
Steroidal Antimetabolites Protect Mice against Trypanosoma brucei


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
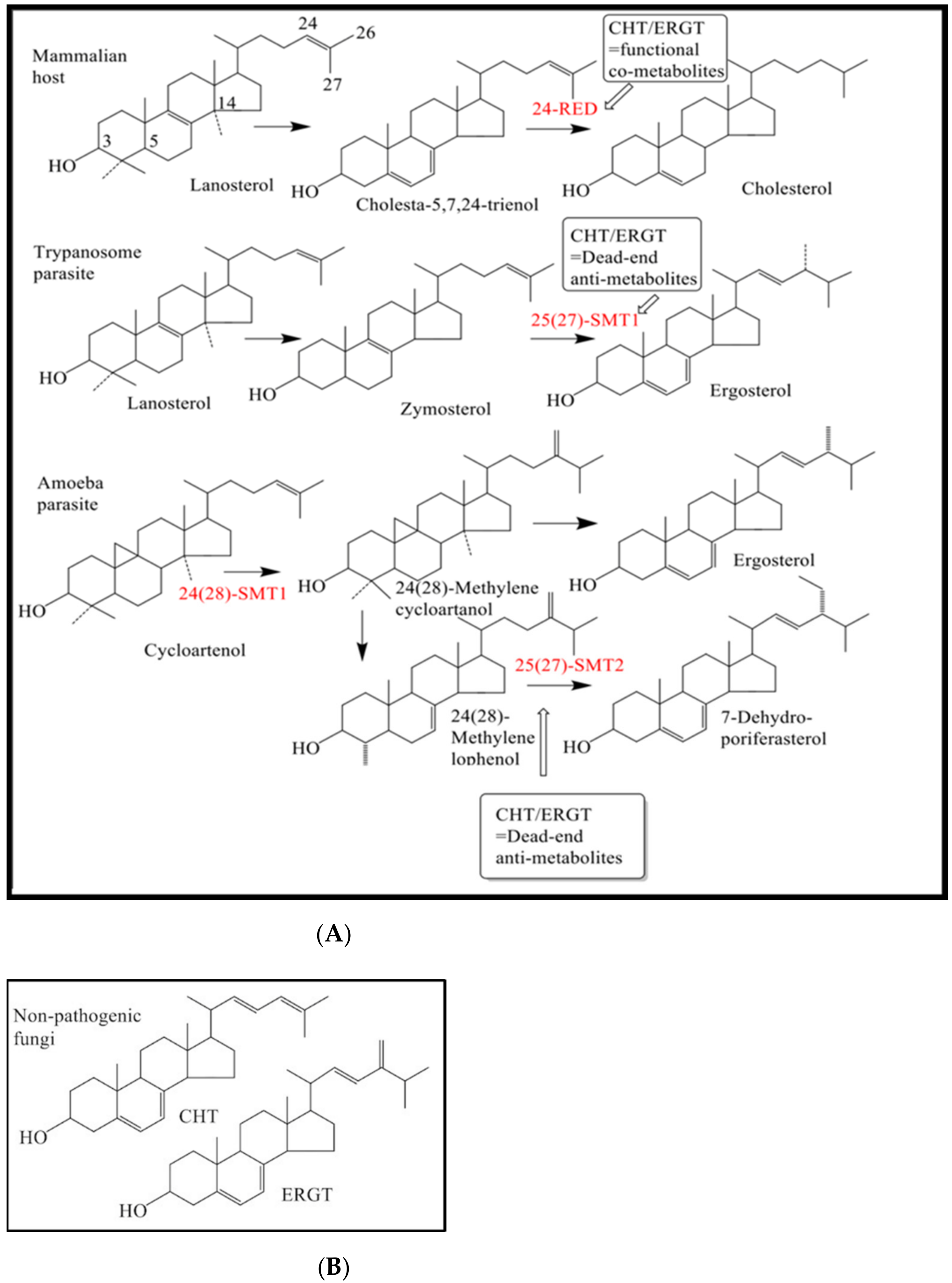
2. Results
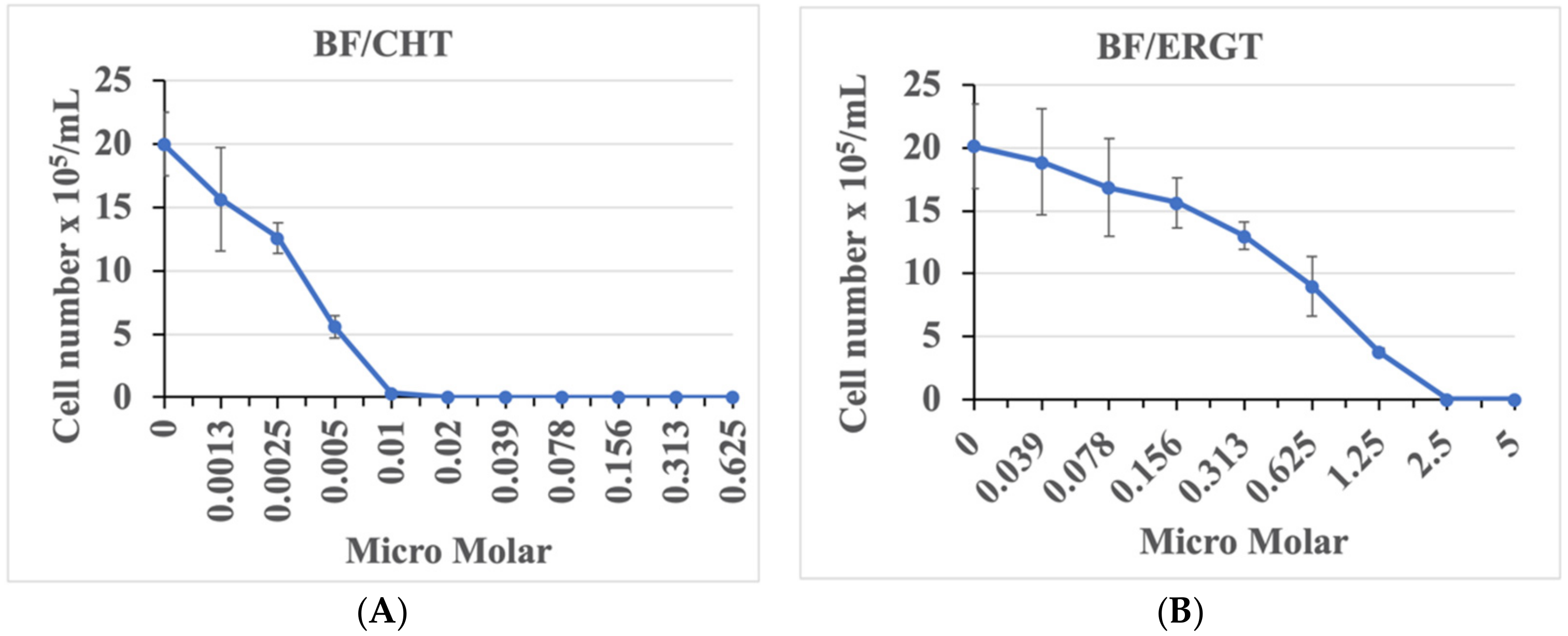
2.1. ERGT/CHT Generates Marked Inhibition of T. brucei BF Growth in Cell Culture
2.2. CHT and ERGT Cause Cell Death within Hours of Treatment
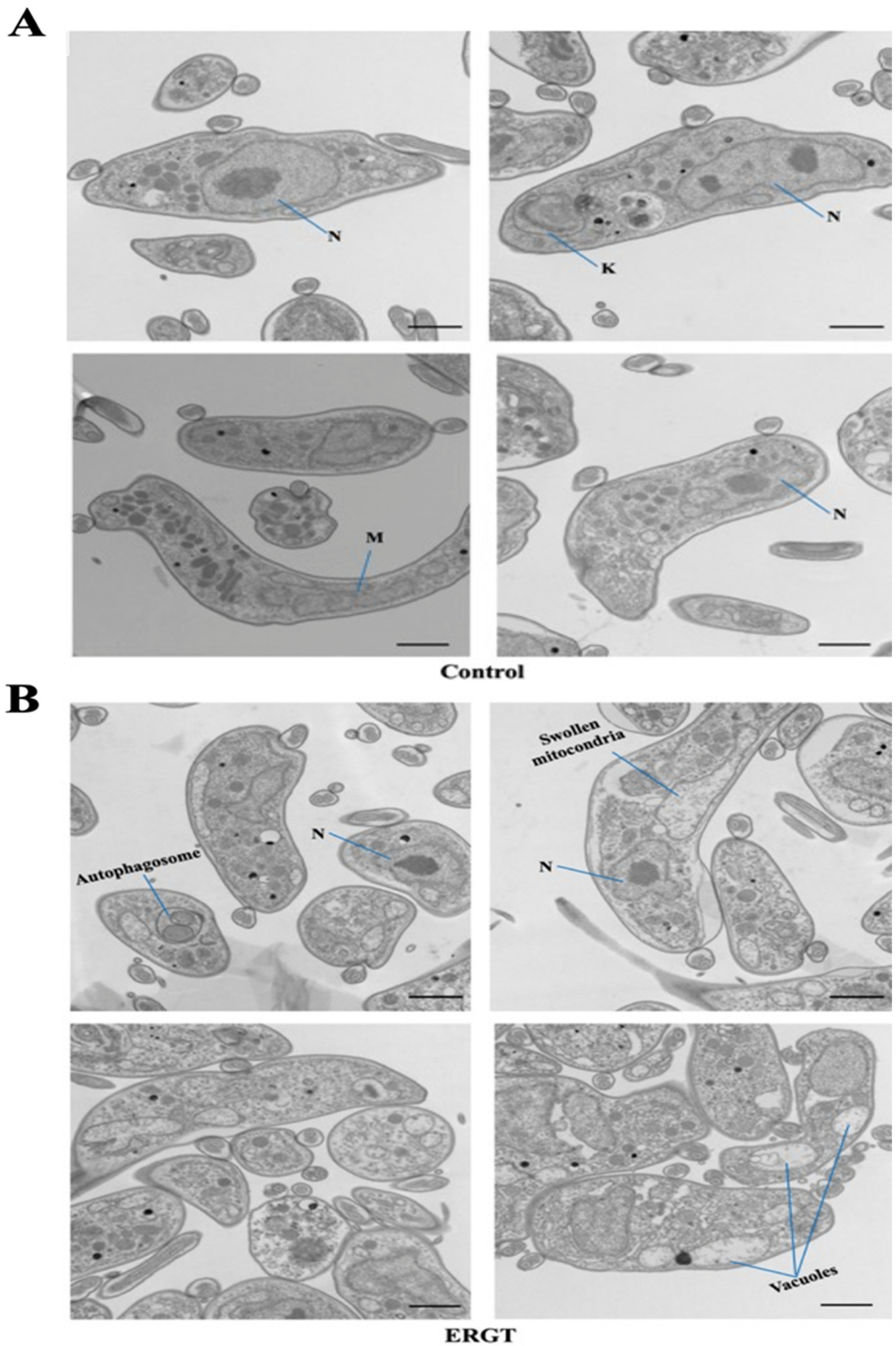
2.3. Antimetabolites Cause Mitochondrial Swelling and Mitophagy
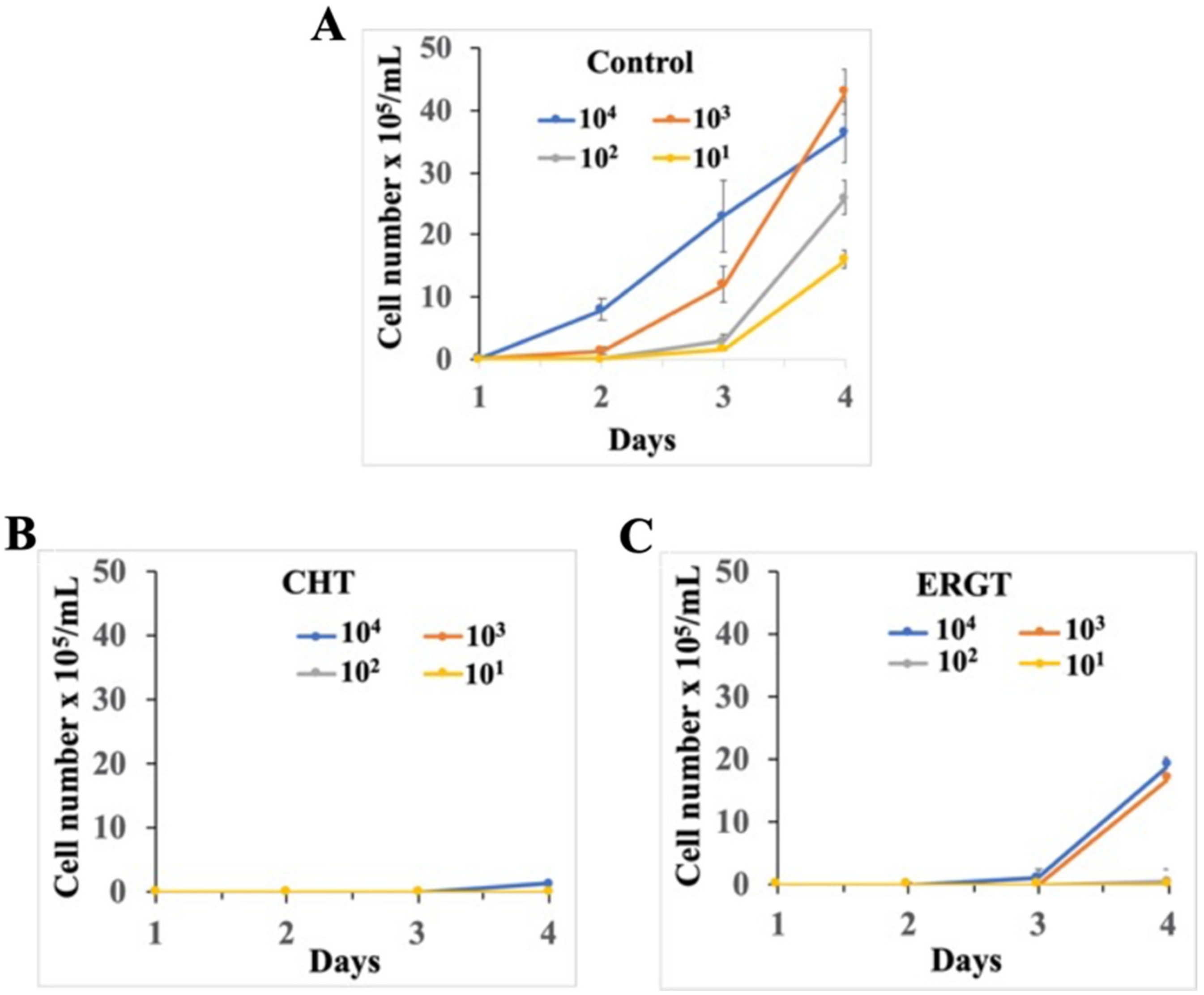
2.4. Washout Experiments of CHT and ERGT Reveal Inhibition of BF Growth and by Analogy Ergosterol Production
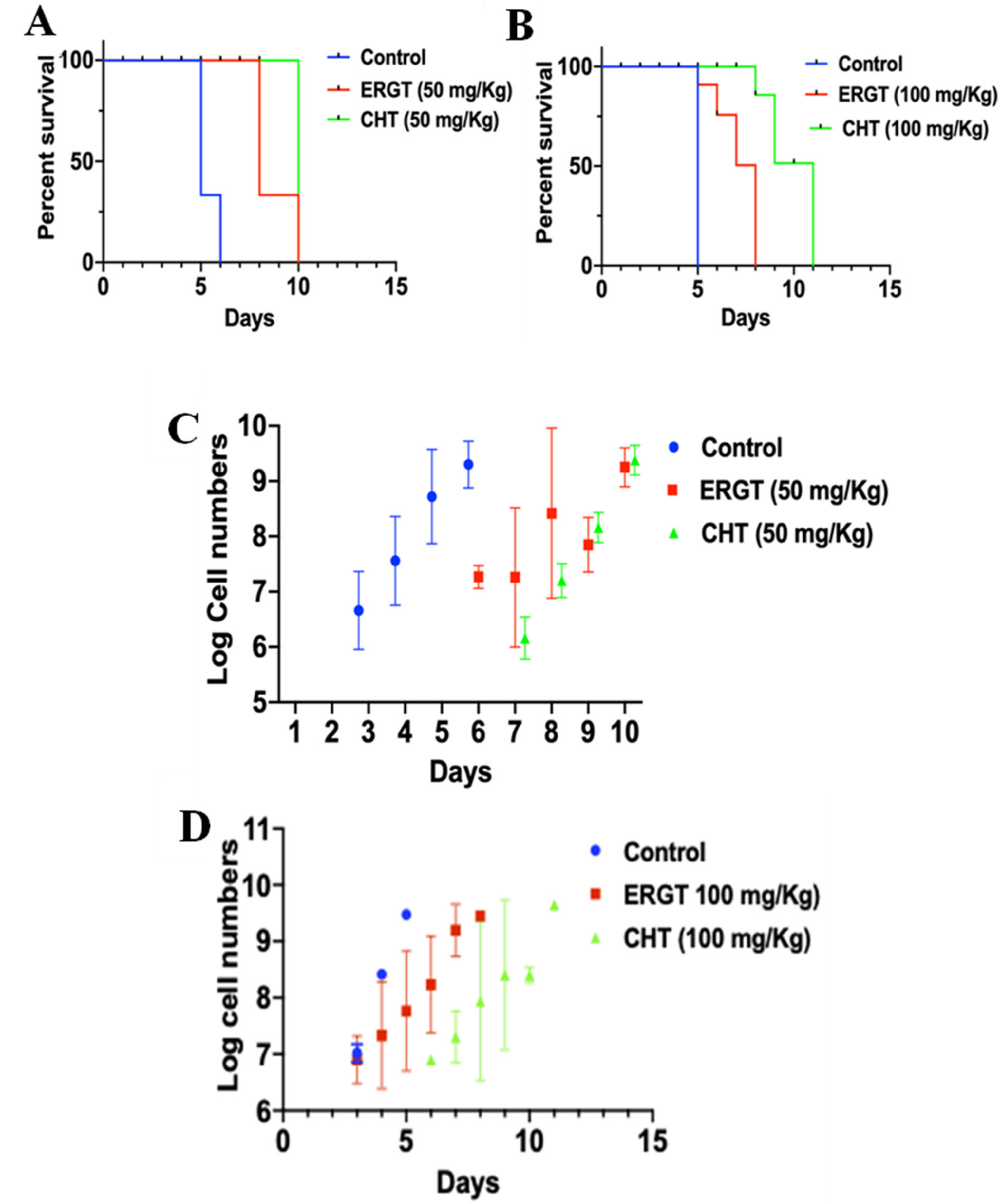
2.5. Protective Role of CHT/ERGT on Mice Infected with T. brucei
3. Discussion
4. Material and Methods
4.1. Tb Cell Culture
4.2. Preparation of ERGT and CHT for Treatment and Sterol Analytics
4.3. Inhibition Studies in Culture
4.4. Giemsa Staining
4.5. Electron Microscopy
4.6. Animal Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sternberg, J.M.; MacLean, L. A spectrum of disease in Human African trypanosomiasis: The host and parasite genetics of virulence. Parasitology 2010, 137, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Silvester, E.; McWilliam, K.; Matthews, K. The Cytological Events and Molecular Control of Life Cycle Development of Trypanosoma brucei in the Mammalian Bloodstream. Pathogens 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekhar, G.N.; Watson, C.P.; Fidanboylu, M.; Sanderson, L.; Thomas, S.A. Delivery of antihuman African trypanosomiasis drugs across the blood-brain and blood-CSF barriers. Adv. Pharmacol. 