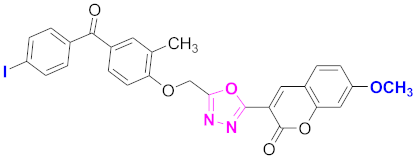 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-methylphenyl)(4-iodophenyl)methanone (CD-30) | With basic skeleton, coumarin contains methoxy group at 7th position, methyl group is present at ortho position of phenyl ring of benzophenone and iodo group is present at para position of benzoyl ring of benzophenone |
| 31 | 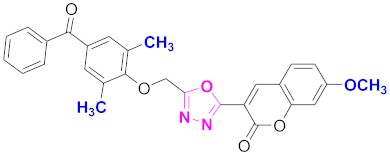 (4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(phenyl)methanone (CD-31) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone, and no substitutions at benzoyl ring of benzophenone |
| 32 | 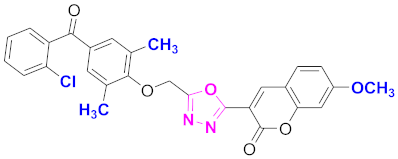 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-chlorophenyl)methanone (CD-32) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and chloro group is present at ortho position of benzoyl ring of benzophenone |
| 33 | 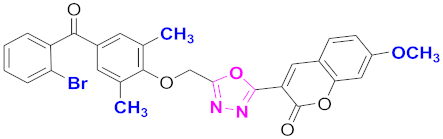 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-bromophenyl)methanone (CD-33) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and bromo group is present at ortho position of benzoyl ring of benzophenone |
| 34 | 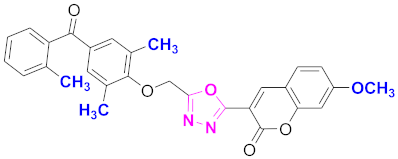 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-methylphenyl)methanone (CD-34) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and methyl group is present at ortho position of benzoyl ring of benzophenone |
| 35 | 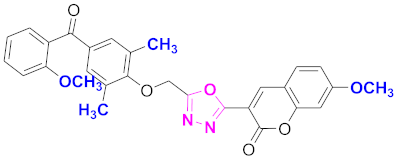 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-methoxyphenyl)methanone (CD-35) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 36 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(2-nitrophenyl)methanone (CD-36) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and nitro group is present at ortho position of benzoyl ring of benzophenone |
| 37 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-chlorophenyl)methanone (CD-37) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and chloro group is present at para position of benzoyl ring of benzophenone |
| 38 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-t-butylphenyl)methanone (CD-38) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and t-butyl group is present at para position of benzoyl ring of benzophenone |
| 39 | 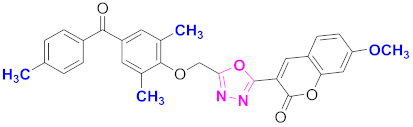 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-methylphenyl)methanone (CD-39) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and methyl group is present at para position of benzoyl ring of benzophenone |
