
Prognostic Ability of Enhancer RNAs in Metastasis of Non-Small Cell Lung Cancer

 , ,
, ,
Abstract
1. Introduction
2. Results
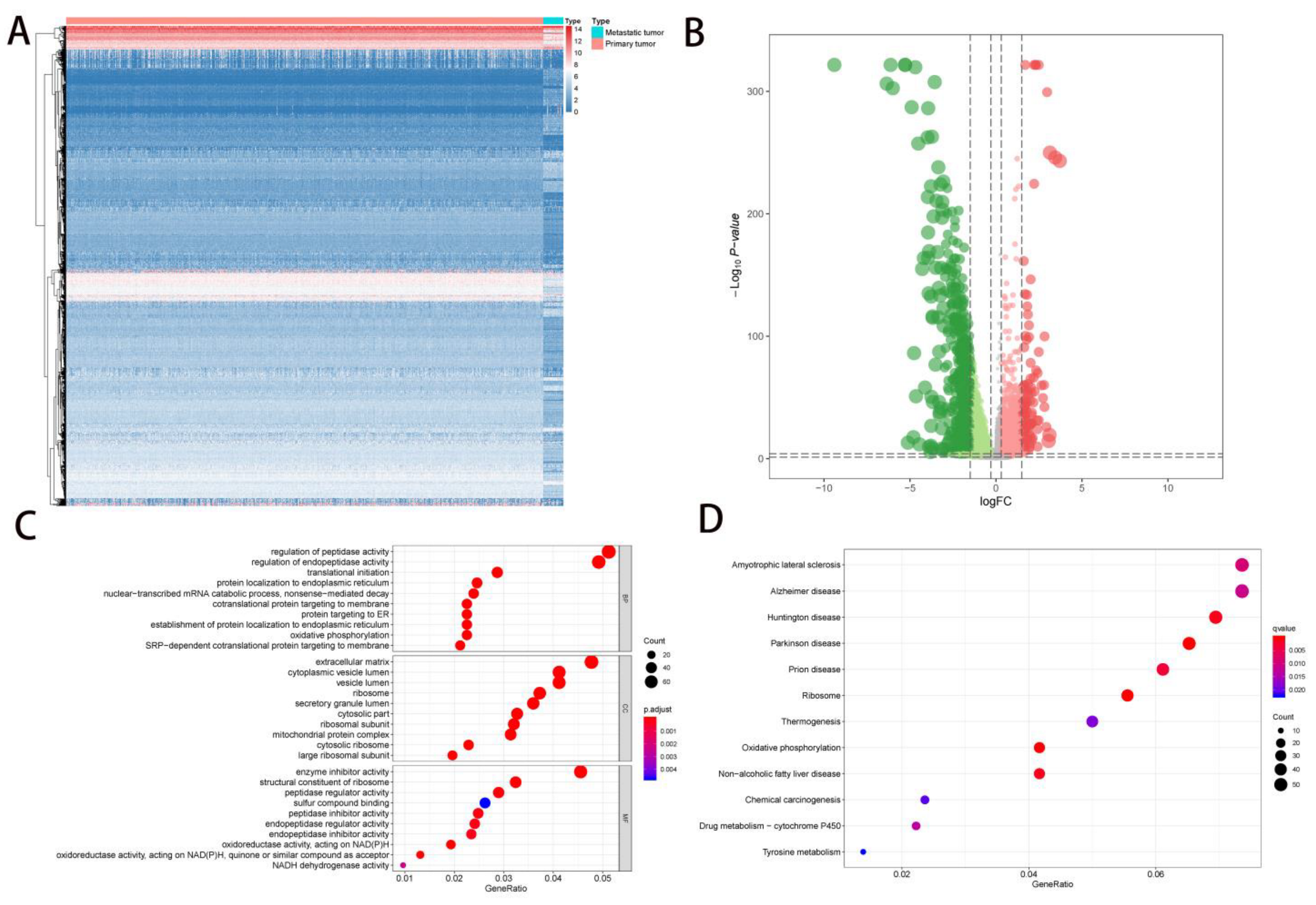
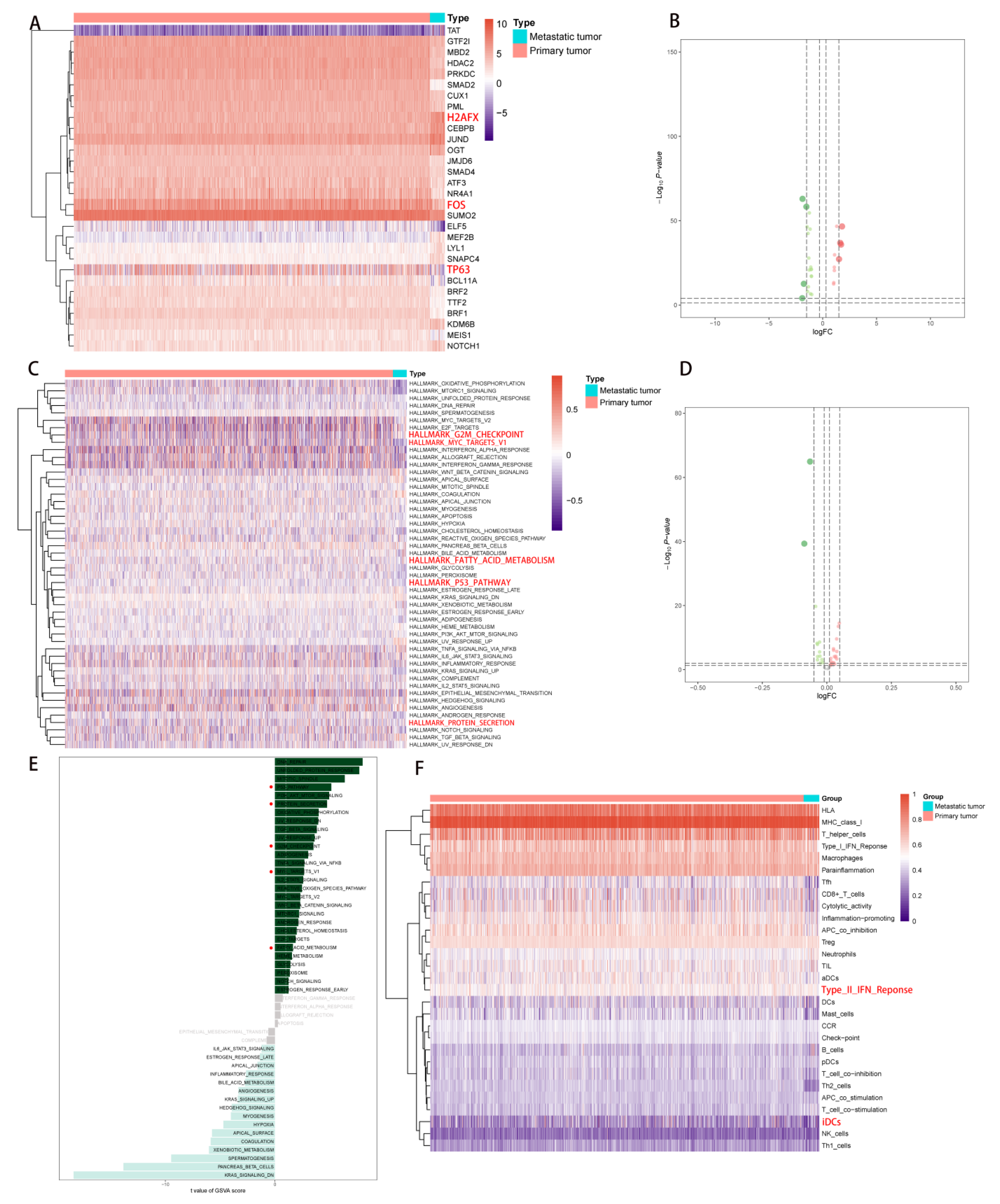
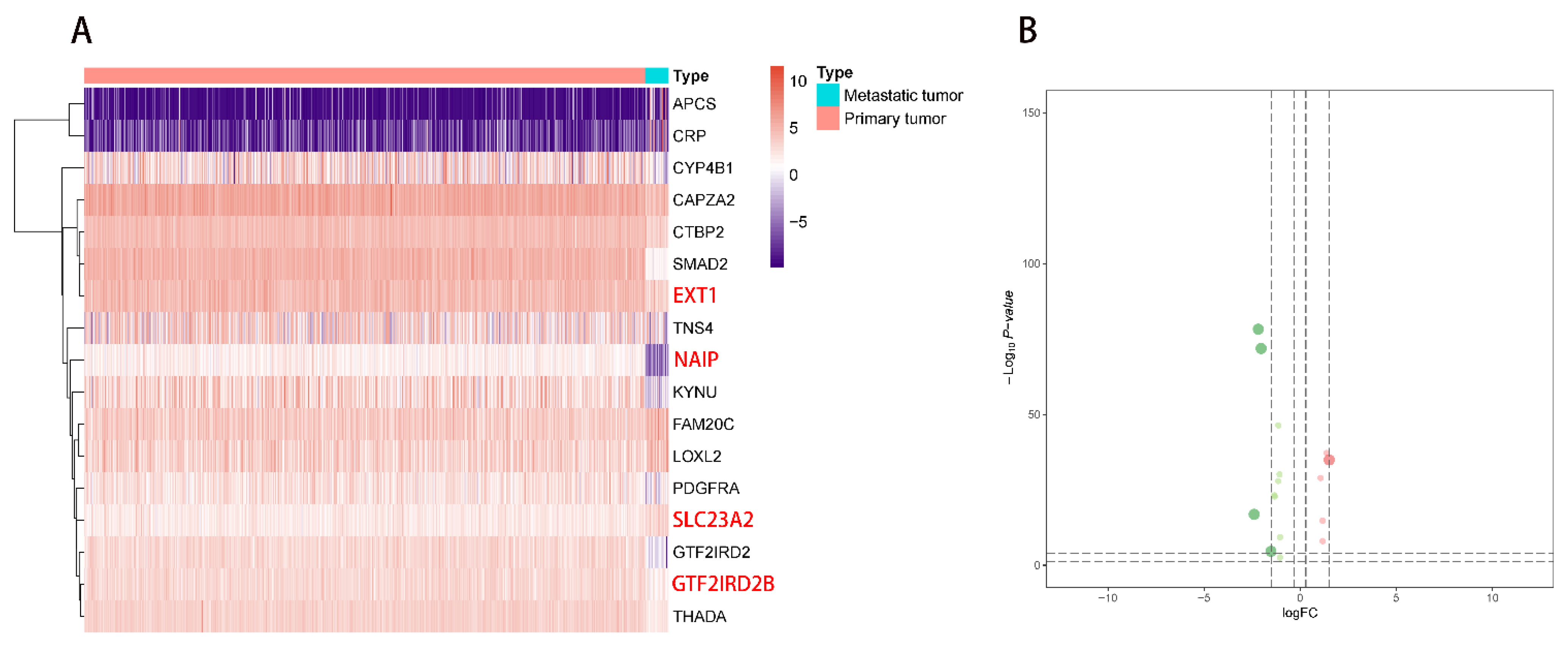
2.1. DEG Identification and Functional Enrichment Analysis
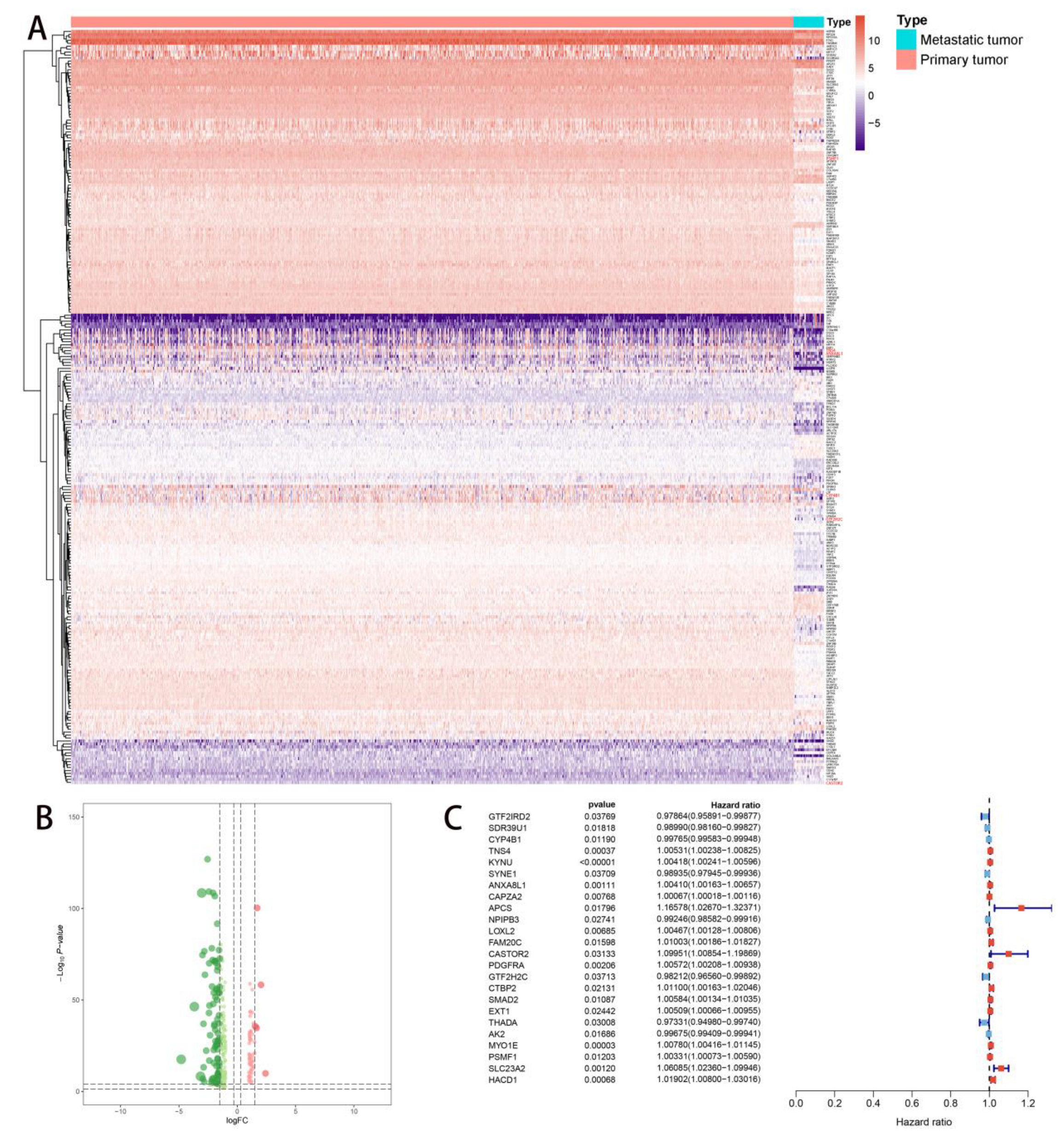
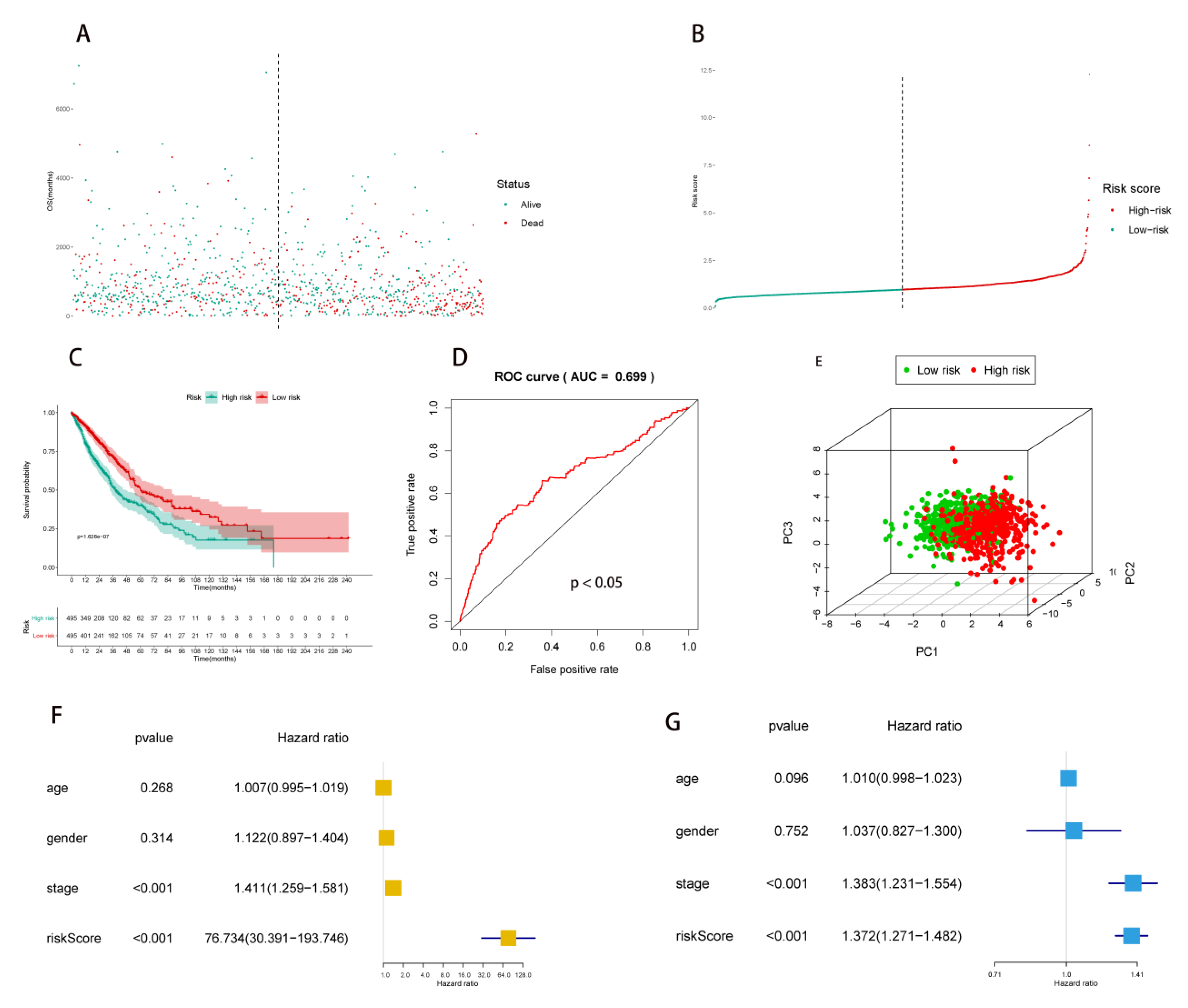
2.2. Multivariate Prognostic Model Construction and Independent Prognostic Factors Identification
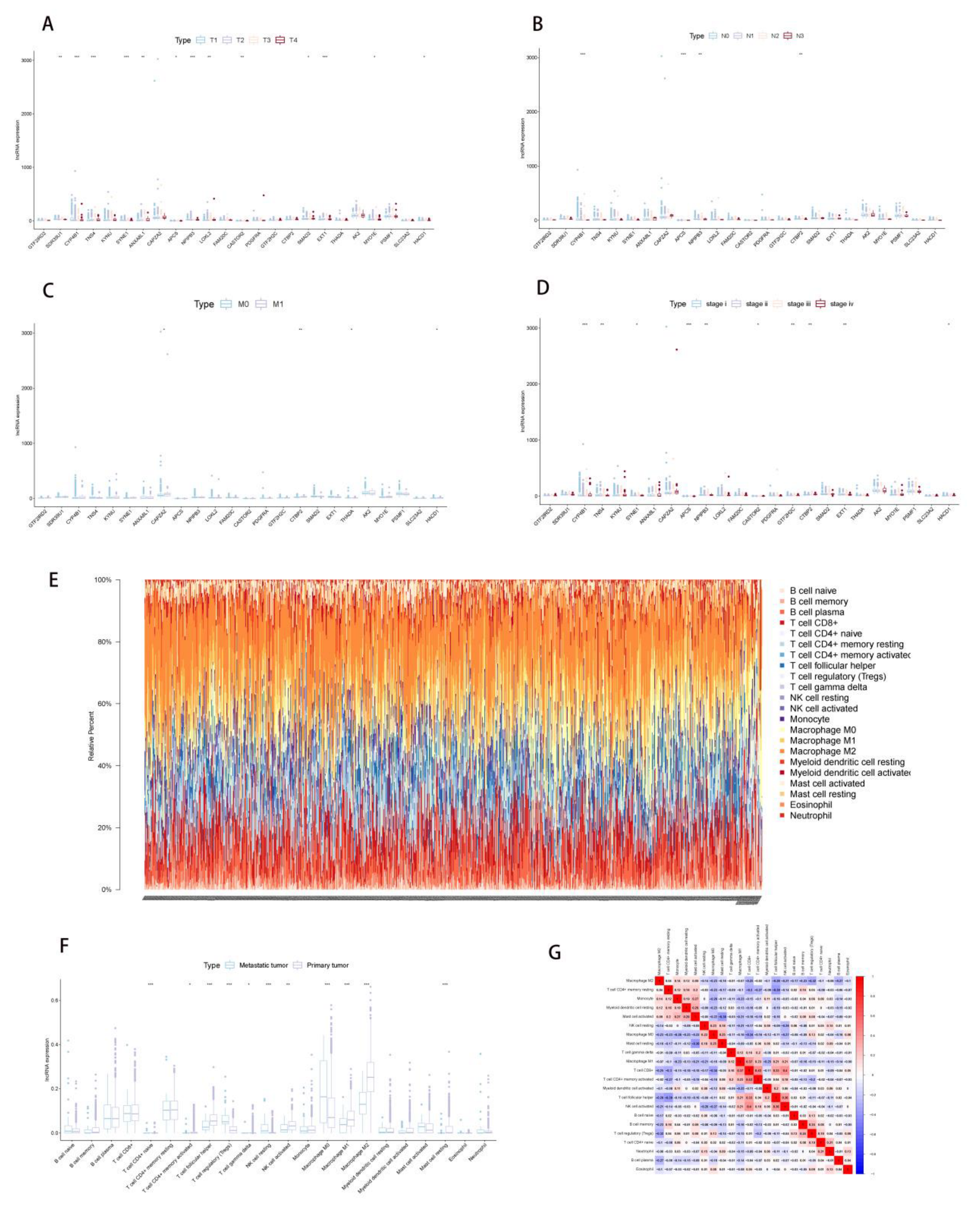
2.3. Correlation Analysis of PDEEs and Immune Cells
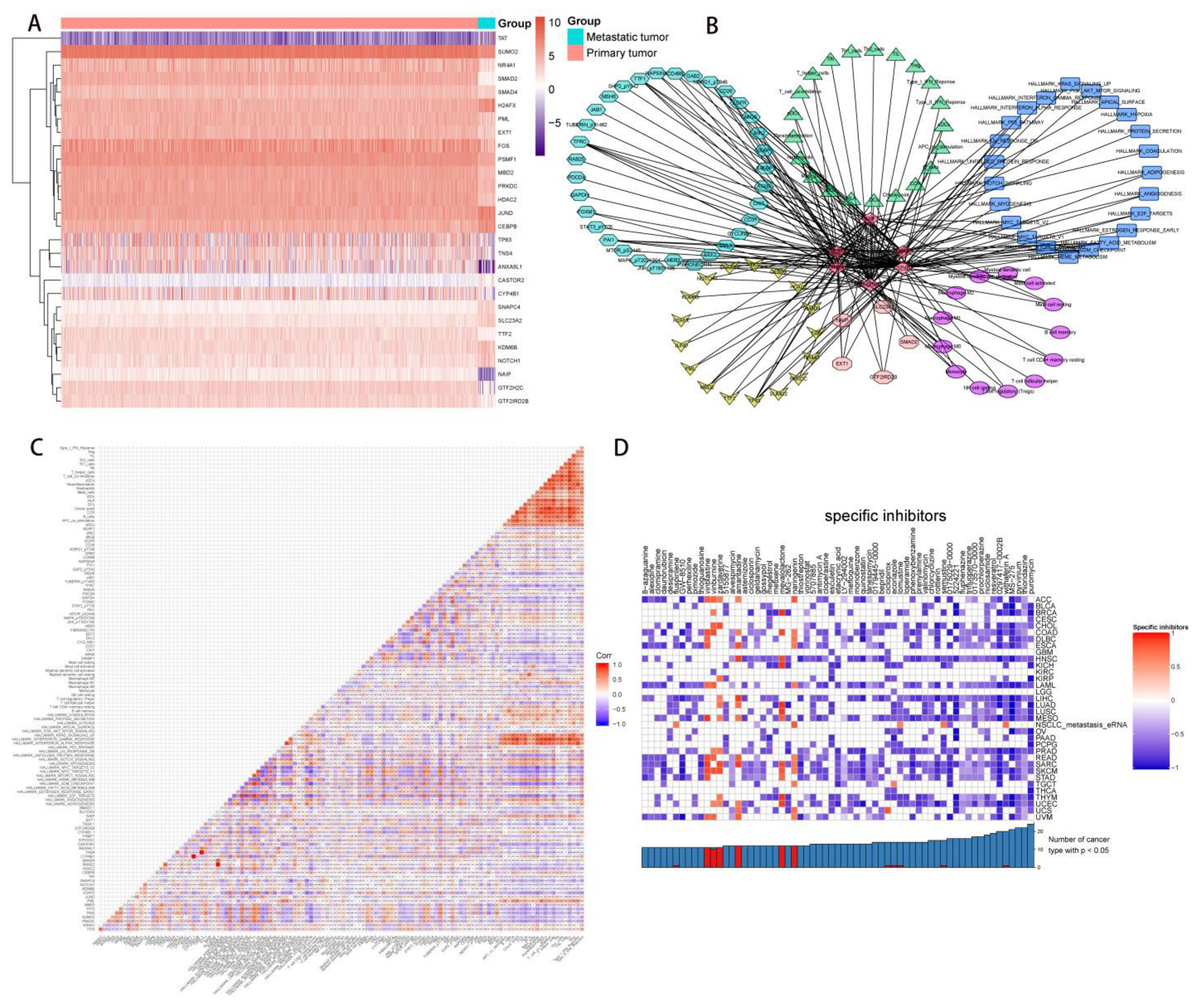
2.4. Correlation Analysis of PDEEs, DETFs, Immune-Related Gene Sets, and Hallmark Pathways
2.5. Correlation Analysis of PDEEs, DETGs, and RPPA Protein Chips
2.6. The Construction of NSCLC Metastasis-Specific eRNA Regulation Network
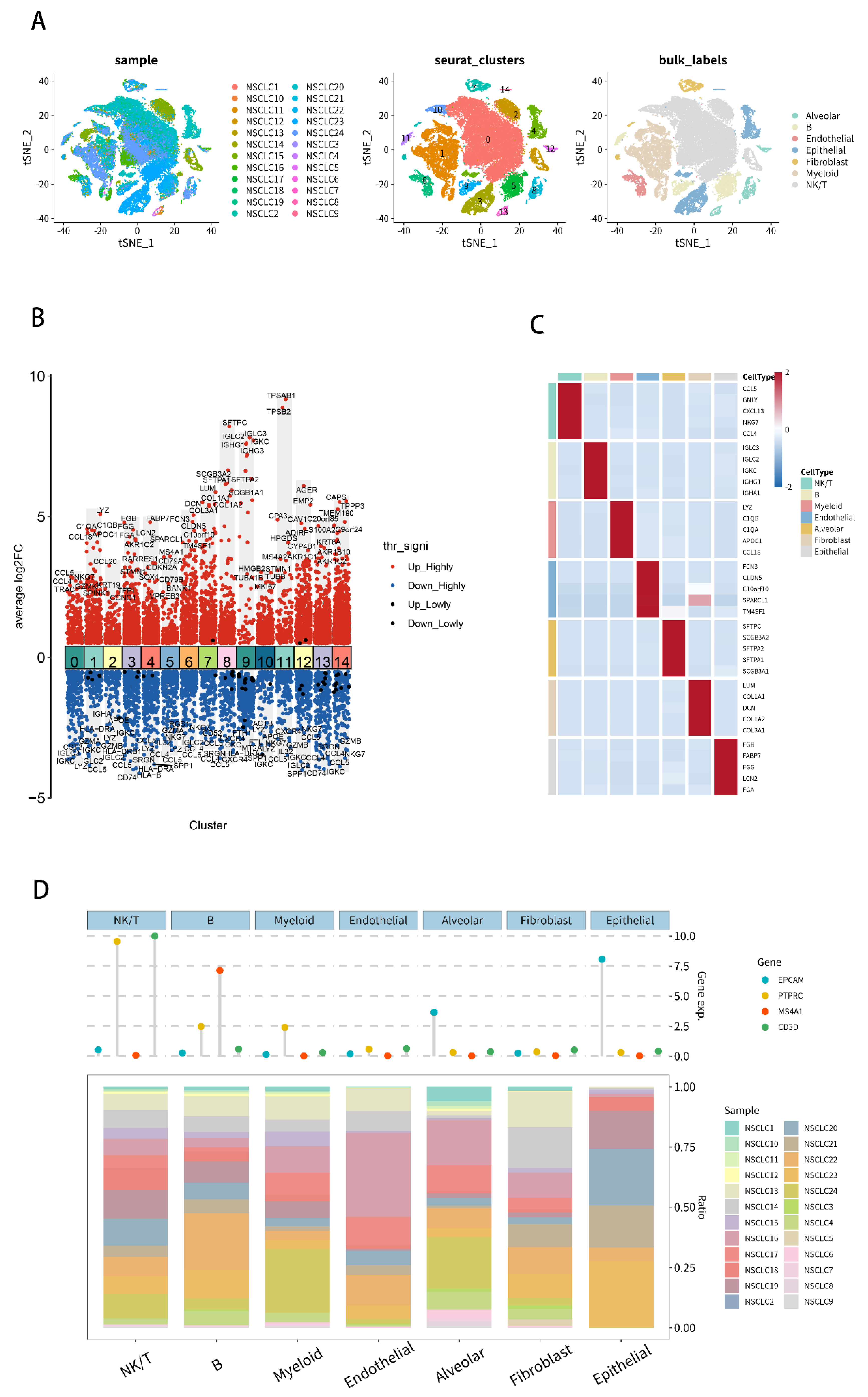