2014, 71, 245–275. [Google Scholar] [PubMed]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole—A new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, S.; Bernardo, J.F.; Kelner, A.; Sokolova, A.Y.; Berriman, M.; Fairlamb, A.H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; The International Natural Products Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 28, 1–17. [Google Scholar] [CrossRef]
- Cockram, P.E.; Smith, T.K. Active natural product scaffolds against trypanosomatid parasites: A review. J. Nat. Prod. 2018, 81, 2138–2154. [Google Scholar] [CrossRef] [Green Version]
- Heby, O.; Roberts, S.C.; Ullman, B. Polyamine biosynthetic enzymes as drug targets in parasitic protozoa. Biochem. Soc. Trans. 2003, 31, 7719–7726. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.W.R.; McCleod, R.; Rice, D.W.; Ginger, M.; Chance, M.L.; Goad, L.J. Fatty acid and sterol metabolism: Potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Pharmacol. 2003, 126, 129–142. [Google Scholar] [CrossRef]
- Schimmel, K.J.; Gelderblom, H.; Guchelaar, H.J. Cyclopentenyl cytosine (CPEC): An overview of its in vitro and in vivo activity. Curr. Cancer Drug Targets 2007, 7, 504–509. [Google Scholar] [CrossRef]
- Peters, G.J. Novel developments in the use of antimetabolites. Nucleosides Nucleotides Nucleic Acids 2014, 33, 358–374. [Google Scholar] [CrossRef]
- Ray, S.; Murkin, A.S. New electrophiles and strategies for mechanism-based and targeted covalent inhibitor design. Biochemistry 2019, 58, 5234–5244. [Google Scholar] [CrossRef]