| 40 | 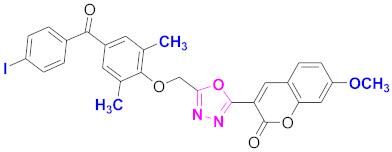 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3,5-dimethylphenyl)(4-iodophenyl)methanone (CD-40) | With basic skeleton, coumarin contains methoxy group at 7th position, 2-methyl groups present at ortho position of phenyl ring of benzophenone and iodo group is present at para position of benzoyl ring of benzophenone |
| 41 | 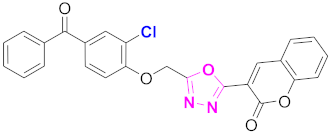 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorolphenyl)(phenyl)methanone (CD-41) | Basic skeleton comprises coumarin, and benzophenone bridged via 1,3,4-oxadiazole, Additional one chloro substituent present at ortho position of phenyl ring of benzophenone, no substituents at benzoyl ring as well as coumarin |
| 42 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-chlorophenyl)methanone (CD-42) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and chloro group is present at ortho position of benzoyl ring of benzophenone |
| 43 | 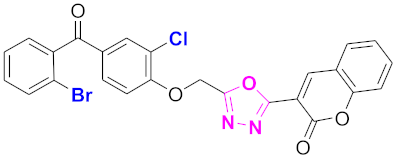 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-bromophenyl)methanone (CD-43) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and bromo group is present at ortho position of benzoyl ring of benzophenone |
| 44 | 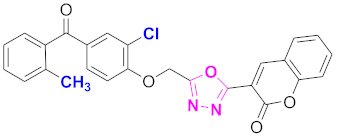 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-methylphenyl)methanone (CD-44) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and methyl group is present at ortho position of benzoyl ring of benzophenone |
| 45 | 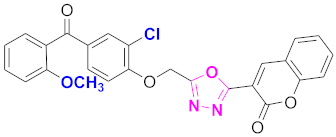 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-methoxyphenyl)methanone (CD-45) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 46 | 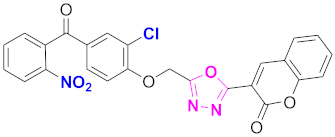 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-nitrophenyl)methanone (CD-46) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and nitro group is present at ortho position of benzoyl ring of benzophenone |
| 47 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-chlorophenyl)methanone (CD-47) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and chloro group is present at para position of benzoyl ring of benzophenone |
| 48 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-t-butylphenyl)methanone (CD-48) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and t-butyl group is present at para position of benzoyl ring of benzophenone |
| 49 | 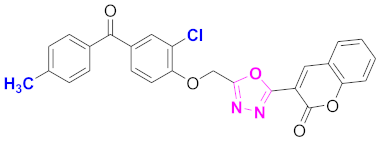 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-methylphenyl)methanone (CD-49) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and methyl group is present at para position of benzoyl ring of benzophenone |
