
Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents



 and
and 
{kind=link}
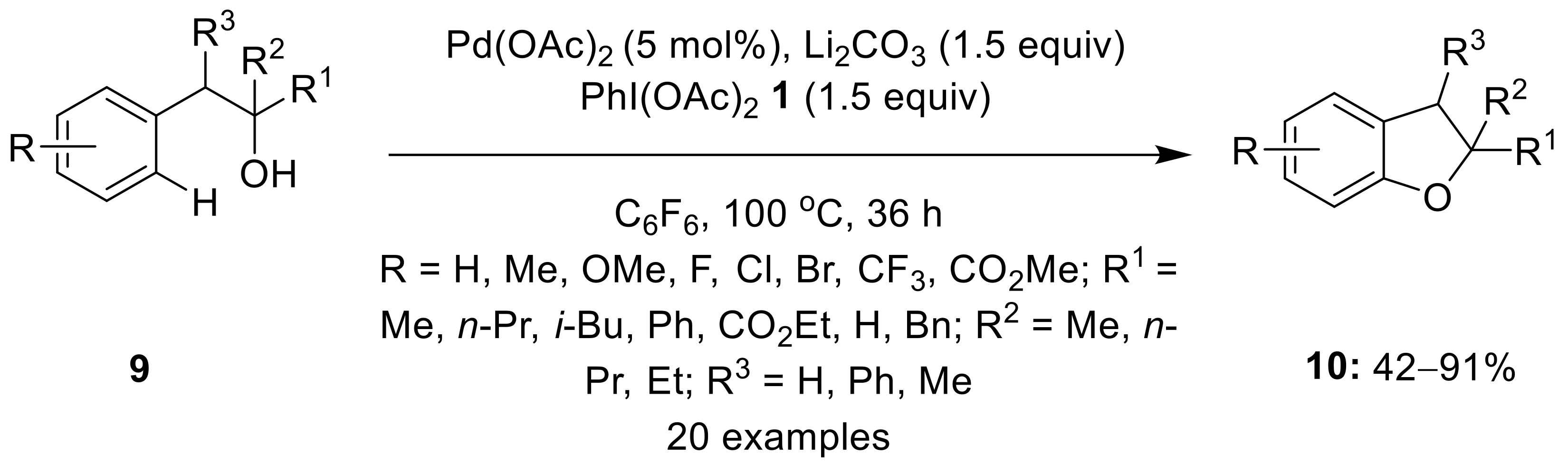{kind=link}
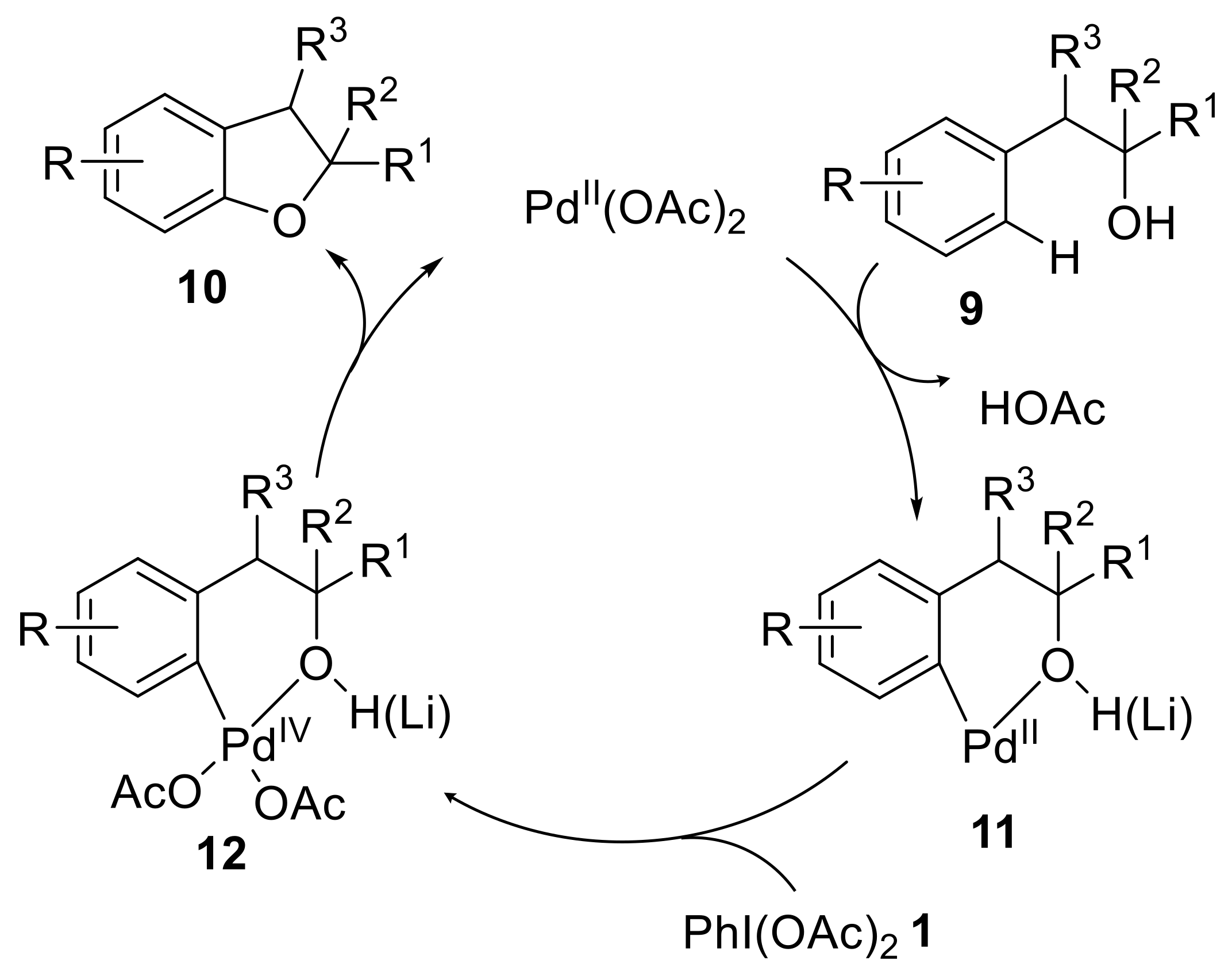{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
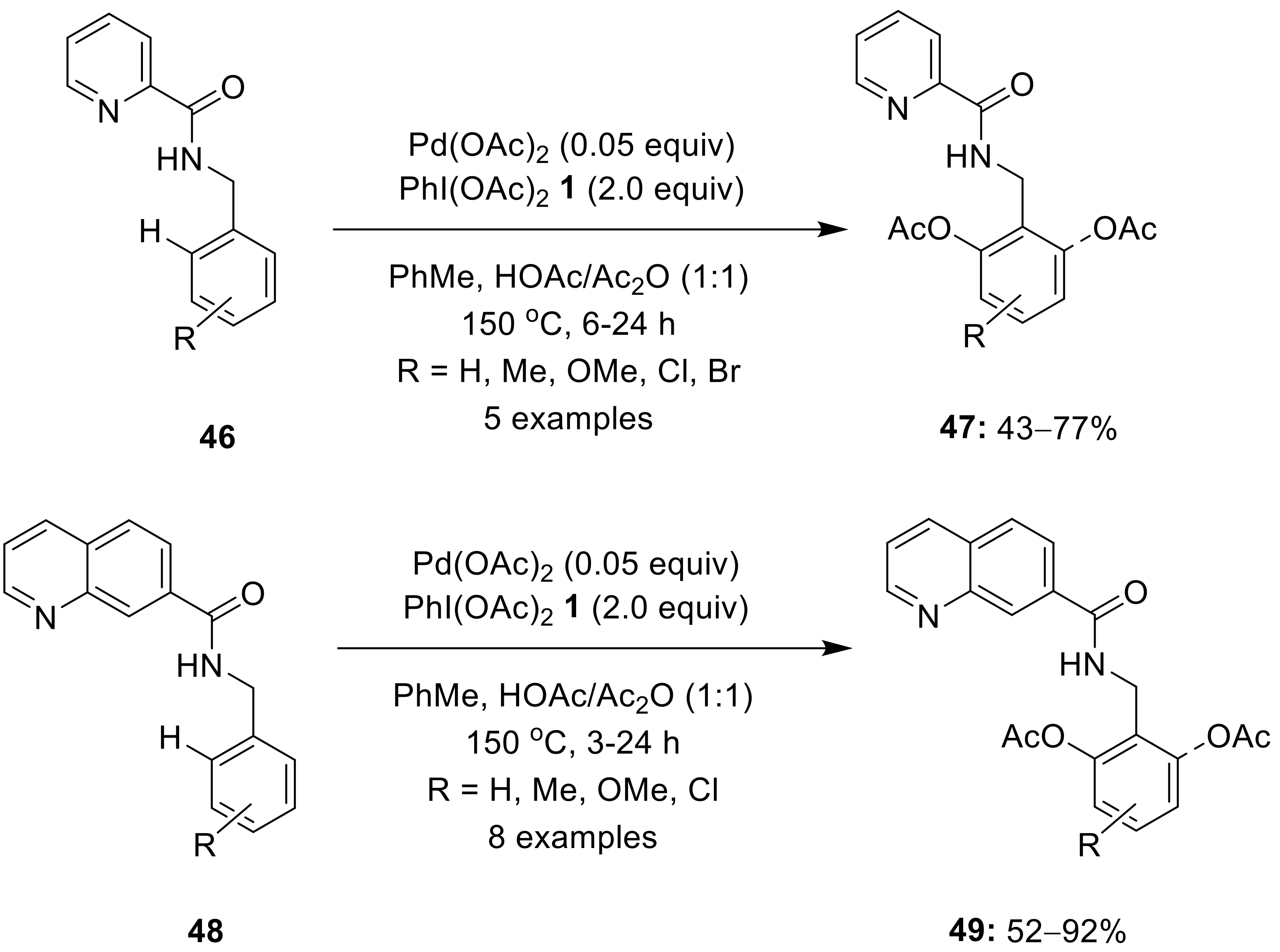{kind=link}
{kind=link}
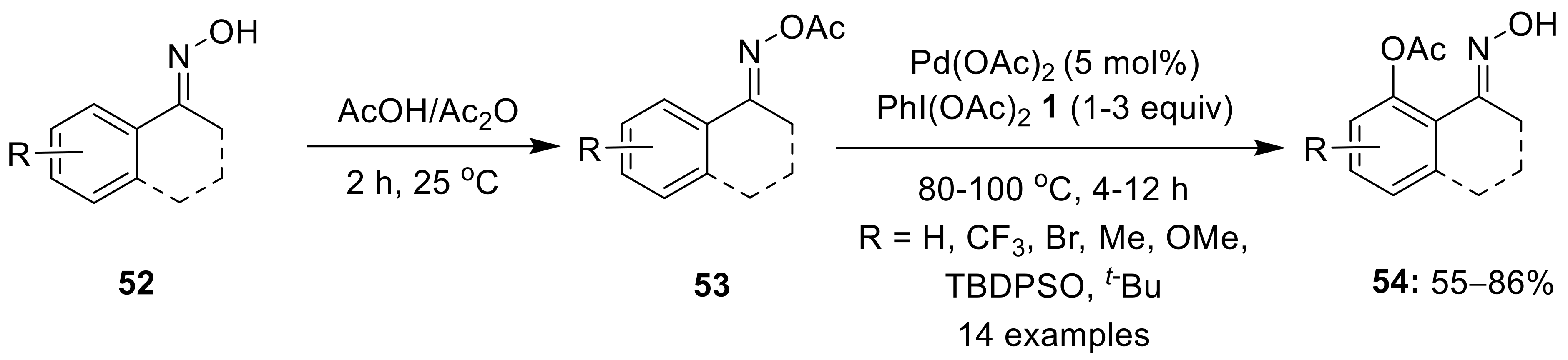{kind=link}
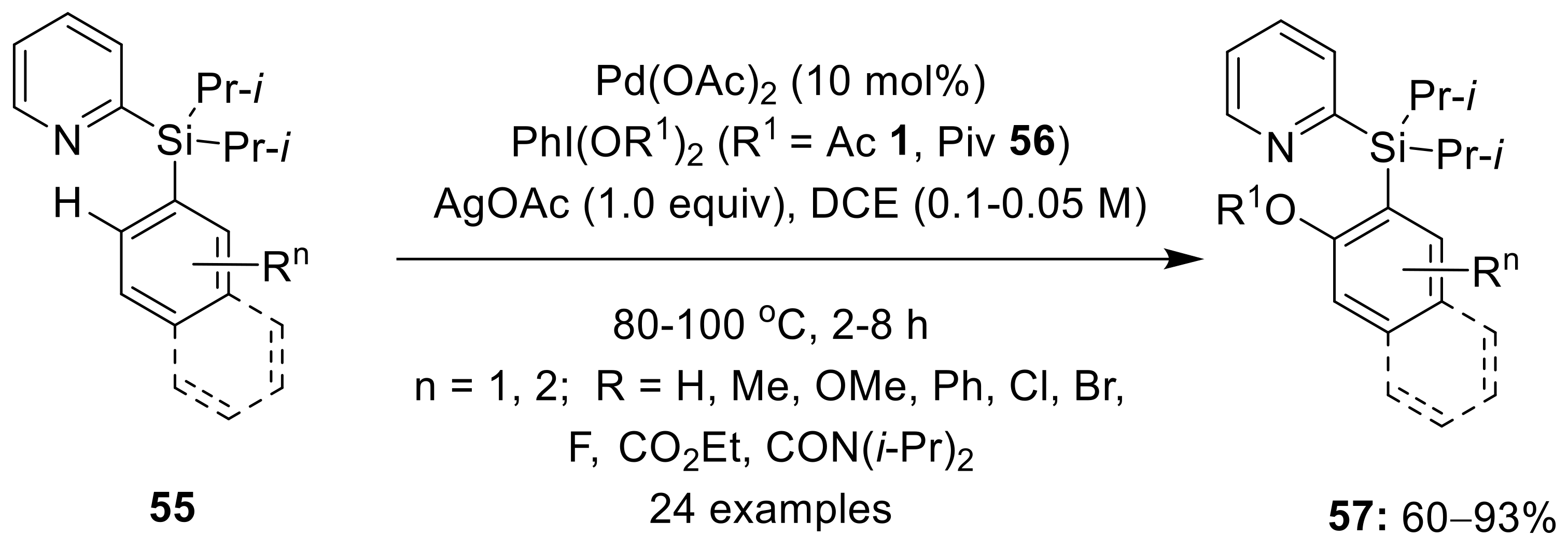{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
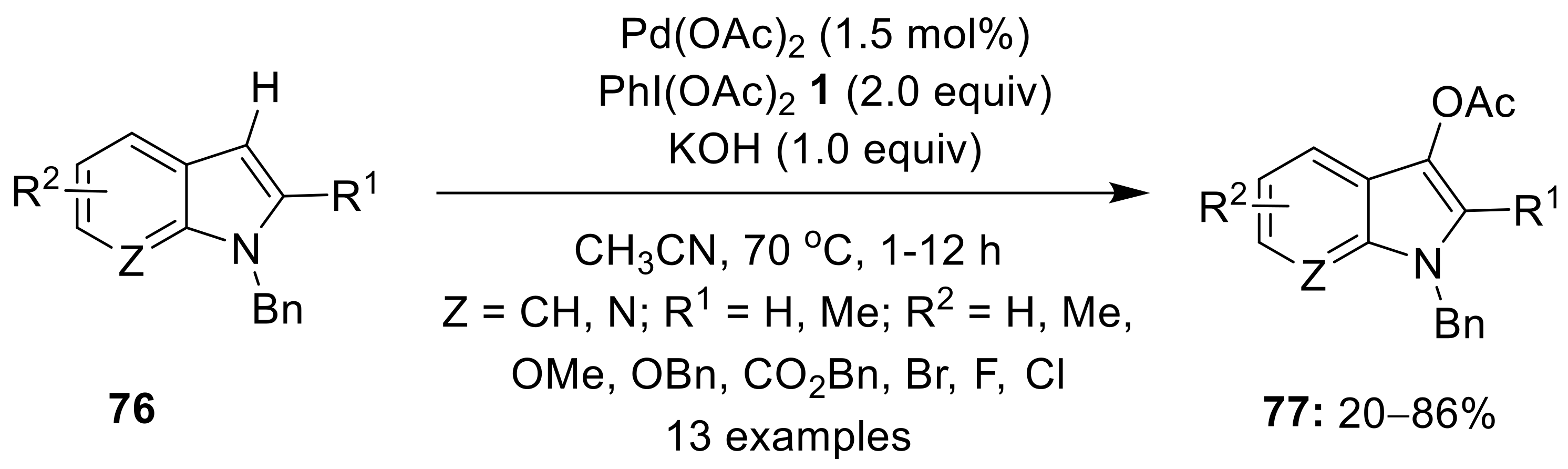{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
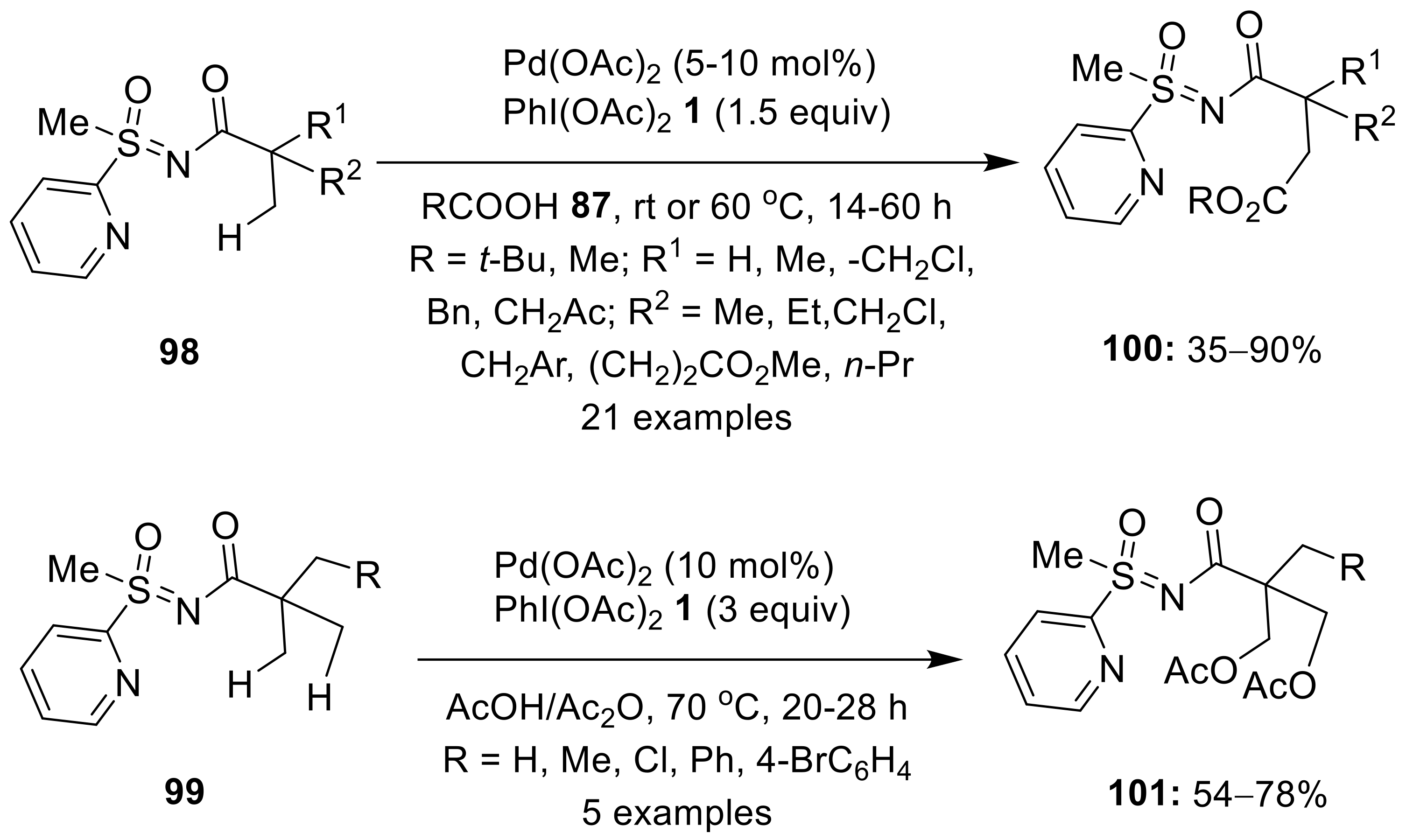{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
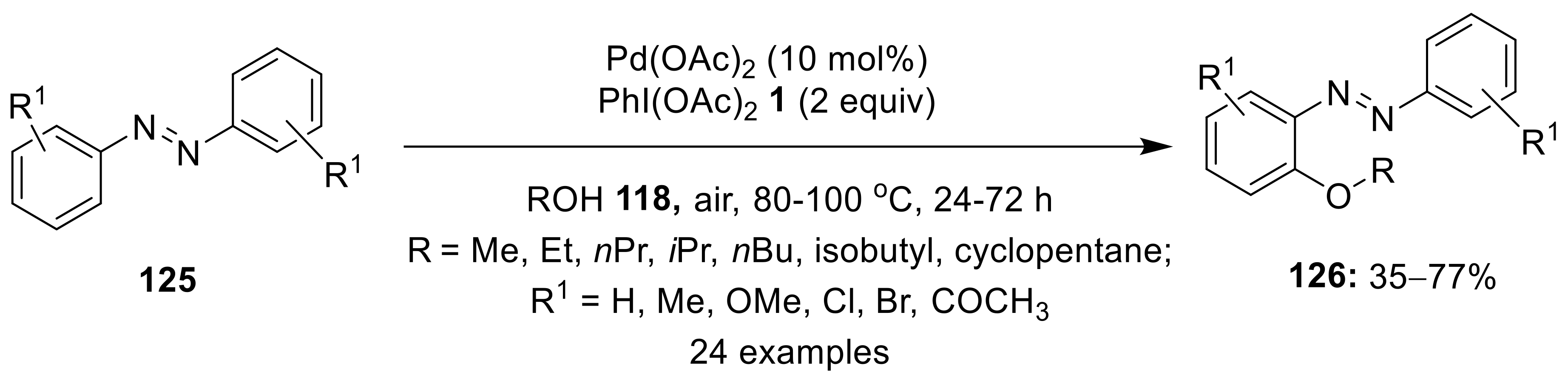{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
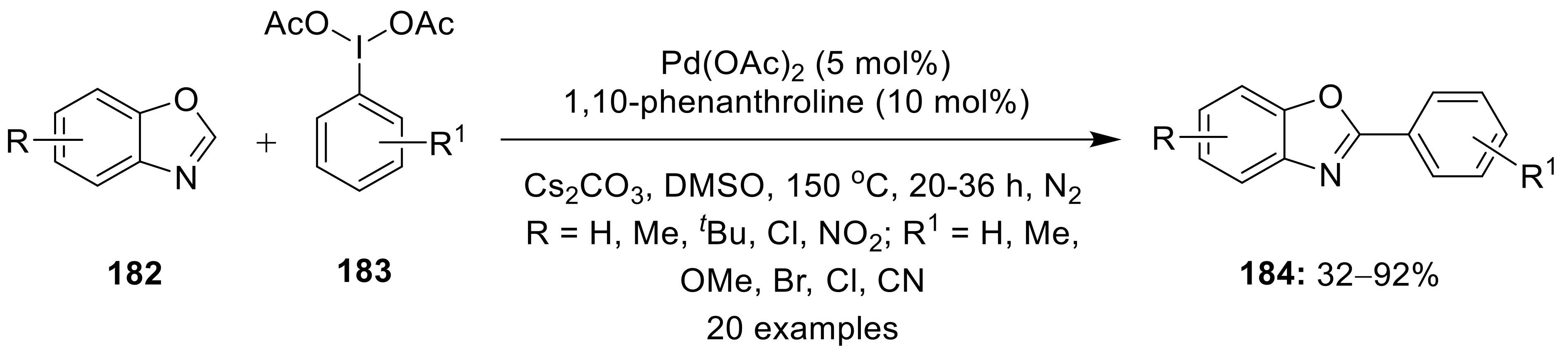{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
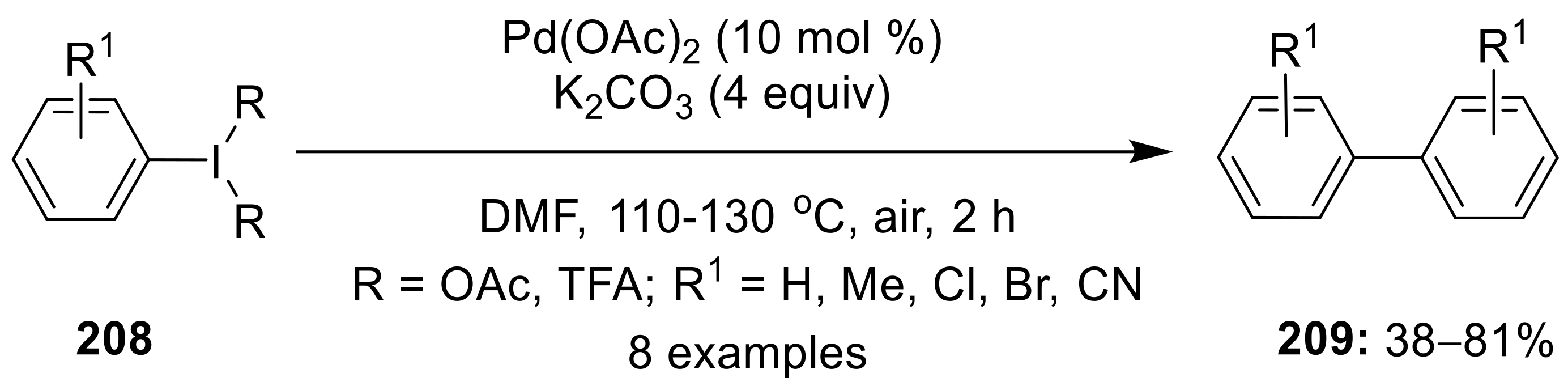{kind=link}
{kind=link}
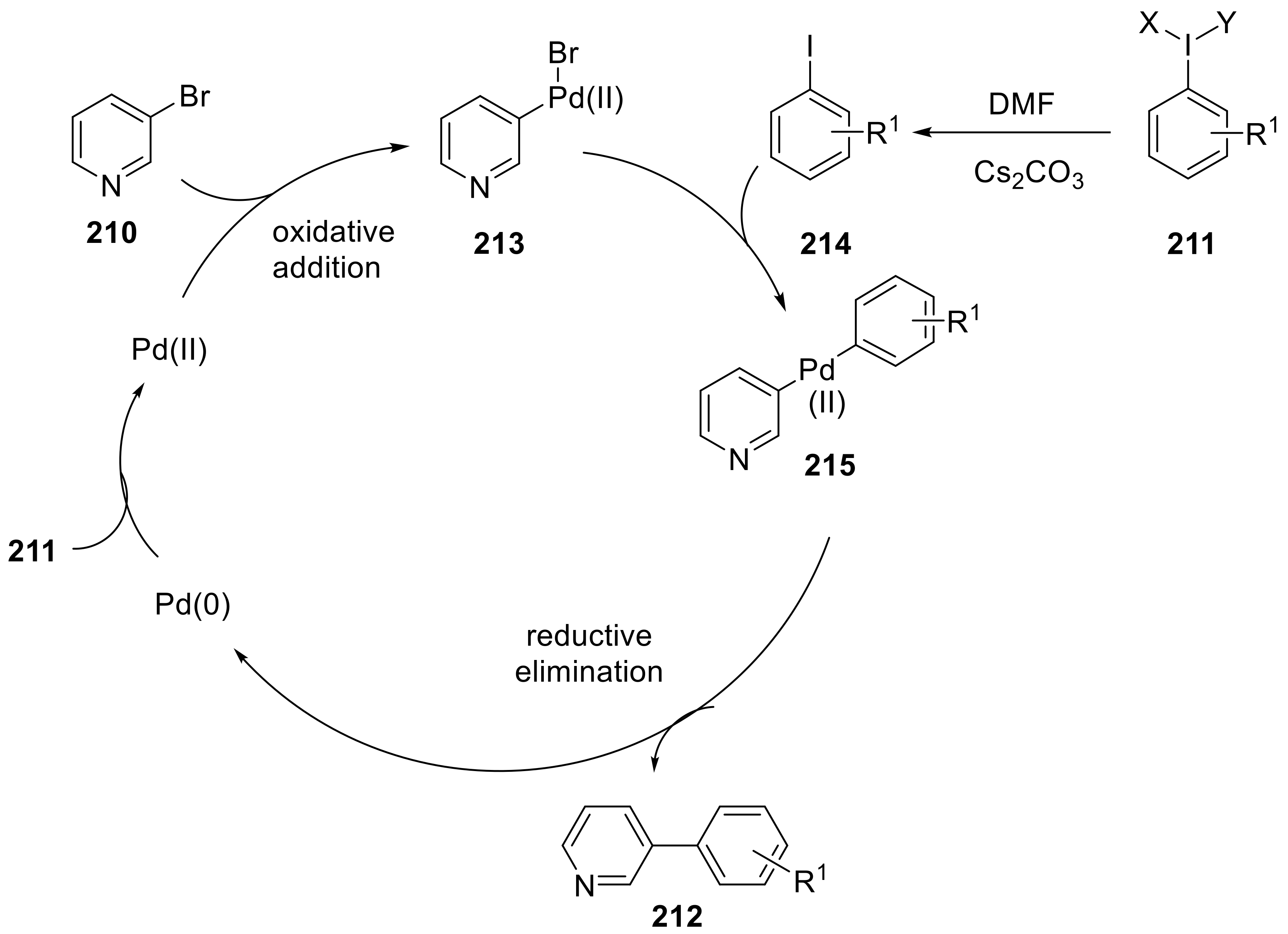{kind=link}
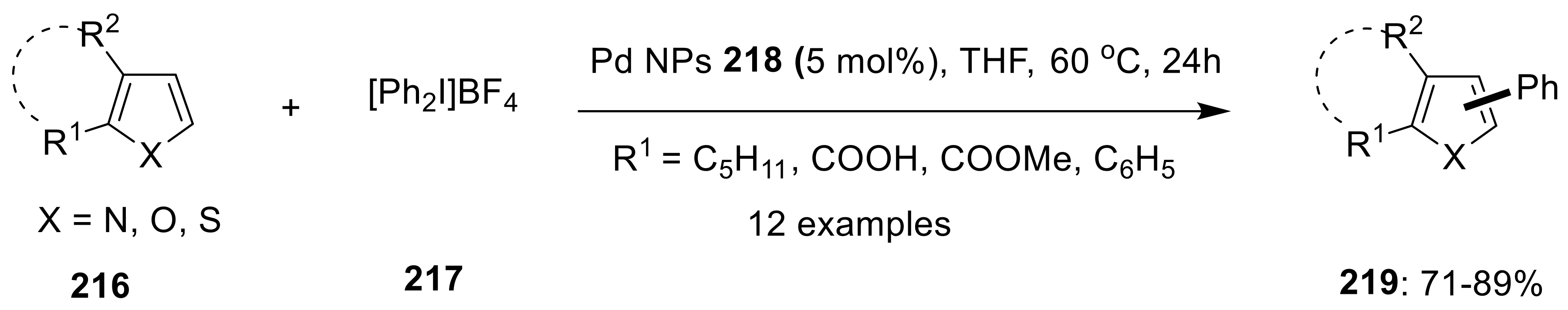{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
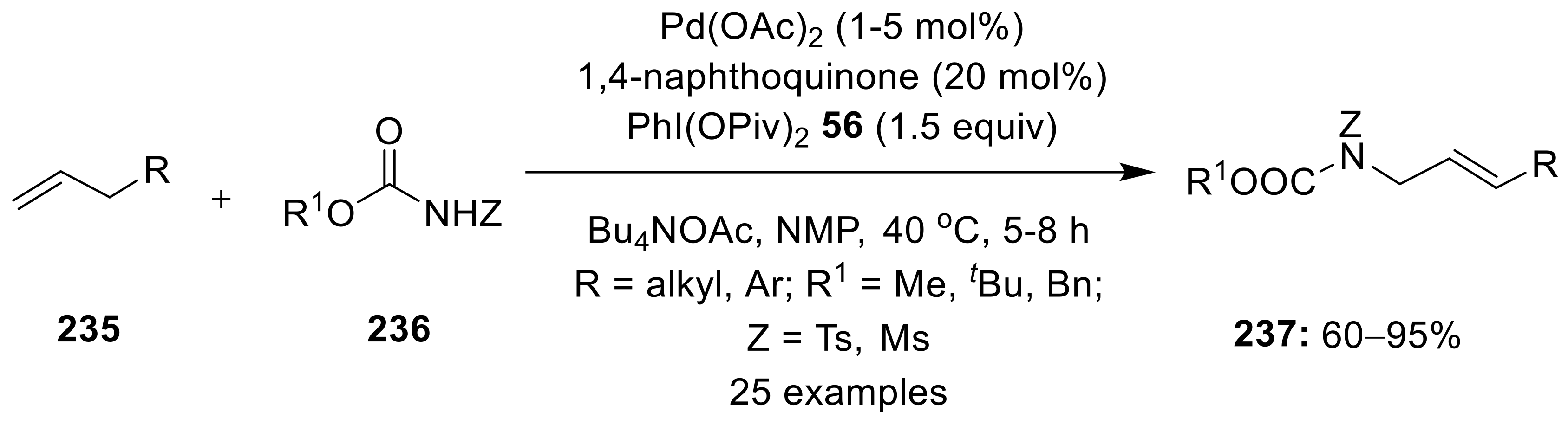{kind=link}
{kind=link}
{kind=link}
{kind=link}
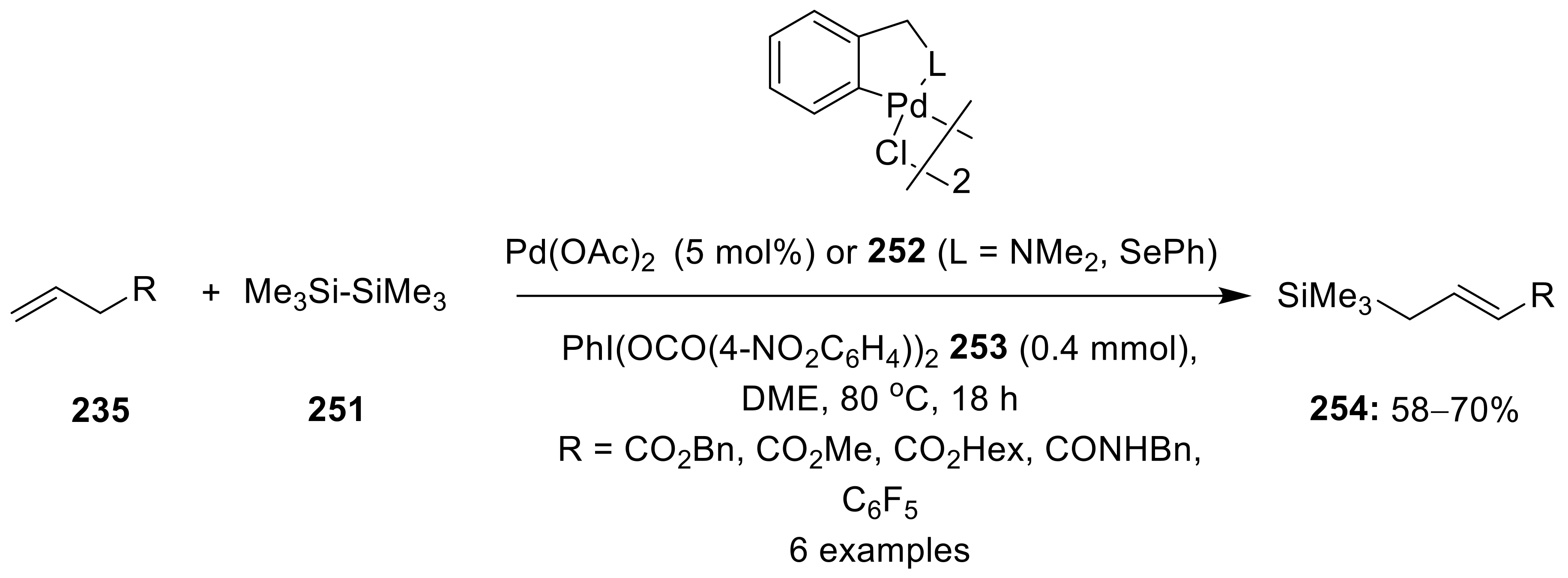{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
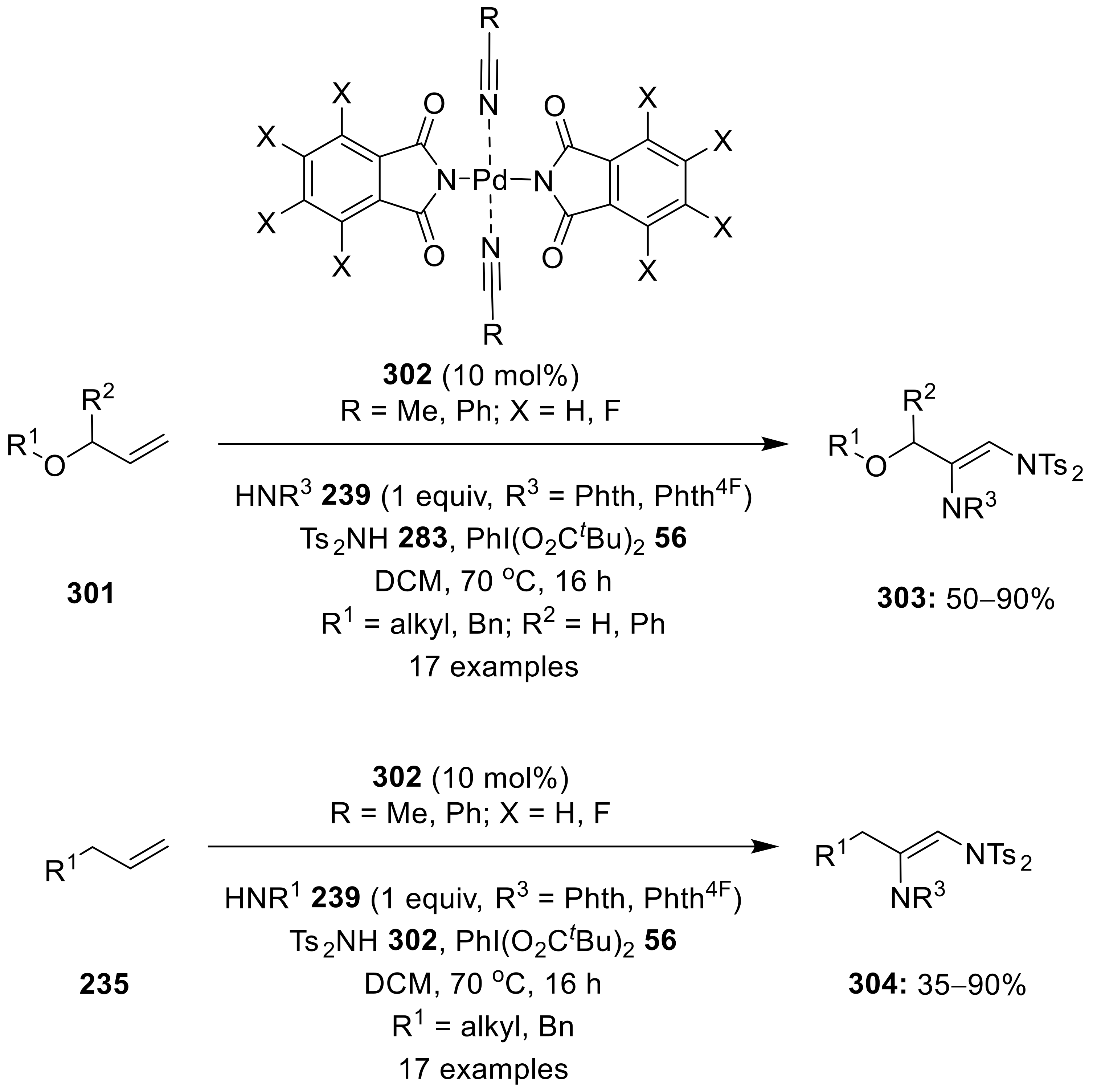{kind=link}
{kind=link}
{kind=link}
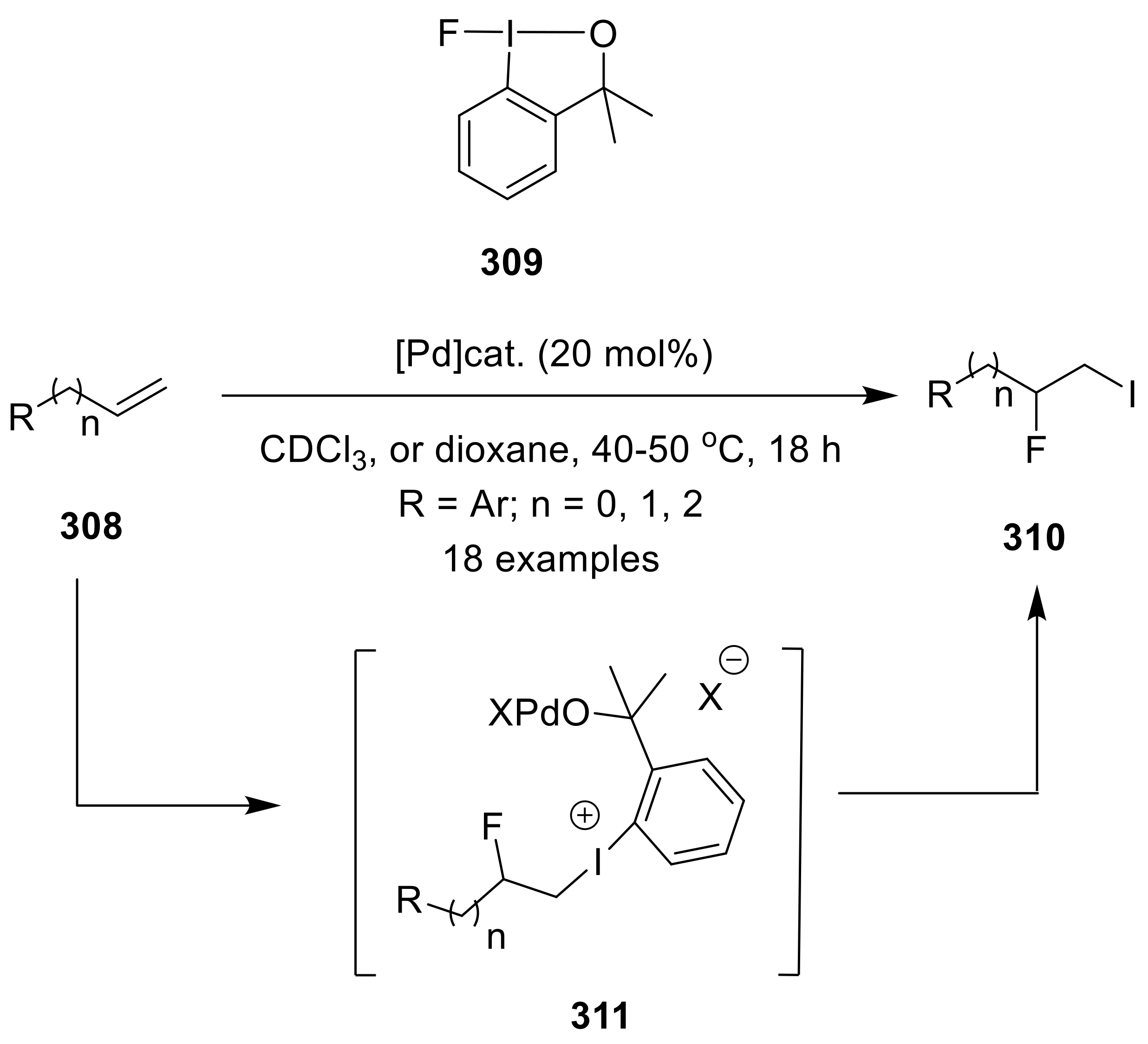{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. C—O Bond Formation
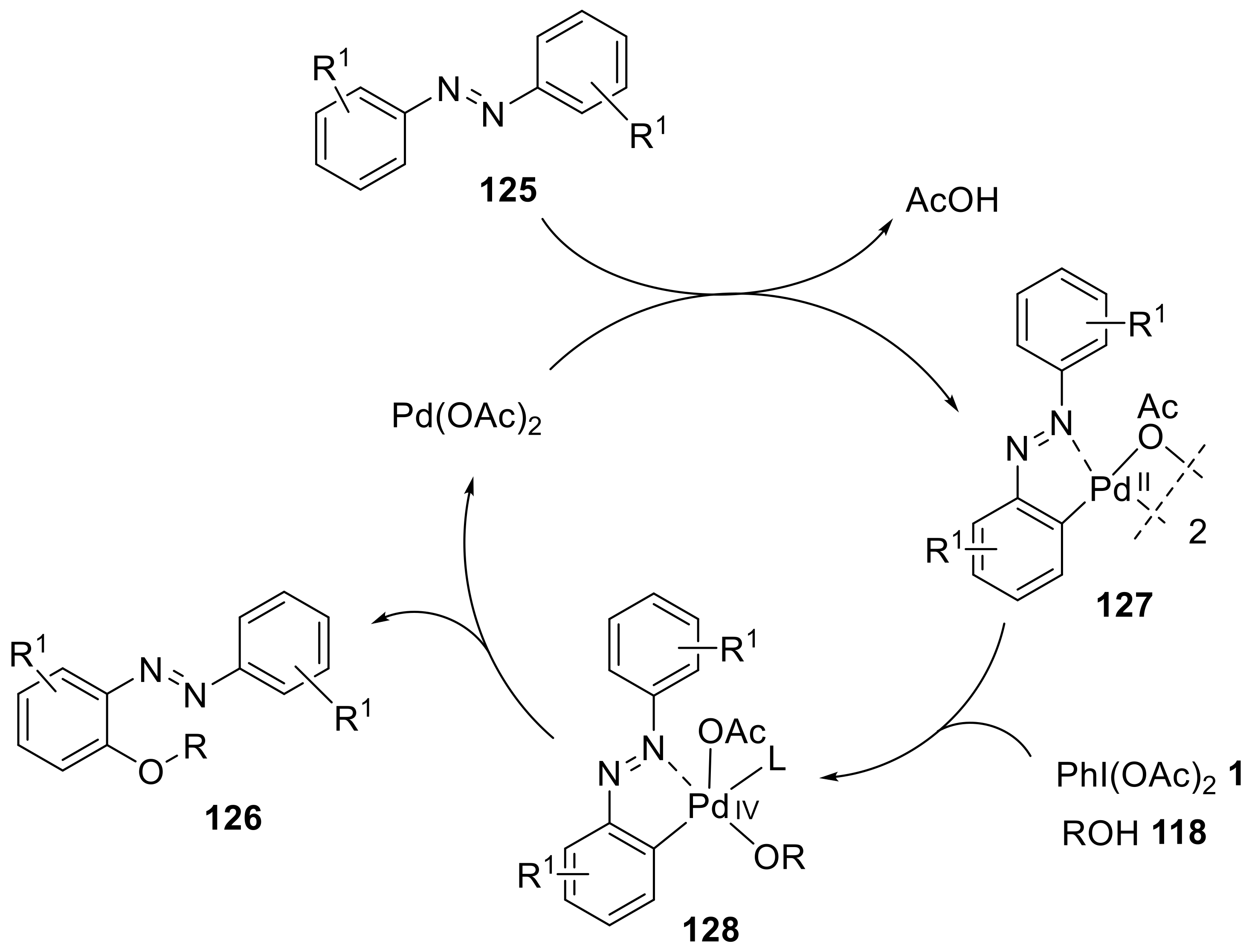
2.1. C—H Cyclization
2.2. C(sp2/sp3)—H Acyloxylation
2.2.1. C(sp2)—H Acyloxylation
2.2.2. C(sp3)—H Acyloxylation
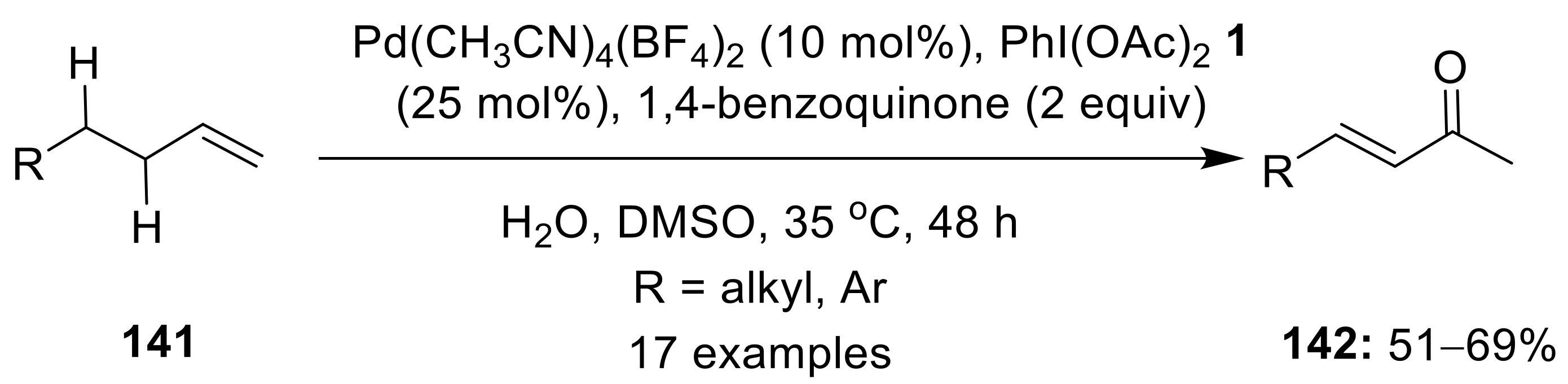
2.3. C(sp2/sp3)—H Alkoxylation
2.4. C(sp2)—H Oxidation
2.5. C—H Phosphorylation/Sulfonation
2.6. Miscellenous
3. C—C Bond Formation
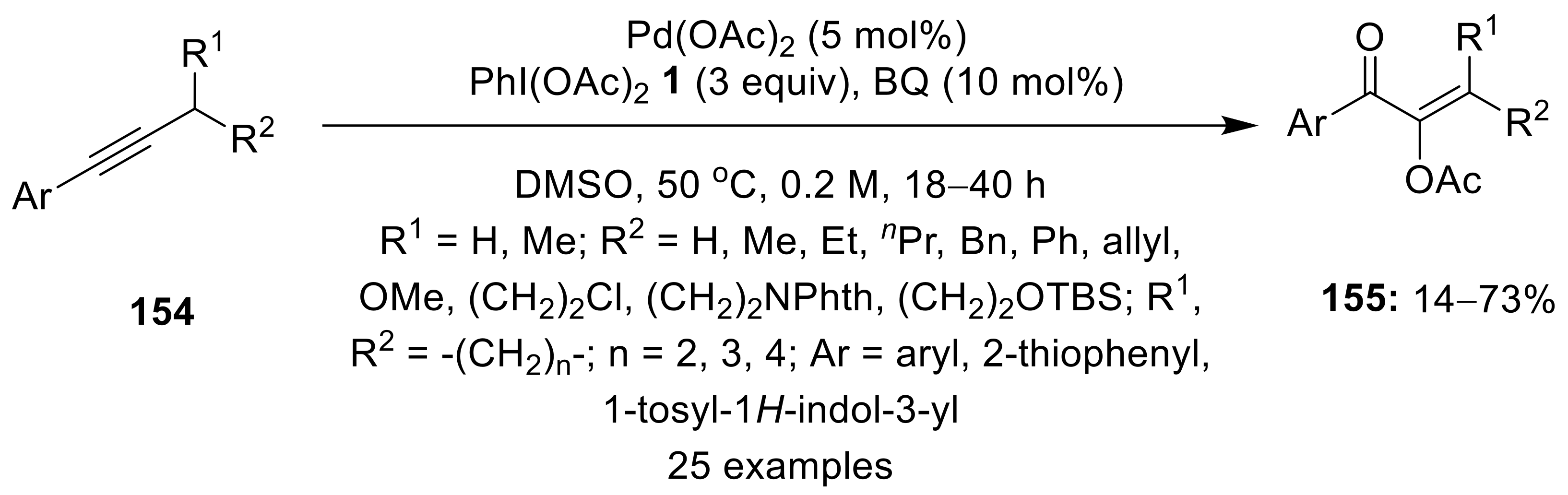
3.1. Via Oxidative Cyclization
3.2. Via C—H Bond Arylation
3.3. Via C—H Fluoroalkenylation
3.4. Via C—H Alkynylation
3.5. Via Coupling
4. C—N Bond Formation
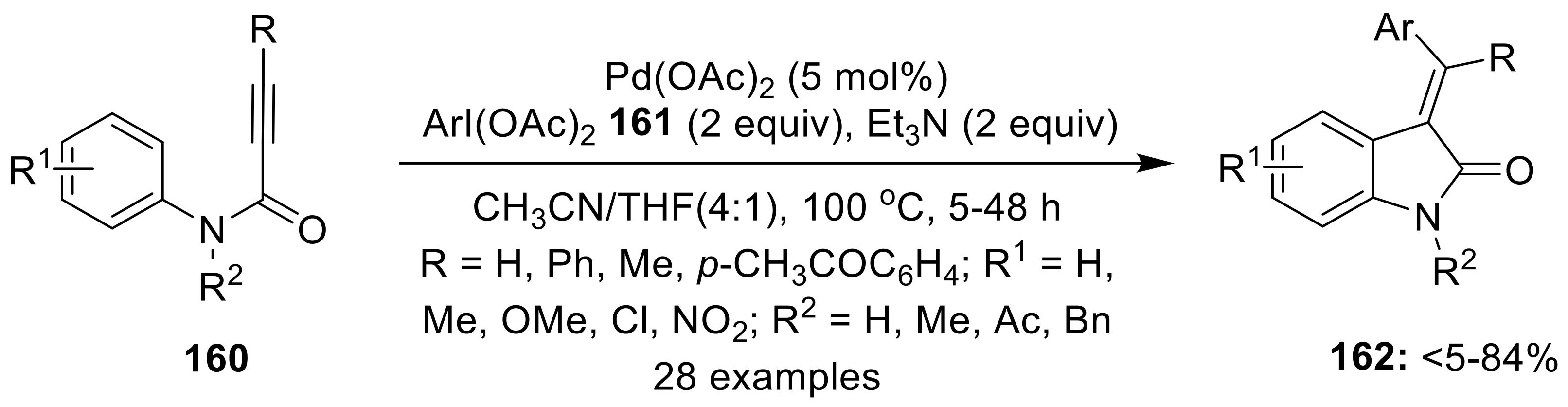
4.1. Via Intramolecular C(sp2/sp3)—H Bond Functionalization
4.2. Via Intermolecular C(sp2)—H Bond Functionalization
5. C—B, C—Si, and C—Halogen Bond Formation
6. Alkene Difunctionalization
6.1. Pd(II)-Catalyzed 1,1-Difunctionalization of Alkenes
6.2. 1,2-Difunctionalization of Alkenes
6.2.1. Intramolecular 1,2-Difunctionalization of Alkenes
6.2.2. Intermolecular 1,2-Difunctionalization of Alkenes
7. Miscellaneous
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Wirth, T. “Stereoselective reactions” Patai’s Chemistry of Functional Groups, 1st ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018. [Google Scholar]
- Yusubov, M.S.; Zhdankin, V.V. Hypervalent iodine reagents and green chemistry. Curr. Org. Synth. 2012, 9, 247–272. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Oxidative functionalization with hypervalent halides. In Comprehensive Organic Synthesis II; Elsevier: Amsterdam, The Netherlands, 2014; pp. 880–933. [Google Scholar]
- Singh, F.V.; Wirth, T. Hypervalent iodine-catalyzed oxidative functionalizations including stereoselective reactions. Chem. Asian J. 2014, 9, 950–971. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Wirth, T. Catalytic oxidation with hypervalent iodine. In Catalytic Oxidation in Organic Synthesis; Thieme: Teningen, Germany, 2017; Volume 1, pp. 29–62. [Google Scholar]
- Singh, F.V.; Mangaonkar, S.R.; Kole, P.B. Ultrasound-assisted rapid synthesis of β-cyanoepoxides using hypervalent iodine reagents. Synth. Commun. 2018, 48, 2169–2176. [Google Scholar] [CrossRef]
- Mangaonkar, S.R.; Singh, F.V. Hypervalent iodine(III)-catalyzed epoxidation of β-cyanostyrenes. Synthesis 2019, 51, 4473–4486. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Hypervalent iodine chemistry and light: Photochemical reactions involving hypervalent iodine chemistry. Arkivoc 2021, 12–47. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Singh, F.V. Hypervalent iodine-mediated synthesis and late-stage functionalization of heterocycles. Arkivoc 2020, 86–161. [Google Scholar] [CrossRef]