- Azijli, K.; van Roosmalen, I.A.; Smit, J.; Pillai, S.; Fukushima, M.; de Jong, S.; Peters, G.J.; Bijnsdorp, I.V.; Kruyt, F.A. The novel thymidylate synthase inhibitor trifluorothymidine (TFT) and TRAIL synergistically eradicate non-small cell lung cancer cells. Cancer Chemother. Pharmacol. 2014, 73, 1273–1283. [Google Scholar] [CrossRef]
- Chavoushi, S.F.; Jharap, B.; Friedrich, P.; Smid, K.; Peters, G.J.; Malingré, M. Thiopurines with low-dose allopurinol (ThiLDA)- a prospective clinical one-way crossover trial. Eur. J. Clin. Pharmacol. 2019, 75, 1669–1674. [Google Scholar] [CrossRef]
- Balboni, B.; El Hassouni, B.; Honeywell, R.J.; Sarkisjan, D.; Giovannetti, E.; Poore, J.; Heaton, C.; Peterson, C.; Benaim, E.; Lee, Y.B.; et al. RX-3117 (fluorocyclopentenyl cytosine): A novel specific antimetabolite for selective cancer treatment. Expert Opin. Investig. Drugs 2019, 28, 311–322. [Google Scholar] [CrossRef]
- Halford, B. Covalent drugs go from fringe to fashionable endeavor. Chem. Eng. News 2020, 98, 28–32. [Google Scholar] [CrossRef]
- World Health Organization. World Health Organization Model List of Essential Medicines: 21st List 2019. Geneva: World Health Organization. hdl: 10665/3225771. License: CC BY-NC-SA 3.0 IGO. Available online: https://hdl.handle.net/10665%2F325771 (accessed on 21 September 2020).
- Villalta, F.; Dobish, M.C.; Nde, P.N.; Kleshchenko, Y.Y.; Hargrove, T.Y.; Johnson, C.A.; Waterman, M.R.; Johnston, J.N.; Lepesheva, G.I. VNI cures acute and chronic experimental Chagas disease. J. Infect. Dis. 2013, 208, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Nes, W.D. Steroidal triterpene: Design of substrate-based inhibitors of ergosterol and sitosterol synthesis. Molecules 2009, 14, 4690–4706. [Google Scholar] [CrossRef] [Green Version]