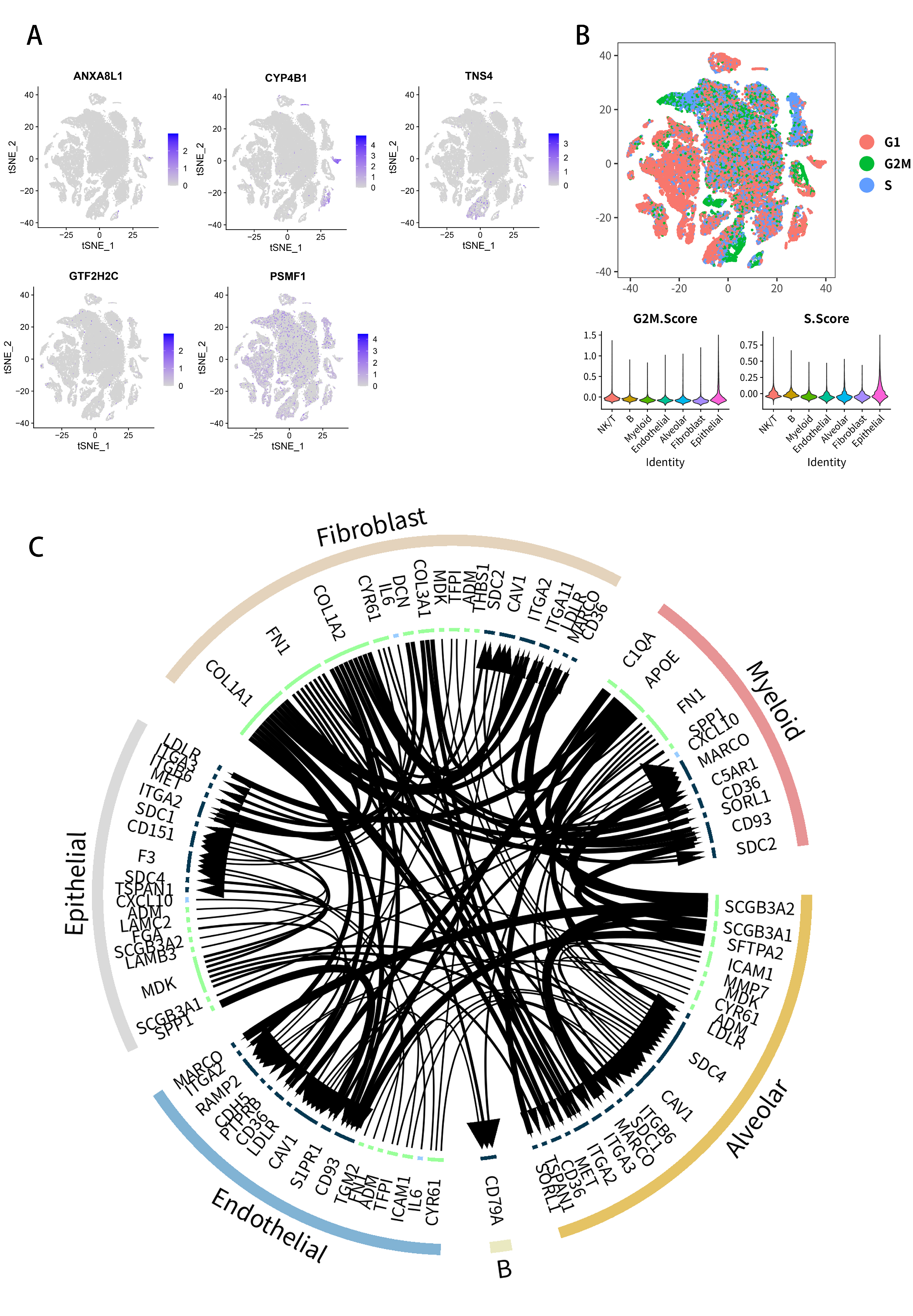
2.7. Analysis of Single-Cell RNA-Seq Transcriptomes
2.8. Multidimensional Validation
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. The eRNA Expression Data
4.3. Differential Expression Analysis
4.4. Multivariate Risk-Prediction Model Construction and Independent Prognostic Factors Identification
4.5. Identification of PDEE-Related Immune Cells and Immune-Reltaed Gene Sets
4.6. Identification of Downstream Hallmark Pathways
4.7. Construction of Metastasis-Specific eRNA Regulation Network for NSCLC
4.8. Analysis of scRNA-Seq Transcriptomes
4.9. Multidimensional Validation
4.10. Statistics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA A Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Strand, T.E.; Rostad, H.; Møller, B.; Norstein, J. Survival after resection for primary lung cancer: A population based study of 3211 resected patients. Thorax 2006, 61, 710–715. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA A Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Bickmore, W.; Dean, A.; Nobrega, M.A.; Bejerano, G. Enhancers: Five essential questions. Nat. Reviews. Genet. 2013, 14, 288–295. [Google Scholar] [CrossRef]
- Mossing, M.C.; Record, M.T., Jr. Upstream operators enhance repression of the lac promoter. Science 1986, 233, 889–892. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Kadonaga, J.T. Going the distance: A current view of enhancer action. Science 1998, 281, 60–63. [Google Scholar] [CrossRef]
- Bulger, M.; Groudine, M. Enhancers: The abundance and function of regulatory sequences beyond promoters. Dev. Biol. 2010, 339, 250–257. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Zare, H.; Dell’orso, S.; Grontved, L.; Gutierrez-Cruz, G.; Derfoul, A.; Hager, G.L.; Sartorelli, V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 2013, 51, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, A.; Mimura, J.; Itoh, K. Non-coding RNA derived from the region adjacent to the human HO-1 E2 enhancer selectively regulates HO-1 gene induction by modulating Pol II binding. Nucleic Acids Res. 2014, 42, 13599–13614. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013, 498, 516–520. [Google Scholar] [CrossRef]
- Li, W.; Hu, Y.; Oh, S.; Ma, Q.; Merkurjev, D.; Song, X.; Zhou, X.; Liu, Z.; Tanasa, B.; He, X.; et al. Condensin I and II Complexes License Full Estrogen Receptor α-Dependent Enhancer Activation. Mol. Cell 2015, 59, 188–202. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.S.; Chen, S.; et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324. [Google Scholar] [CrossRef]