- Dasgupta, A.; Thiehoff, C.; Newman, P.D.; Wirth, T.; Melen, R.L. Reactions promoted by hypervalent iodine reagents and boron Lewis acids. Org. Biomol. Chem. 2021, 19, 4852–4865. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, F.V.; Takenaga, N.; Dohi, T. Asymmetric direct/stepwise dearomatization reactions involving hypervalent iodine reagents. Chem. Asian J. 2022, 17, e202101115. [Google Scholar] [CrossRef]
- Yang, X.-G.; Hu, Z.-N.; Jia, M.-C.; Du, F.-H.; Zhang, C. Recent advances and the prospect of hypervalent iodine chemistry. Synlett 2021, 32, 1289–1296. [Google Scholar]
- Rimi, R.; Soni, S.; Uttam, B.; China, H.; Dohi, T.; Zhdankin, V.; Kumar, R. Recyclable Hypervalent Iodine Reagents in Modern Organic Synthesis. Synthesis 2022, 54, 2731–2748. [Google Scholar]
- Chennaiah, A.; Vankar, Y.D. One-Step TEMPO-Catalyzed and Water-Mediated Stereoselective Conversion of Glycals into 2-Azido-2-deoxysugars with a PIFA–Trimethylsilyl Azide Reagent System. Org. Lett. 2018, 20, 2611–2614. [Google Scholar] [CrossRef]
- Chennaiah, A.; Verma, A.K.; Vankar, Y.D. TEMPO-Catalyzed Oxidation of 3-O-Benzylated/Silylated Glycals to the Corresponding Enones Using a PIFA–Water Reagent System. J. Org. Chem. 2018, 83, 10535–10540. [Google Scholar] [CrossRef]
- Chennaiah, A.; Bhowmick, S.; Vankar, Y.D. Conversion of glycals into vicinal-1,2- diazides and 1,2-(or 2,1)-azidoacetates using hypervalent iodine reagents and Me3SiN3. Application in the synthesis of N-glycopeptides, pseudo-trisaccharides and an iminosugar. RSC Adv. 2017, 7, 41755–41762. [Google Scholar] [CrossRef]
- Merritt, E.A.; Olofsson, B. α-Functionalization of carbonyl compounds using hypervalent iodine reagents. Synthesis 2011, 4, 517–538. [Google Scholar]
- Dong, D.-Q.; Hao, S.-H.; Wang, Z.-L.; Chen, C. Hypervalent iodine: A powerful electrophile for asymmetric α-functionalization of carbonyl compounds. Org. Biomol. Chem. 2014, 12, 4278–4289. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Selenium-catalyzed regioselective cyclization of unsaturated carboxylic acids using hypervalent iodine oxidants. Org. Lett. 2011, 13, 6504–6507. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Hypervalent iodine(III) mediated cyclization of ortho-Stillbenes into benzofurans. Synthesis 2012, 44, 1171–1177. [Google Scholar]
- Singh, F.V.; Mangaonkar, S.R. Hypervalent iodine(III)-catalyzed synthesis of 2-arylbenzofurans. Synthesis 2018, 50, 4940–4948. [Google Scholar] [CrossRef]
- Singh, F.V.; Kole, P.B.; Mangaonkar, S.R.; Shetgaonkar, S.E. Synthesis of spirocyclic scaffolds using hypervalent iodine reagents. Beilstein J. Org. Chem. 2018, 14, 1778–1805. [Google Scholar] [CrossRef]
- Singh, F.V.; Rehbein, J.; Wirth, T. Facile oxidative rearrangements using hypervalent iodine reagents. Chem. Open 2012, 1, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Wirth, T. Oxidative rearrangements with hypervalent iodine reagents. Synthesis 2013, 45, 2499–2511. [Google Scholar]
- Maertens, G.; Canesi, S. Rearrangements induced by hypervalent iodine. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2015; Volume 373, pp. 223–241. [Google Scholar]
- Tan, Y.; Wang, J.; Zhang, H.-Y.; Zhang, Y.; Zhao, J. The C3-H bond functionalization of quinoxalin-2(1H)-ones with hypervalent iodine(III) reagents. Front. Chem. 2020, 8, 582. [Google Scholar]
- Li, X.; Chen, P.; Liu, G. Recent advances in hypervalent iodine(III)-catalyzed functionalization of alkenes. Beilstein J. Org. Chem. 2018, 14, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, S.; Hong, K.B. Alkene difunctionalization using hypervalent iodine reagents: Progress and developments in the past ten years. Molecules 2019, 24, 2634. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.-Z.; Zhang, C.-P. Fluorination and fluoroalkylation reactions mediated by hypervalent iodine reagents. Adv. Synth. Catal. 2020, 362, 4256–4292. [Google Scholar] [CrossRef]
- Li, Y.; Hari, D.P.; Vita, M.V.; Waser, J. Cyclic hypervalent iodine reagents for atom-transfer reactions: Beyond trifluoromethylation. Angew. Chem. Int. Ed. 2016, 55, 4436–4454. [Google Scholar] [CrossRef]