- De Macedo-Silva, S.T.; de Souza, W.; Rodrigues, J.C.F. Sterol biosynthesis pathway as an alternative for the anti-protozoan parasite chemotherapy. Cur. Med. Chem. 2015, 22, 2186–2198. [Google Scholar] [CrossRef]
- Zhou, W.; Warrilow, A.G.S.; Thomas, C.D.; Ramos, E.; Parker, J.E.; Price, C.L.; Vanderloop, B.H.; Fisher, P.M.; Loftis, M.D.; Kelly, D.E.; et al. Functional importance for developmental regulation of sterol biosynthesis in Acanthameoba castelanii. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids. 2018, 1863, 1164–1178. [Google Scholar] [CrossRef]
- Zhou, W.; Lepesheva, G.I.; Waterman, M.R.; Nes, W.D. Mechanistic analysis of a multiple product sterol methyltransferase implicated in ergosterol biosynthesis in Trypanosoma brucei. J. Biol. Chem. 2006, 281, 6290–6296. [Google Scholar] [CrossRef] [Green Version]
- Nes, C.R.; Singha, U.K.; Liu, J.; Ganapathy, K.; Villalta, F.; Waterman, M.R.; Lepesheva, G.I.; Chaudhuri, M.; Nes, W.D. Novel sterol metabolic network of Trypanosoma brucei procyclic and bloodstream forms. Biochem. J. 2012, 443, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Haubrich, B.A.; Singha, U.K.; Miller, M.B.; Nes, C.R.; Anyatonwu, H.; Lecordier, L.; Patkar, P.; Leaver, D.J.; Villalta, F.; Vanhollebeke, B.; et al. Discovery of an ergosterol-signaling factor that regulates Trypanosoma brucei growth. J. Lipid Res. 2015, 56, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Kidane, M.E.; Vanderloop, B.H.; Zhou, W.; Thomas, C.D.; Ramos, E.; Singha, U.; Chaudhuri, M.; Nes, W.D. Sterol methyltransferase a target for anti-ameoba therapy: Towards transition state analog and suicide substrate drug design. J. Lipid Res. 2017, 58, 2310–2323. [Google Scholar] [CrossRef] [Green Version]
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef]
- Zhou, W.; Ramos, E.; Zhu, X.; Fisher, P.M.; Kidane, M.E.; Vanderloop, B.H.; Thomas, C.D.; Yan, J.; Singha, U.; Chaudhuri, M.; et al. Steroidal antibiotics are antimetabolites of Acanthameoba steroidogenesis with phylogenetic implications. J. Lipid Res. 2019, 60, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ganapathy, K.; Wyyial, E.; Buknicki, J.M.; Nwogwugwu, C.A.; Nes, W.D. Effect of substrate features and mutagenesis of the active site tyrosine residues on the reaction course catalyzed by Trypanosma brucei sterol C-24-methyltransferase. Biochem. J. 2011, 439, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Nes, W.D.; Marshall, J.A.; Jia, Z.; Jaradat, T.T.; Song, Z.; Jaysimha, P. Active site mapping and substrate channeling in the sterol methyltransferase pathway. J. Biol. Chem. 2002, 277, 42459–42565. [Google Scholar] [CrossRef] [Green Version]
- Soape, M. Protein Chemistry, Peptide Mapping, and Preliminary Structural Characterization of Saccharomyces cerevisiae Sterol C-24-Methyltransferase Expressed in Escherichia coli. Master’s Thesis, Texas Tech University, Lubbock, TX, USA, 2006; pp. 1–106. [Google Scholar]