| 50 | 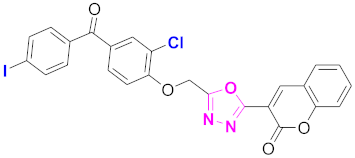 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-iodophenyl)methanone (CD-50) | With basic skeleton, additional one chloro substituent present at ortho position of phenyl ring of benzophenone, and iodo group is present at para position of benzoyl ring of benzophenone |
| 51 | 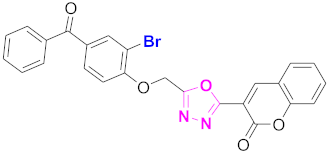 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(phenyl)methanone (CD-51) | Basic skeleton comprises coumarin, and benzophenone bridged via 1,3,4-oxadiazole, Additional one bromo group is present at ortho position of phenyl ring of benzophenone, no substituents at benzoyl ring as well as coumarin |
| 52 | 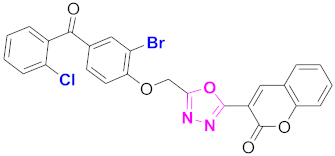 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-chlorophenyl)methanone (CD-52) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and chloro group is present at ortho position of benzoyl ring of benzophenone |
| 53 | 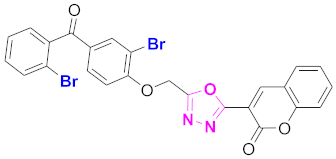 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-bromophenyl)methanone (CD-53) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and bromo group is present at ortho position of benzoyl ring of benzophenone |
| 54 | 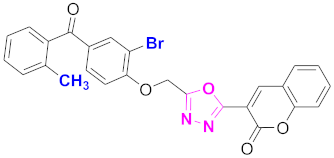 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-methylphenyl)methanone (CD-54) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and methyl group is present at ortho position of benzoyl ring of benzophenone |
| 55 | 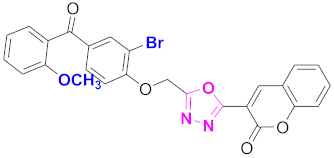 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-methoxyphenyl)methanone (CD-55) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 56 | 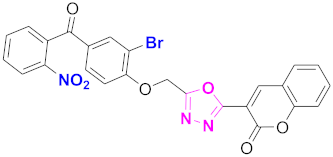 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-nitrophenyl)methanone (CD-56) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and nitro group is present at ortho position of benzoyl ring of benzophenone |
| 57 | 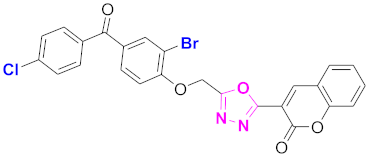 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-chlorophenyl)methanone (CD-57) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and chloro group is present at para position of benzoyl ring of benzophenone |
| 58 |  (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-t-butylphenyl)methanone (CD-58) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and t-butyl group is present at para position of benzoyl ring of benzophenone |
| 59 | 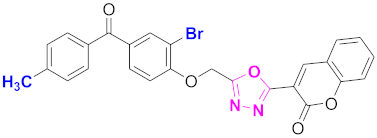 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-methylphenyl)methanone (CD-59) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and methyl group is present at para position of benzoyl ring of benzophenone |