- Mangaonkar, S.R.; Kole, P.B.; Singh, F.V. Oxidation of Organosulfides to Organosulfones with Trifluoromethyl 3-Oxo-1λ3,2-benziodoxole-1(3H)-carboxylate as an Oxidant. Synlett 2018, 29, 199–202. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Krishnan, M.; Singh, F.V. Hypervalent Iodine Reagents for Oxidative Rearrangements. Mini Rev. Org. Chem. 2021, 18, 138–158. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Zotto, A.D.; Zecca, M. Pd metal catalysts for cross-couplings and related reactions in the 21st Century: A critical review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Deprez, N.R.; Sanford, M.S. Reactions of hypervalent iodine reagents with palladium: Mechanisms and applications in organic synthesis. Inorg. Chem. 2007, 46, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.C.S.; Tierno, A.F.; Wengryniuk, S.E. Hypervalent iodine reagents in high valent transition metal chemistry. Molecules 2017, 22, 780. [Google Scholar] [CrossRef] [PubMed]
- Shetgaonkar, S.E.; Singh, F.V. Hypervalent iodine reagent in palladium-catalyzed oxidative cross-coupling reactions. Front. Chem. 2020, 8, 705. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Y.; Dai, H.-X.; Yu, J.-Q. Pd(II)-catalyzed hydroxyl-directed C—H activation/C—O cyclization: Expedient construction of dihydrobenzofurans. J. Am. Chem. Soc. 2010, 132, 12203–12205. [Google Scholar] [CrossRef]
- Huang, C.; Ghavtadze, N.; Chattopadhyay, B.; Gevorgyan, V. Synthesis of catechols from phenols via Pd-catalyzed silanol-directed C—H oxygenation. J. Am. Chem. Soc. 2011, 133, 17630–17633. [Google Scholar] [CrossRef]
- Huang, C.; Ghavtadze, N.; Godoi, B.; Gevorgyan, V. Pd-catalyzed modifiable silanol-directed aromatic C—H oxygenation. Chem. Eur. J. 2012, 18, 9789–9792. [Google Scholar] [CrossRef]
- Thompson, S.J.; Thach, D.Q.; Dong, G. Cyclic ether synthesis via palladium-catalyzed directed dehydrogenative annulation at unactivated terminal positions. J. Am. Chem. Soc. 2015, 137, 11586–11589. [Google Scholar] [CrossRef]
- Yang, M.; Jiang, X.; Shi, W.-J.; Zhu, Q.-L.; Shi, Z.-J. Direct lactonization of 2-arylacetic acids through Pd(II)-catalyzed C—H activation/C—O formation. Org. Lett. 2013, 15, 690–693. [Google Scholar] [CrossRef]
- Cheng, X.-F.; Li, Y.; Su, Y.-M.; Yin, F.; Wang, J.-Y.; Sheng, J.; Vora, H.U.; Wang, X.-S.; Yu, J.-Q. Pd(II)-catalyzed enantioselective C—H activation/C—O bond formation: Synthesis of chiral benzofuranones. J. Am. Chem. Soc. 2013, 135, 1236–1239. [Google Scholar] [CrossRef]
- Li, Y.; Ding, Y.-J.; Wang, J.-Y.; Su, Y.-M.; Wang, X.-S. Pd-Catalyzed C—H lactonization for expedient synthesis of biaryl lactones and total synthesis of Cannabinol. Org. Lett. 2013, 15, 2574–2577. [Google Scholar] [CrossRef]
- Liu, B.; Shi, B.-F. γ-Lactone synthesis via palladium(II)-catalyzed lactonization of unactivated methylene C(sp3)—H Bonds. Synlett 2016, 27, 2396–2400. [Google Scholar]
- Gu, S.; Chen, C.; Chen, W. Ortho-functionalization of 2-phenoxypyrimidines via palladium-catalyzed C—H bond activation. J. Org. Chem. 2009, 74, 7203–7206. [Google Scholar] [CrossRef]
- Gou, F.-R.; Wang, X.-C.; Huo, P.-F.; Bi, H.-P.; Guan, Z.-H.; Liang, Y.-M. Palladium-catalyzed aryl C—H bonds activation/acetoxylation utilizing a bidentate system. Org. Lett. 2009, 11, 5726–5729. [Google Scholar] [CrossRef]
- Neufeldt, S.R.; Sanford, M.S. O-Acetyl oximes as transformable directing groups for Pd-catalyzed C—H bond functionalization. Org. Lett. 2010, 12, 532–535. [Google Scholar] [CrossRef]
- Chernyak, N.; Dudnik, A.S.; Huang, C.; Gevorgyan, V. PyDipSi: A general and easily modifiable/traceless Si-tethered directing group for C—H acyloxylation of arenes. J. Am. Chem. Soc. 2010, 132, 8270–8272. [Google Scholar] [CrossRef][Green Version]
- Gulevich, A.V.; Melkonyan, F.S.; Sarkar, D.; Gevorgyan, V. Double-Fold C—H oxygenation of arenes using PyrDipSi: A general and efficient traceless/modifiable silicon-tethered directing group. J. Am. Chem. Soc. 2012, 134, 5528–5531. [Google Scholar] [CrossRef][Green Version]