- Magaraci, F.; Jimenez, C.J.; Rodrigues, C.; Rodrigues, J.C.; Braga, M.V.; Yardley, V.; de Luca-Fradley, K.; Croft, S.L.; de Souza, W.; Ruiz-Perez, L.M.; et al. Azasterol as inhibitors of sterol 24-methyltransferase in Leishmania species and Trypanosoma cruzi. J. Med. Chem. 2003, 46, 4714–4727. [Google Scholar] [CrossRef]
- Zhou, W.; Cross, A.M.; Nes, W.D. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol synthesis and cell proliferation of Trypanosoma brucei. J. Lipid Res. 2007, 48, 665–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaver, D.J.; Patkar, P.; Singha, U.K.; Miller, M.B.; Haubrich, B.A.; Chaudhuri, M.; Nes, W.D. Fluorinated sterols are suicide inhibitors of ergosterol biosynthesis and growth in Trypanosoma brucei. Chem. Biol. 2015, 22, 1374–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppens, I.; Courtoy, P.J. Host plasma low density lipoprotein particles as an essential source of lipids for the bloodstream forms of Trypanosoma brucei. J. Biol. Chem. 1995, 270, 5736–5741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbina, J.A.; Vivas, J.; Lazardi, K.; Molina, J.; Payares, G.; Piras, M.M.; Piras, R. Antiproliferative effects of delta24(25) sterol methyl transferase inhibitors on Trypanosoma (Schizotrypanum) cruzi: In vitro and in vivo studies. Chemotherapy 1996, 42, 294–307. [Google Scholar] [CrossRef]
- Mensa-Wilmot, K.; Hoffman, B.; Wiedeman, J.; Sullenberger, C.; Sharma, A. Kinetoplast Division Factors in a Trypanosome. Trends Parasitol. 2019, 35, 119–128. [Google Scholar] [CrossRef]
- Matthews, K.R. Trypanosome signaling-quorum sensing. Ann. Rev. Microbiol. 2021, 75, 495–514. [Google Scholar] [CrossRef]