| 60 | 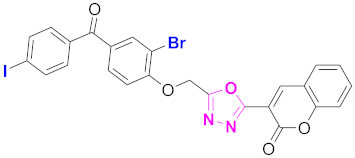 (4-((5-(2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-iodophenyl)methanone (CD-60) | With basic skeleton, additional one bromo substituent present at ortho position of phenyl ring of benzophenone, and iodo group is present at para position of benzoyl ring of benzophenone |
| 61 | 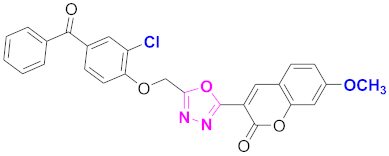 (4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(phenyl)methanone (CD-61) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and no substitutions at benzoyl ring of benzophenone |
| 62 | 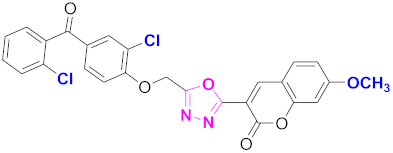 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-chlorophenyl)methanone (CD-62) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and chloro group is present at ortho position of benzoyl ring of benzophenone |
| 63 | 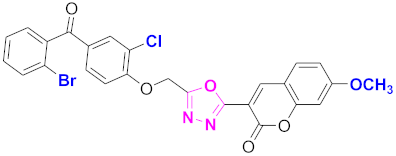 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-bromophenyl)methanone (CD-63) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and bromo group is present at ortho position of benzoyl ring of benzophenone |
| 64 | 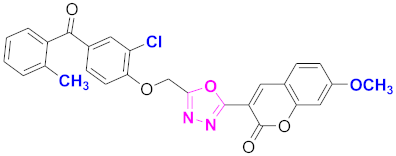 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-methylphenyl)methanone (CD-64) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone, and methyl group is present at ortho position of benzoyl ring of benzophenone |
| 65 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-methoxyphenyl)methanone (CD-65) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 66 | 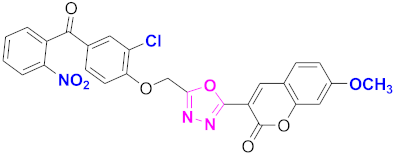 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(2-nitrophenyl)methanone (CD-66) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone, and nitro group is present at ortho position of benzoyl ring of benzophenone |
| 67 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-chlorophenyl)methanone (CD-67) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and chloro group is present at para position of benzoyl ring of benzophenone |
| 68 | 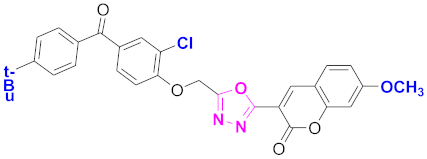 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-t-butylphenyl)methanone (CD-68) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and t-butyl group is present at para position of benzoyl ring of benzophenone |
| 69 | 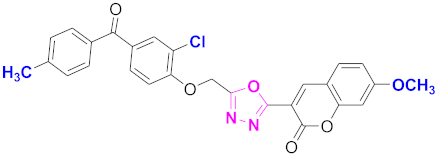 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-methylphenyl)methanone (CD-69) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone, and methyl group is present at para position of benzoyl ring of benzophenone |