- Ye, X.; He, Z.; Ahmed, T.; Weise, K.; Akhmedov, N.G.; Petersen, J.L.; Shi, X. 1,2,3-triazoles as versatile directing group for selective sp2 and sp3 C—H activation: Cyclization vs substitution. Chem. Sci. 2013, 4, 3712–3716. [Google Scholar] [CrossRef]
- Ren, Z.; Schulz, J.E.; Dong, G. Catalytic ortho-acetoxylation of masked benzyl alcohols via an exo-directing mode. Org. Lett. 2015, 17, 2696–2699. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Yang, T.; Li, L.; Li, D. Palladium catalyzed ortho-C—H-benzoxylation of 2-arylpyridines using iodobenzene dibenzoates. Tetrahedron Lett. 2015, 56, 6136–6141. [Google Scholar] [CrossRef]
- Dick, A.R.; Kampf, J.W.; Sanford, M.S. Unusually Stable Palladium(IV) Complexes: Detailed Mechanistic Investigation of C—O Bond-Forming Reductive Elimination. J. Am. Chem. Soc. 2005, 127, 12790–12791. [Google Scholar] [CrossRef]
- Emmert, M.H.; Cook, A.K.; Xie, Y.J.; Sanford, M.S. Remarkably high reactivity of Pd(OAc)2/pyridine catalysts: Nondirected C—H oxygenation of arenes. Angew. Chem. Int. Ed. 2011, 50, 9409–9412. [Google Scholar] [CrossRef]
- Cook, A.K.; Emmert, M.H.; Sanford, M.S. Steric control of site selectivity in the Pd-catalyzed C—H acetoxylation of simple arenes. Org. Lett. 2013, 15, 5428–5431. [Google Scholar] [CrossRef]
- Mutule, I.; Suna, E.; Olofsson, K.; Pelcman, B. Catalytic direct acetoxylation of indoles. J. Org. Chem. 2009, 74, 7195–7198. [Google Scholar] [CrossRef]
- Choy, P.Y.; Lau, C.P.; Kwong, F.Y. Palladium-catalyzed direct and regioselective C—H bond functionalization/oxidative acetoxylation of indoles. J. Org. Chem. 2011, 76, 80–84. [Google Scholar] [CrossRef]
- Liu, Q.; Li, G.; Yi, H.; Wu, P.; Liu, J.; Lei, A. Pd-catalyzed direct and selective C—H functionalization: C 3-acetoxylation of indoles. Chem. Eur. J. 2011, 17, 2353–2357. [Google Scholar] [CrossRef]
- Pilarski, L.T.; Selander, N.; Bose, D.; Szabó, K.J. Catalytic allylic C—H acetoxylation and benzoyloxylation via suggested (η3-allyl)palladium(IV) intermediates. Org. Lett. 2009, 11, 5518–5521. [Google Scholar] [CrossRef]
- Alam, R.; Pilarski, L.T.; Pershagen, E.; Szabó, K.J. Stereoselective intermolecular allylic C—H trifluoroacetoxylation of functionalized alkenes. J. Am. Chem. Soc. 2012, 134, 8778–8781. [Google Scholar] [CrossRef]
- Zhang, S.; Luo, F.; Wang, W.; Jia, X.; Hu, M.; Cheng, J. Chelation-assisted palladium-catalyzed acyloxylation of benzyl sp3 C—H bonds using PhI(OAc)2 as oxidant. Tetrahedron Lett. 2010, 51, 3317–3319. [Google Scholar] [CrossRef]
- Ren, Z.; Mo, F.; Dong, G. Catalytic functionalization of unactivated sp3 C—H bonds via exo-directing groups: Synthesis of chemically differentiated 1,2-diols. J. Am. Chem. Soc. 2012, 134, 16991–16994. [Google Scholar] [CrossRef] [PubMed]
- Rit, R.K.; Yadav, M.R.; Sahoo, A.K. Pd(II)-Catalyzed primary-C(sp3)—H acyloxylation at room temperature. Org. Lett. 2012, 14, 3724–3727. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Yao, J.; Wu, Z.; Liu, Z.; Zhang, Y. Palladium-catalyzed oxidative acetoxylation of benzylic C—H bond using bidentate auxiliary. J. Org. Chem. 2013, 78, 10821–10831. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, S.-Y.; He, G.; Nack, W.A.; Chen, G. Palladium-catalyzed picolinamide-directed acetoxylation of unactivated γ-C(sp3)—H bonds of alkylamines. Adv. Synth. Catal. 2014, 356, 1544–1548. [Google Scholar] [CrossRef]
- Check, C.T.; Henderson, W.H.; Wray, B.C.; Eynden, M.J.V.; Stambuli, J.P. Oxidant-controlled stereoselectivity in the Pd-catalyzed allylic oxidation of cis-vinylsilanes. J. Am. Chem. Soc. 2011, 133, 18503–18505. [Google Scholar] [CrossRef]
- Vijaykumar, M.; Punji, B. Pd(II)-Catalyzed Chemoselective Acetoxylation of C(sp2)—H and C(sp3)—H Bonds in Tertiary Amides. J. Org. Chem. 2021, 86, 8172–8181. [Google Scholar] [CrossRef]
- Abdolalian, P.; Tizhoush, S.K.; Farshadfar, K.; Ariafard, A. The role of hypervalent iodine (III) reagents in promoting alkoxylation of unactivated C (sp3)—H bonds catalyzed by palladium (II) complexes. Chem. Sci. 2021, 12, 7185–7195. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; He, G.; Zhao, Y.; Wright, K.; Nack, W.A.; Chen, G. Efficient alkyl ether synthesis via palladium-catalyzed, picolinamide-directed alkoxylation of unactivated C(sp3)—H and C(sp2)—H bonds at remote positions. J. Am. Chem. Soc. 2012, 134, 7313–7316. [Google Scholar] [CrossRef]