- Bitoni, A.J.; Dumont, J.A.; McCann, P.P. Characterization of Trypanosoma brucei brucei S-adenosyl-l-methionine decarboxylase and its inhibition by berenil, pentamidine and methylglyoxal (guanyhydrazone). Biochem. J. 1986, 237, 518–521. [Google Scholar]
- Kipkorir, L.W.; John, T.K.; Owino, O.B.; John, O.; Robert, S.; Daniel, M.; Owino, A.V. Mouse experiments demonstrate differential pathogenicity and virulence of Trypanosoma brucei rhodesiense strains. Exp. Parasitol. 2021, 228, 108135. [Google Scholar] [CrossRef]
- Darnet, S.; Blary, A.; Chevalier, Q.; Schaller, H. Phytosterol profiles, genomes and enzymes—An overview. Front. Plant Sci. 2021, 12, 1–31. [Google Scholar] [CrossRef]
- Nes, W.D. Control of Sterol biosynthesis and its importance to developmental regulation. Rec.Adv. Phytochem. 1990, 24, 283–327. [Google Scholar]
- Neelakandan, A.K.; Song, Z.; Wang, J.; Richards, M.H.; Wu, X.; Valliyodan, B.; Nguyen, H.T.; Nes, W.D. Cloning, functional expression and phylogenetic analysis of sterol C24-methyltransferases involved in sitosterol biosynthesis. Phytochemistry 2009, 70, 1982–1998. [Google Scholar] [CrossRef]
- Nes, W.D.; Song, A.L.; Dennis, W.; Zhou, W.; Nam, J.; Miller, M.B. Biosynthesis of phytosterols: Kinetic mechanism for the enzymatic C-methylation of sterols. J. Biol. Chem. 2003, 278, 34505–34516. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Nes, W.D. Cyclobranol: A substrate for C25-methyl sterol side chains and potent mechanistic-based inactivator of plant sterol methyltransferase. Bioorg. Med. Chem. Lett. 2008, 18, 3878–3881. [Google Scholar] [CrossRef]