| 70 | 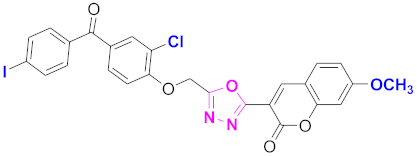 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-chlorophenyl)(4-iodophenyl)methanone (CD-70) | With basic skeleton, coumarin contains methoxy group at 7th position, and chloro group is present at ortho position of phenyl ring of benzophenone and iodo group is present at para position of benzoyl ring of benzophenone |
| 71 | 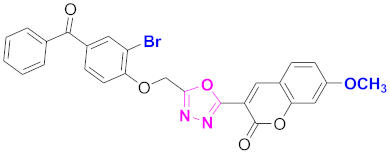 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(phenyl)methanone (CD-71) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone and no substitutions at benzoyl ring of benzophenone |
| 72 | 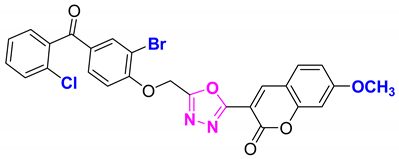 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-chlorophenyl)methanone (CD-72) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone and chloro group is present at ortho position of benzoyl ring of benzophenone |
| 73 | 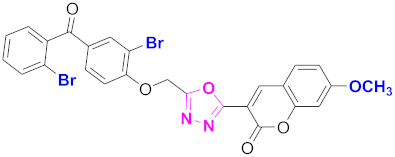 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-bromophenyl)methanone (CD-73) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone and bromo group is present at ortho position of benzoyl ring of benzophenone |
| 74 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-methylphenyl)methanone (CD-74) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone, and methyl group is present at ortho position of benzoyl ring of benzophenone |
| 75 | 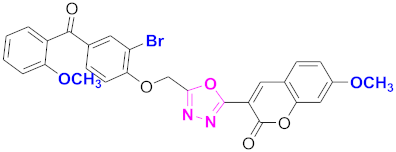 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-methoxyphenyl)methanone (CD-75) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone and methoxy group is present at ortho position of benzoyl ring of benzophenone |
| 76 | 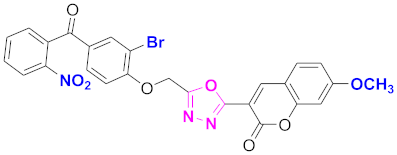 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(2-nitrophenyl)methanone (CD-76) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone, and nitro group is present at ortho position of benzoyl ring of benzophenone |
| 77 |  4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-chlorophenyl)methanone (CD-77) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone and chloro group is present at para position of benzoyl ring of benzophenone |
| 78 | 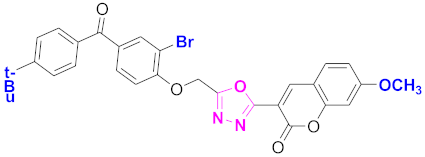 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-t-butylphenyl)methanone (CD-78) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone and t-butyl group is present at para position of benzoyl ring of benzophenone |
| 79 | 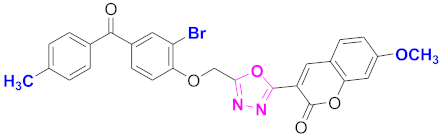 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-methylphenyl)methanone (CD-79) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone, and methyl group is present at para position of benzoyl ring of benzophenone |