- Chen, F.-J.; Zhao, S.; Hu, F.; Chen, K.; Zhang, Q.; Zhang, S.-Q.; Shi, B.-F. Pd(II)-catalyzed alkoxylation of unactivated C(sp3)—H and C(sp2)—H bonds using a removable directing group: Efficient synthesis of alkyl ethers. Chem. Sci. 2013, 4, 4187–4192. [Google Scholar] [CrossRef]
- Yin, Z.; Jiang, X.; Sun, P. Palladium-catalyzed direct ortho alkoxylation of aromatic azo compounds with alcohols. J. Org. Chem. 2013, 78, 10002–10007. [Google Scholar] [CrossRef]
- Shan, G.; Yang, X.; Zong, Y.; Rao, Y. An efficient palladium-catalyzed C—H alkoxylation of unactivated methylene and methyl groups with cyclic hypervalent iodine (I3+) oxidants. Angew. Chem. Int. Ed. 2013, 52, 13606–13610. [Google Scholar] [CrossRef]
- Zong, Y.; Rao, Y. Developing Pd(II) catalyzed double sp3 C—H alkoxylation for synthesis of symmetric and unsymmetric Acetal. Org. Lett. 2014, 16, 5278–5281. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, P. Palladium-catalyzed direct C(sp2)—H alkoxylation of 2-aryloxypyridines using 2-pyridyloxyl as the directing group. J. Org. Chem. 2014, 79, 8457–8461. [Google Scholar] [CrossRef] [PubMed]
- Bigi, M.A.; White, M.C. Terminal olefins to linear α,β-unsaturated ketones: Pd(II)/hypervalent iodine co-catalyzed Wacker oxidation–dehydrogenation. J. Am. Chem. Soc. 2013, 135, 7831–7834. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, D.A.; Fernandes, R.A. Hypervalent iodine as a terminal oxidant in Wacker-type oxidation of terminal olefins to methyl ketones. J. Org. Chem. 2016, 81, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, L.; Xie, L.; Liu, P.; Wei, Q.; Mao, F.; Zhang, X.; Huang, J.; Chen, S.; Huang, C. Palladium-catalyzed C—H bond functionalization reactions using phosphate/sulfonate hypervalent iodine reagents. J. Org. Chem. 2019, 84, 10088–10101. [Google Scholar] [CrossRef] [PubMed]
- Gondo, K.; Oyamada, J.; Kitamura, T. Palladium-catalyzed desilylative acyloxylation of silicon–carbon bonds on (trimethylsilyl)arenes: Synthesis of phenol derivatives from trimethylsilylarenes. Org. Lett. 2015, 17, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Quan, X.; Zhu, C.; Andersson, P.G.; Backvall, J.-E. Palladium-catalyzed oxidative synthesis of α-acetoxylated enones from alkynes. Angew. Chem. Int. Ed. 2016, 55, 5824–5828. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, S.; Zhu, G. Divergent synthesis of 2-aminofurans via palladium-catalyzed acetoxylative, alkoxylative, and hydroxylative cycloisomerization of homoallenyl amides. J. Org. Chem. 2015, 80, 7604–7612. [Google Scholar] [CrossRef]
- Tang, S.; Peng, P.; Zhong, P.; Li, J. Palladium-catalyzed C—H functionalization of N-arylpropiolamides with aryliodonium salts: Selective synthesis of 3-(1-arylmethylene)oxindoles. J. Org. Chem. 2008, 73, 5476–5480. [Google Scholar] [CrossRef]
- Tang, S.; Peng, P.; Pi, S.; Liang, Y.; Wang, N.; Li, J. Sequential intermolecular aminopalladation/ortho-arene C—H activation reactions of N-phenylpropiolamides with phthalimide. Org. Lett. 2008, 10, 1179–1182. [Google Scholar] [CrossRef]
- Tang, S.; Peng, P.; Wang, Z.; Tang, B.; Deng, C.; Li, J.; Zhong, P.; Wang, N. Synthesis of (2-oxoindolin-3-ylidene)methyl acetates involving a C—H functionalization process. Org. Lett. 2008, 10, 1875–1878. [Google Scholar] [CrossRef]
- Tong, X.; Beller, M.; Tse, M.K. A Palladium-catalyzed cyclization-oxidation sequence: Synthesis of bicyclo [3.1.0]hexanes and evidence for SN2 C—O bond formation. J. Am. Chem. Soc. 2007, 129, 4906–4907. [Google Scholar] [CrossRef]
- Welbes, L.L.; Lyons, T.W.; Cychosz, K.A.; Sanford, M.S. Synthesis of cyclopropanes via Pd(II/IV)-catalyzed reactions of enynes. J. Am. Chem. Soc. 2007, 129, 5836–5837. [Google Scholar] [CrossRef]