- Miller, M.B.; Patkar, P.; Singha, U.K.; Chaudhuri, M.; Nes, W.D. 24-Methylenecyclopropane steroidal inhibitors: A Trojan horse in ergosterol biosynthesis that prevents growth of Trypanosoma brucei. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids 2017, 1862, 305–313. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. In vitro cultivation of Trypanosoma congolense bloodstream forms in the absence of feeder cell layers. Parasitology 1991, 112, 225. [Google Scholar] [CrossRef]
- Nebesarova, J.; Hozák, P.; Frank, L.; Štěpan, P.; Vancová, M. The cutting of ultrathin sections with the thickness less than 20 nm from biological specimens embedded in resin blocks. Microsc. Res. Tech. 2016, 79, 512–517. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhuri, M.; Singha, U.K.; Vanderloop, B.H.; Tripathi, A.; Nes, W.D. Steroidal Antimetabolites Protect Mice against Trypanosoma brucei. Molecules 2022, 27, 4088. https://doi.org/10.3390/molecules27134088
Chaudhuri M, Singha UK, Vanderloop BH, Tripathi A, Nes WD. Steroidal Antimetabolites Protect Mice against Trypanosoma brucei. Molecules. 2022; 27(13):4088. https://doi.org/10.3390/molecules27134088
Chicago/Turabian StyleChaudhuri, Minu, Ujjal K. Singha, Boden H. Vanderloop, Anuj Tripathi, and W. David Nes. 2022. "Steroidal Antimetabolites Protect Mice against Trypanosoma brucei" Molecules 27, no. 13: 4088. https://doi.org/10.3390/molecules27134088
APA StyleChaudhuri, M., Singha, U. K., Vanderloop, B. H., Tripathi, A., & Nes, W. D. (2022). Steroidal Antimetabolites Protect Mice against Trypanosoma brucei. Molecules, 27(13), 4088. https://doi.org/10.3390/molecules27134088