| 80 | 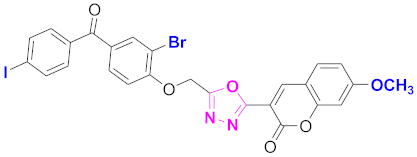 4-((5-(7-methoxy-2H-chromen-2-one)-1,3,4-oxadiazol-2-yl)methoxy)-3-bromophenyl)(4-iodophenyl)methanone (CD-80) | With basic skeleton, coumarin contains methoxy group at 7th position, and bromo group is present at ortho position of phenyl ring of benzophenone, and iodo group is present at para position of benzoyl ring of benzophenone |
| Coumarin Derivative | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds | |||
|---|---|---|---|---|---|---|
| α-Glucosidase | α-Amylase | α-Glucosidase | α-Amylase | α-Glucosidase | α-Amylase | |
| 58 | −11.9 | −11.1 | 16 | 16 | 5 | 2 |
| 78 | −11.8 | −10.7 | 18 | 14 | 6 | 2 |
| 68 | −11.8 | −10.6 | 16 | 17 | 6 | 1 |
| 18 | −11.8 | −11.3 | 10 | 16 | 2 | 1 |
| 28 | −11.7 | −10.7 | 11 | 20 | 4 | 2 |
| 38 | −11.7 | −10.8 | 10 | 17 | 2 | 3 |
| 59 | −11.6 | −11.3 | 14 | 16 | 4 | 3 |
| 8 | −11.6 | −11.2 | 11 | 15 | 2 | 2 |
| 48 | −11.5 | −11.1 | 11 | 14 | 2 | 1 |
| 29 | −11.4 | −10.7 | 14 | 13 | 5 | 1 |
| 11 | −11.4 | −11 | 13 | 12 | 2 | 1 |
| 19 | −11.4 | −11.4 | 11 | 16 | 2 | 1 |
| 49 | −11.3 | −11.3 | 16 | 17 | 3 | 2 |
| 47 | −11.3 | −11.1 | 14 | 17 | 4 | 3 |
| 67 | −11.3 | −10.9 | 14 | 16 | 5 | 2 |
| 10 | −11.3 | −11.2 | 13 | 13 | 4 | 1 |
| 27 | −11.3 | −10.9 | 13 | 13 | 3 | 1 |
| 69 | −11.3 | −11 | 12 | 14 | 3 | 1 |
| 62 | −11.2 | −10.7 | 14 | 11 | 4 | 1 |
| 74 | −11.2 | −10.7 | 14 | 13 | 4 | 3 |
| 77 | −11.2 | −10.5 | 14 | 14 | 3 | 3 |
| 79 | −11.2 | −10.5 | 14 | 15 | 3 | 1 |
| 22 | −11.2 | −10.8 | 13 | 11 | 5 | 1 |
| 64 | −11.2 | −10.7 | 13 | 11 | 4 | 2 |
| 24 | −11.2 | −10.5 | 11 | 17 | 2 | 1 |
| 1 | −11.2 | −11.3 | 9 | 14 | 1 | 1 |
| 32 | −11.1 | −11 | 16 | 11 | 6 | 1 |
| 33 | −11.1 | −11 | 16 | 12 | 5 | 1 |
| 80 | −11.1 | −10.8 | 16 | 15 | 5 | 2 |
| 70 | −11.1 | −10.9 | 15 | 14 | 4 | 1 |
| 34 | −11.1 | −11 | 14 | 9 | 6 | 1 |
| 39 | −11.1 | −10.7 | 14 | 14 | 2 | 3 |
| 72 | −11.1 | −10.6 | 13 | 13 | 3 | 2 |
| 76 | −11.1 | −10.2 | 13 | 18 | 5 | 4 |
| 26 | −11.1 | −10.4 | 12 | 13 | 4 | 4 |
| 41 | −11.1 | −10.9 | 11 | 15 | 2 | 2 |
| 9 | −11.1 | −10.7 | 11 | 9 | 2 | 1 |
| 30 | −11.1 | −11 | 10 | 14 | 3 | 1 |
| 2 | −11.1 | −10.9 | 10 | 10 | 4 | 1 |
| 42 | −11.1 | −11 | 9 | 10 | 4 | 3 |
| 14 | −11.1 | −10.6 | 9 | 10 | 1 | - |
| 66 | −11 | −10.4 | 17 | 11 | 8 | 2 |
| 23 | −11 | −10.7 | 15 | 11 | 2 | 2 |
| Acarbose | −7.9 | −7.7 | 5 | 3 | 5 | 3 |
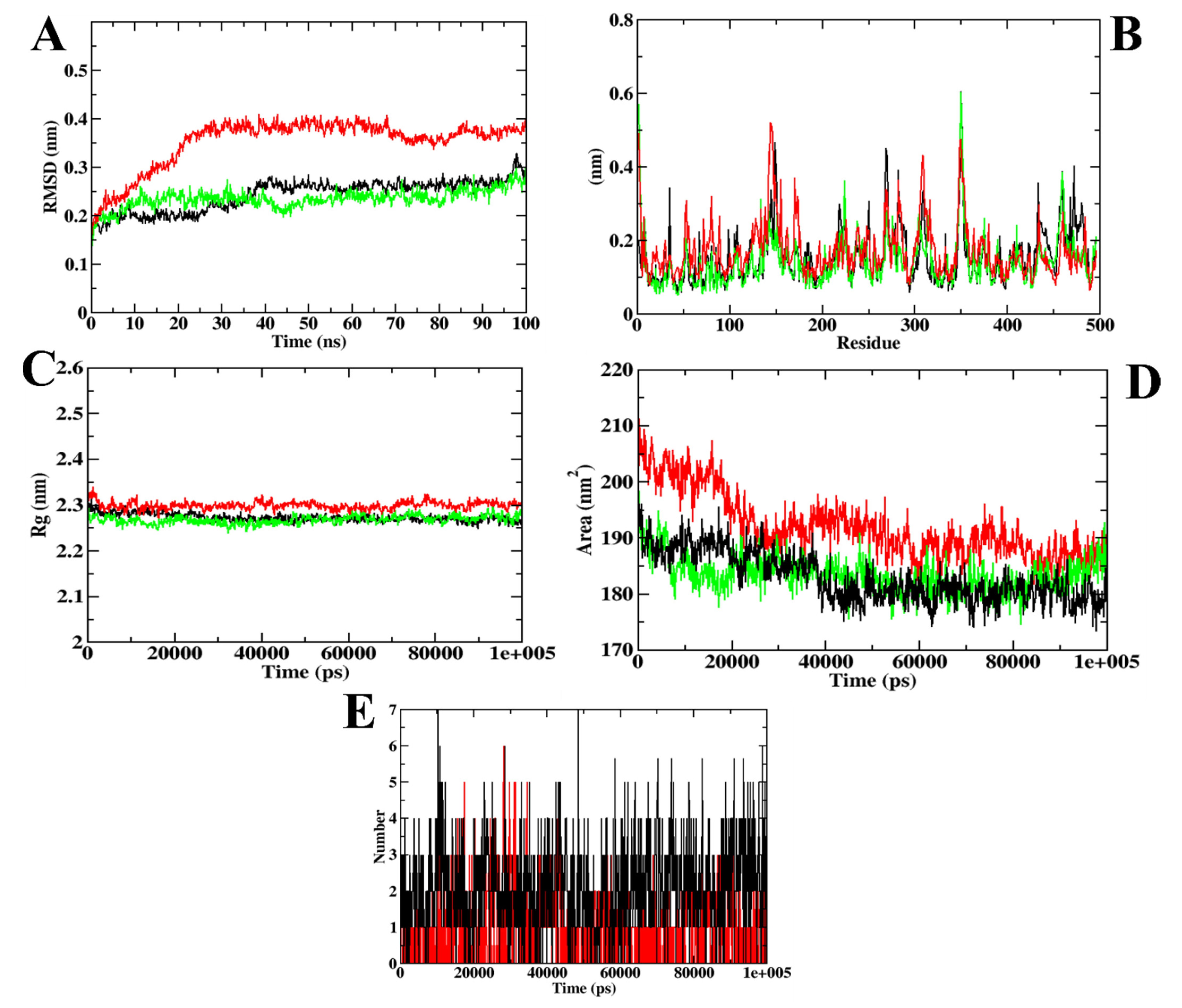
| MD Trajectory Values | Apoprotein | Protein–Acarbose Complex | Protein–ZINC02789441 Complex |