- Liu, H.; Yu, J.; Wang, L.; Tong, X. Cyclization–oxidation of 1,6-enyne derivatived from Baylis–Hillman adducts via Pd(II)/Pd(IV)-catalyzed reactions: Stereoselective synthesis of multi-substituted bicyclo [3.1.0] hexanes and insight into reaction pathways. Tetrahedron Lett. 2008, 49, 6924–6928. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium (II/IV) catalyzed cyclopropanation reactions: Scope and mechanism. Tetrahedron 2009, 65, 3211–3221. [Google Scholar] [CrossRef]
- Tsujihara, T.; Takenaka, K.; Onitsuka, K.; Hatanaka, M.; Sasai, H. PdII/PdIV Catalytic enantioselective synthesis of bicyclo [3.1.0]hexanes via oxidative cyclization of enynes. J. Am. Chem. Soc. 2009, 131, 3452–3453. [Google Scholar] [CrossRef]
- Mu, X.; Wu, T.; Wang, H.; Guo, Y.-L.; Liu, G. Palladium-catalyzed oxidative aryltrifluoromethylation of activated alkenes at room temperature. J. Am. Chem. Soc. 2012, 134, 878–881. [Google Scholar] [CrossRef]
- Xu, T.; Wang, D.; Tong, X. Pd(II)-catalyzed intramolecular acetoxylative (3 + 2) annulation of propargylic alcohol and alkene: Polycyclic oxa-heterocycle synthesis and mechanistic insight. Org. Lett. 2019, 21, 5368–5372. [Google Scholar] [CrossRef]
- Qu, X.; Sun, P.; Li, T.; Mao, J. Ligand-free reusable palladium-catalyzed Heck-type coupling reactions of hypervalent iodine reagents under mild conditions. Adv. Synth. Catal. 2011, 353, 1061–1066. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Kornienko, A.; Magedov, I.V. Aryliodine(III) diacetates as substrates for Pd–Ag catalyzed arylation of alkenes. Tetrahedron Lett. 2011, 52, 4327–4329. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, G.; Chen, F.; Cheng, J. Direct arylation of benzoxazole C—H bonds with iodobenzene diacetates. Tetrahedron Lett. 2012, 53, 4588–4590. [Google Scholar] [CrossRef]
- Fu, Z.; Xiong, Q.; Zhang, W.; Li, Z.; Cai, H. Pd-catalyzed direct arylation of electron-deficient polyfluoroarenes with aryliodine(III) diacetates. Tetrahedron Lett. 2015, 56, 123–126. [Google Scholar] [CrossRef]
- Mu, X.; Chen, S.; Zhen, X.; Liu, G. Palladium-catalyzed oxidative trifluoromethylation of indoles at room temperature. Chem. Eur. J. 2011, 17, 6039–6042. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Cao, C.K.; Ge, C.; Qu, H.; Chen, C. The Pd-catalyzed synthesis of difluoroethyl and difluorovinyl compounds with a chlorodifluoroethyl iodonium salt (CDFI). Chin. Chem. Lett. 2022, 33, 1541–1544. [Google Scholar]
- Tóth, B.L.; Sályi, G.; Domján, A.; Egyed, O.; Bényei, A.; Gonda, Z.; Novák, Z. Z-Selective Fluoroalkenylation of (Hetero)Aromatic Systems by Iodonium Reagents in Palladium-Catalyzed Directed C—H Activation. Adv. Synth. Catal. 2022, 364, 348–354. [Google Scholar] [CrossRef]
- Ruyet, L.; Lapuh, M.; Koshti, V.; Földesi, T.; Jubault, P.; Poisson, T.; Novák, Z.; Besset, T. Z-SelectivePd-catalyzed 2,2,2-trifluoroethylation of acrylamides at room temperature. Chem. Commun. 2021, 57, 6241–6244. [Google Scholar]
- Tolnai, G.L.; Ganss, S.; Brand, J.P.; Waser, J. C2-selective direct alkynylation of indoles. Org. Lett. 2013, 15, 112–115. [Google Scholar] [CrossRef]
- Xiong, Q.; Fu, Z.; Li, Z.; Cai, H. Synthesis of symmetrical biaryls through palladium-catalyzed ligand-free homocoupling of aryliodine(III) diacetates. Synlett 2015, 26, 975–979. [Google Scholar] [CrossRef]
- Wang, X.; He, Y.; Ren, M.; Liu, S.; Liu, H.; Huang, G. Pd-Catalyzed ligand-free synthesis of arylated heteroaromatics by coupling of N-heteroaromatic bromides with iodobenzene diacetate, iodosobenzene, or diphenyliodonium salts. J. Org. Chem. 2016, 81, 7958–7962. [Google Scholar] [CrossRef]
- Shin, T.; Kim, M.; Jung, Y.; Cho, S.J.; Kim, H.; Song, H. Characterization of heterogeneous aryl–Pd(II)–oxo clusters as active species for C—H arylation. Chem. Commun. 2020, 56, 14404–14407. [Google Scholar] [CrossRef]
- Jordan.-Hore, J.A.; Johansson, C.C.C.; Gulias, M.; Beck, E.M.; Gaunt, M.J. Oxidative Pd(II)-catalyzed C—H bond amination to carbazole at ambient temperature. J. Am. Chem. Soc. 2008, 130, 16184–16186. [Google Scholar] [CrossRef]
- Mei, T.; Leow, D.; Xiao, H.; Laforteza, B.N.; Yu, J. Synthesis of indolines via Pd(II)-catalyzed amination of C—H bonds using PhI(OAc)2 as the bystanding oxidant. Org. Lett. 2013, 15, 3058–3061. [Google Scholar] [CrossRef]