- Lee, J.H.; Xiong, F.; Li, W. Enhancer RNAs in cancer: Regulation, mechanisms and therapeutic potential. RNA Biol. 2020, 17, 1550–1559. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.H.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 4562. [Google Scholar] [CrossRef]
- Léveillé, N.; Melo, C.A.; Agami, R. Enhancer-associated RNAs as therapeutic targets. Expert Opin. Biol. Ther. 2015, 15, 723–734. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, Z.; Zhu, X.; Yan, P.; Song, D.; Yin, H.; Hu, P.; Lin, R.; Wu, S.; Meng, T.; et al. Collagen Type III Alpha 1 chain regulated by GATA-Binding Protein 6 affects Type II IFN response and propanoate metabolism in the recurrence of lower grade glioma. J. Cell. Mol. Med. 2020, 24, 10803–10815. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Houston, K.A.; Henley, S.J.; Li, J.; White, M.C.; Richards, T.B. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004-2009. Lung Cancer 2014, 86, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Kalantari-Dehaghi, M.; Anhalt, G.J.; Camilleri, M.J.; Chernyavsky, A.I.; Chun, S.; Felgner, P.L.; Jasinskas, A.; Leiferman, K.M.; Liang, L.; Marchenko, S.; et al. Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS ONE 2013, 8, e57587. [Google Scholar] [CrossRef]
- Ding, H.; Xiong, X.X.; Fan, G.L.; Yi, Y.X.; Chen, Y.R.; Wang, J.T.; Zhang, W. The New Biomarker for Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (CESC) Based on Public Database Mining. BioMed Res. Int. 2020, 2020, 5478574. [Google Scholar] [CrossRef]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef]
- Xia, J.; Wang, R.; Zhang, T.; Ding, J. Structural insight into the arginine-binding specificity of CASTOR1 in amino acid-dependent mTORC1 signaling. Cell Discov. 