|---|---|---|---|
| RMSD | 0.30–0.35 nm | 0.35–0.40 nm | 0.30–0.35 nm |
| Rg | 2.35–2.40 nm | 2.40–2.45 nm | 2.35–2.40 nm |
| SASA | 220–230 nm2 | 230–235 nm2 | 220–230 nm2 |
| Ligand H-bonds | - | 9 | 7 |
| MD Trajectory Values | Apoprotein | Protein–Acarbose Complex | Protein–CD-59 Complex |
|---|---|---|---|
| RMSD | 0.25–0.30 nm | 0.35–0.40 nm | 0.25–0.30 nm |
| Rg | 2.25–2.30 nm | 2.30 nm | 2.25–2.30 nm |
| SASA | 185–190 nm2 | 185–190 nm2 | 185–190 nm2 |
| Ligand H-bonds | - | 7 | 6 |
| Coumarin Derivative | Name | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds |
|---|---|---|---|---|
| 32 | ZINC13496808 | −11.8 | 14 | 6 |
| 9 | ZINC59502854 | −11.6 | 14 | 5 |
| 52 | ZINC15109170 | −11.6 | 13 | 3 |
| 5 | ZINC02789441 | −11.4 | 15 | 4 |
| 20 | ZINC12779729 | −11.4 | 12 | 2 |
| 68 | ZINC41013463 | −11.4 | 13 | 4 |
| 16 | ZINC01641963 | −11 | 12 | 4 |
| 91 | ZINC09781623 | −11 | 13 | 5 |
| 108 | Acarbose | −7.9 | 5 | 5 |
| Coumarin Derivative | Name | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds |
|---|---|---|---|---|
| 51 | ZINC68601297 | −11.4 | 4 | - |
| 7 | ZINC03144061_1 | −11.4 | 4 | - |
| 23 | ZINC08721887_4 | −11.4 | 4 | - |
| 24 | ZINC08721887_5 | −11.4 | 4 | - |
| 43 | ZINC15799331_4 | −11.4 | 4 | - |
| 44 | ZINC15799331_5 | −11.4 | 4 | - |
| 10 | ZINC03144061_4 | −11.3 | 4 | - |
| 11 | ZINC03144061_5_1 | −11.3 | 4 | - |
| 5 | ZINC02929288 | −10.6 | 8 | 1 |
| 28 | ZINC08901231 | −10.5 | 8 | - |
| 49 | ZINC21884078_3 | −10.1 | 14 | - |
| 55 | ZINC09781623 | −10.1 | 13 | 3 |
| 9 | ZINC03144061_3 | −10 | 13 | - |
| 42 | ZINC15799331_3 | −10 | 14 | - |
| 46 | ZINC20143660 | −10 | 9 | 1 |
| 50 | ZINC25576471 | −10 | 18 | 4 |
| 8 | ZINC03144061_2 | −9.7 | 14 | 2 |
| 17 | ZINC05350534_1_2 | −9.7 | 14 | 2 |
| 21 | ZINC08721887_2 | −9.7 | 14 | 2 |
| 22 | ZINC08721887_3 | −9.7 | 15 | - |
| 31 | ZINC13120435_2 | −9.7 | 15 | 2 |
| 41 | ZINC15799331_2 | −9.7 | 14 | 2 |
| 48 | ZINC21884078_2 | −9.7 | 14 | 2 |
| 6 | ZINC02933910 | −9.6 | 13 | 6 |
| 18 | ZINC05350534_2 | −9.6 | 14 | 3 |
| 32 | ZINC13120435_3 | −9.6 | 15 | - |
| 52 | ZINC02789441 | −9.5 | 16 | 1 |
| 56 | Acarbose | −7.7 | 2 | 2 |
| Compounds | α-Glucosidase | α-Amylase | ||||
|---|---|---|---|---|---|---|
| Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrogen Bonds | |
| ZINC02789441 | −11.4 | 15 | 4 | −9.5 | 16 | 1 |
| ZINC40949448 | −10.5 | 14 | 5 | −8.2 | 19 | 1 |
| ZINC13496808 | −11.8 | 14 | 6 | −9.2 | 18 | 2 |
| ZINC09781623 | −11 | 13 | 5 | −10.1 | 13 | 3 |
| Acarbose | −7.9 | 5 | 5 | −7.7 | 2 | 2 |