- Nadres, E.T.; Daugulis, O. Heterocycle Synthesis via direct C—H/N—H coupling. J. Am. Chem. Soc. 2012, 134, 7–10. [Google Scholar] [CrossRef]
- He, G.; Lu, C.; Zhao, Y.; Nack, W.A.; Chen, G. Improved protocol for indoline synthesis via palladium-catalyzed intramolecular C(sp2)—H amination. Org. Lett. 2012, 14, 2944–2947. [Google Scholar] [CrossRef]
- He, G.; Zhao, Y.; Zhang, S.; Lu, C.; Chen, G. Highly efficient syntheses of azetidines, pyrrolidines, and indolines via palladium catalyzed intramolecular amination of C(sp3)—H and C(sp2)—H bonds at γ and δ positions. J. Am. Chem. Soc. 2012, 134, 3–6. [Google Scholar] [CrossRef]
- Yin, G.; Wu, Y.; Liu, G. Scope and mechanism of allylic C—H amination of terminal alkenes by the palladium/PhI(OPiv)2 catalyst system: Insights into the effect of naphthoquinone. J. Am. Chem. Soc. 2010, 132, 11978–11987. [Google Scholar] [CrossRef]
- Shrestha, R.; Mukherjee, P.; Tan, Y.; Litman, Z.C.; Hartwig, J.F. Sterically controlled, palladium-catalyzed intermolecular amination of arenes. J. Am. Chem. Soc. 2013, 135, 8480–8483. [Google Scholar] [CrossRef]
- Selander, N.; Willy, B.; Szabó, K.J. Selective C—H borylation of alkenes by palladium pincer complex catalyzed oxidative functionalization. Angew. Chem. Int. Ed. 2010, 49, 4051–4053. [Google Scholar] [CrossRef]
- Larsson, J.M.; Zhao, T.S.N.; Szabó, K.J. Palladium-catalyzed oxidative allylic C—H silylation. Org. Lett. 2011, 13, 1888–1891. [Google Scholar] [CrossRef]
- McMurtrey, K.B.; Racowski, J.M.; Sanford, M.S. Pd-catalyzed C—H fluorination with nucleophilic fluoride. Org. Lett. 2012, 14, 4094–4097. [Google Scholar] [CrossRef]
- Sun, X.; Yao, X.; Zhang, C.; Rao, Y. Pd(II) catalyzed ortho C—H iodination of phenylcarbamates at room temperature using cyclic hypervalent iodine reagents. Chem. Commun. 2015, 51, 10014–10017. [Google Scholar] [CrossRef]
- Rodriguez, A.; Moran, W.J. Palladium-catalyzed three-component coupling reactions: 1,1-difunctionalization of activated alkenes. Eur. J. Org. Chem. 2009, 2009, 1313–1316. [Google Scholar] [CrossRef]
- Satterfield, A.D.; Kubota, A.; Sanford, M.S. Palladium-catalyzed 1,1-aryloxygenation of terminal olefins. Org. Lett. 2011, 13, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Desai, L.V.; Sanford, M.S. Construction of tetrahydrofurans by PdII/PdIV-catalyzed aminooxygenation of alkenes. Angew. Chem. Int. Ed. 2007, 46, 5737–5740. [Google Scholar] [CrossRef] [PubMed]
- Streuff, J.; Hovelmann, C.H.; Nieger, M.; Muniz, K. Palladium(II)-catalyzed intramolecular diamination of unfunctionalized alkenes. J. Am. Chem. Soc. 2005, 127, 14586. [Google Scholar] [CrossRef]
- Muñiz, K. Advancing palladium-catalyzed C—N bond formation: Bisindoline construction from successive amide transfer to internal alkenes. J. Am. Chem. Soc. 2007, 129, 14542–14543. [Google Scholar] [CrossRef]
- Muñiz, K.; Hovelmann, C.H.; Streuff, J. Oxidative diamination of alkenes with ureas as nitrogen sources: Mechanistic pathways in the presence of a high oxidation state palladium catalyst. J. Am. Chem. Soc. 2008, 130, 763–773. [Google Scholar] [CrossRef]
- Fujino, D.; Yorimitsu, H.; Oshima, K. Palladium-catalyzed intramolecular carboacetoxylation of 4-pentenyl-substituted malonate esters with iodobenzene diacetate. Chem. Asian J. 2010, 5, 1758–1760. [Google Scholar] [CrossRef]
- Jaegli, S.; Dufour, J.; Wei, H.; Piou, T.; Duan, X.; Vors, J.; Neuville, L.; Zhu, J. Palladium-catalyzed carbo-heterofunctionalization of alkenes for the synthesis of oxindoles and spirooxindoles. Org. Lett. 2010, 12, 4498–4501. [Google Scholar] [CrossRef]
- Nicolai, S.; Erard, S.; Gonzalez, D.F.; Waser, J. Pd-catalyzed intramolecular oxyalkynylation of alkenes with hypervalent iodine. Org. Lett. 2010, 12, 384–387. [Google Scholar] [CrossRef]
- Nicolai, S.; Piemontesi, C.; Waser, J. A palladium-catalyzed aminoalkynylation strategy towards bicyclic heterocycles: Synthesis of (±)-Trachelanthamidine. Angew. Chem. Int. Ed. 2011, 50, 4680–4683. [Google Scholar] [CrossRef]
- Wu, T.; Yin, G.; Liu, G. Palladium-catalyzed intramolecular aminofluorination of unactivated alkenes. J. Am. Chem. Soc. 2009, 131, 16354–16355. [Google Scholar] [CrossRef]
- Iglesias, A.; Perez, E.G.; Muñiz Muniz, K. An intermolecular palladium-catalyzed diamination of unactivated alkenes. Angew. Chem. Int. Ed. 2010, 49, 8109–8111. [Google Scholar] [CrossRef]
- Martinez, C.; Muñiz Muniz, K. Palladium-catalyzed vicinal difunctionalization of internal alkenes: Diastereoselective synthesis of diamines. Angew. Chem. Int. Ed. 2012, 51, 7031–7034. [Google Scholar] [CrossRef]
- Muñiz, K.; Kirsch, J.; Chavez, P. Intermolecular regioselective 1,2-diamination of allylic ethers. Adv. Synth. Catal. 2011, 353, 689–694. [Google Scholar] [CrossRef]
- Martinez, C.; Muñiz, K. Defined palladium–phthalimidato catalysts for improved oxidative amination. Chem. Eur. J. 2016, 22, 7367–7370. [Google Scholar] [CrossRef]
- Martnez, C.; Perez, E.G.; Iglesias, A.; Escudero-Adan, E.C.; Muñiz., K. Regioselective intermolecular diamination and aminooxygenation of alkenes with saccharin. Org. Lett. 2016, 18, 2998–3001. [Google Scholar] [CrossRef]
- Li, Y.; Song, D.; Dong, V.M. Palladium-catalyzed olefin dioxygenation. J. Am. Chem. Soc. 2008, 130, 2962–2964. [Google Scholar] [CrossRef]
- Wang, W.; Wang, F.; Shi, M. Bis(NHC)-palladium(II) complex-catalyzed dioxygenation of alkenes. Organometallics 2010, 29, 928–933. [Google Scholar] [CrossRef]
- Neufeldt, S.R.; Sanford, M.S. Asymmetric chiral ligand-directed alkene dioxygenation. Org. Lett. 2013, 15, 46–49. [Google Scholar] [CrossRef]
- Alexanian, E.J.; Lee, C.; Sorensen, E.J. Palladium-catalyzed ring-forming aminoacetoxylation of alkenes. J. Am. Chem. Soc. 2005, 127, 7690–7691. [Google Scholar] [CrossRef]
- Liu, G.; Stahl, S.S. Highly regioselective Pd-catalyzed intermolecular aminoacetoxylation of alkenes and evidence for cis-aminopalladation and SN2 C—O bond formation. J. Am. Chem. Soc. 2006, 128, 7179–7181. [Google Scholar] [CrossRef]
- Martinez, C.; Wu, Y.; Weinstein, A.B.; Stahl, S.S.; Liu, G.; Muñiz, K. Palladium-catalyzed intermolecular aminoacetoxylation of alkenes and the influence of PhI(OAc)2 on aminopalladation stereoselectivity. J. Org. Chem. 2013, 78, 6309–6315. [Google Scholar] [CrossRef] [PubMed]
- Ilchenko, N.O.; Cortes, M.A.; Szabó, K.J. Palladium-catalyzed iodofluorination of alkenes using fluoro-iodoxole reagent. ACS Catal. 2016, 6, 447–450. [Google Scholar] [CrossRef]
- Cheng, J.; Qi, X.; Li, M.; Chen, P.; Liu, G. Palladium-catalyzed intermolecular aminocarbonylation of alkenes: Efficient access of β-amino acid derivatives. J. Am. Chem. Soc. 2015, 137, 2480–2483. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, F.; Qi, X.; Chen, P.; Liu, G. A cooperative strategy for the highly selective intermolecular oxycarbonylation reaction of alkenes using a palladium catalyst. Angew. Chem. Int. Ed. 2016, 55, 13843–13848. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.; Das, A.R. Hypervalent iodine promoted ortho diversification: 2-aryl benzimidazole, quinazoline and imidazopyridine as directing templates. Org. Biomol. Chem. 2020, 18, 941. [Google Scholar] [CrossRef] [PubMed]




































































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shetgaonkar, S.E.; Mamgain, R.; Kikushima, K.; Dohi, T.; Singh, F.V. Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents. Molecules 2022, 27, 3900. https://doi.org/10.3390/molecules27123900
Shetgaonkar SE, Mamgain R, Kikushima K, Dohi T, Singh FV. Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents. Molecules. 2022; 27(12):3900. https://doi.org/10.3390/molecules27123900
Chicago/Turabian StyleShetgaonkar, Samata E., Ritu Mamgain, Kotaro Kikushima, Toshifumi Dohi, and Fateh V. Singh. 2022. "Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents" Molecules 27, no. 12: 3900. https://doi.org/10.3390/molecules27123900
APA StyleShetgaonkar, S. E., Mamgain, R., Kikushima, K., Dohi, T., & Singh, F. V. (2022). Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents. Molecules, 27(12), 3900. https://doi.org/10.3390/molecules27123900