| MD Trajectory Values | Apoprotein | Protein–Acarbose Complex | Protein–ZINC02789441 Complex |
|---|---|---|---|
| RMSD | 0.40 nm | 0.30–0.35 nm | 0.30–0.35 nm |
| Rg | 2.4 nm | 2.4 nm | 2.4 nm |
| SASA | 220–235 nm2 | 220–235 nm2 | 220–235 nm2 |
| Ligand H-bonds | - | 7 | 9 |
| MD Trajectory Values | Apoprotein | Protein–Acarbose Complex | Protein–ZINC02789441 Complex |
|---|---|---|---|
| RMSD | 0.30 nm | 0.30 nm | 0.25–0.30 nm |
| Rg | 2.25–2.30 nm | 0.30 nm | 2.25–2.30 nm |
| SASA | 185–190 nm2 | 185–190 nm2 | 185–190 nm2 |
| Ligand H-bonds | - | 6 | 7 |
| Types of Binding Free Energy | ZINC02789441-α-Glucosidase Complex | Acarbose-α-Glucosidase Complex | ZINC02789441-α-Amylase Complex | Acarbose-α-Amylase Complex |
|---|---|---|---|---|
| Values (kj/mol) | Values (kj/mol) | Values (kj/mol) | Values (kj/mol) | |
| Van der Waal’s energy | −316.391 ± 15.473 | −218.605 ± 23.706 | −169.669 ± 17.479 | −151.112 ± 19.233 |
| Electrostatic energy | −21.871± 5.801 | −4.761 ± 6.221 | −6.992 ± 11.374 | −10.911 ± 6.801 |
| Polar solvation energy | 106.897 ± 13.989 | −103.307 ± 55.952 | 79.945 ± 50.793 | 61.951 ± 25.681 |
| SASA energy | −22.576 ± 0.997 | −17.835 ± 13.498 | −12.899 ± 7.329 | −14.929 ± 6.997 |
| Binding energy | −115.796 ± 61.774 | −137.894 ± 70.951 | −213.410 ± 59.230 | −112.119 ± 58.114 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, S.M.; Martiz, R.M.; Satish, A.M.; Shbeer, A.M.; Ageel, M.; Al-Ghorbani, M.; Ranganatha, L., V; Parameswaran, S.; Ramu, R. Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches. Molecules 2022, 27, 3888. https://doi.org/10.3390/molecules27123888
Patil SM, Martiz RM, Satish AM, Shbeer AM, Ageel M, Al-Ghorbani M, Ranganatha L V, Parameswaran S, Ramu R. Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches. Molecules. 2022; 27(12):3888. https://doi.org/10.3390/molecules27123888
Chicago/Turabian StylePatil, Shashank M., Reshma Mary Martiz, A. M. Satish, Abdullah M. Shbeer, Mohammed Ageel, Mohammed Al-Ghorbani, Lakshmi Ranganatha, V, Saravanan Parameswaran, and Ramith Ramu. 2022. "Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches" Molecules 27, no. 12: 3888. https://doi.org/10.3390/molecules27123888
APA StylePatil, S. M., Martiz, R. M., Satish, A. M., Shbeer, A. M., Ageel, M., Al-Ghorbani, M., Ranganatha, L., V, Parameswaran, S., & Ramu, R. (2022). Discovery of Novel Coumarin Derivatives as Potential Dual Inhibitors against α-Glucosidase and α-Amylase for the Management of Post-Prandial Hyperglycemia via Molecular Modelling Approaches. Molecules, 27(12), 3888. https://doi.org/10.3390/molecules27123888