2016, 2, 16035. [Google Scholar] [CrossRef]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ju, E.; Gao, S.J. Kaposi sarcoma-associated herpesvirus miRNAs suppress CASTOR1-mediated mTORC1 inhibition to promote tumorigenesis. J. Clin. Investig. 2019, 129, 3310–3323. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; De, M.J.A.J.P.P. De, M. Association of cytochrome P450 with cancer induced by Betel quid (BQ): A review. Am. J. Pharmacol. Pharmacother. 2015, 2, 81–94. [Google Scholar] [CrossRef]
- Baer, B.R.; Rettie, A.E. CYP4B1: An enigmatic P450 at the interface between xenobiotic and endobiotic metabolism. Drug Metab. Rev. 2006, 38, 451–476. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Alshagga, M.; Ong, C.E.; Chieng, J.Y.; Pan, Y. Cytochrome P450 4B1 (CYP4B1) as a target in cancer treatment. Hum. Exp. Toxicol. 2020, 39, 785–796. [Google Scholar] [CrossRef]
- Czerwinski, M.; McLemore, T.L.; Gelboin, H.V.; Gonzalez, F.J.J.C.R. Quantification of CYP2B7, CYP4B1, and CYPOR messenger RNAs in normal human lung and lung tumors. Cancer Res. 1994, 54, 1085–1091. [Google Scholar]
- Chhabra, D.; Sharma, S.; Kho, A.T.; Gaedigk, R.; Vyhlidal, C.A.; Leeder, J.S.; Morrow, J.; Carey, V.J.; Weiss, S.T.; Tantisira, K.G.J.E. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics 2014, 9, 1473–1484. [Google Scholar] [CrossRef]
- Ren, P.; Rosas, I.O.; Macdonald, S.D.; Wu, H.P.; Billings, E.M.; Gochuico, B.R. Impairment of alveolar macrophage transcription in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 1151–1157. [Google Scholar] [CrossRef]
- McCutchen-Maloney, S.L.; Matsuda, K.; Shimbara, N.; Binns, D.D.; Tanaka, K.; Slaughter, C.A.; DeMartino, G.N. cDNA cloning, expression, and functional characterization of PI31, a proline-rich inhibitor of the proteasome. J. Biol. Chem. 2000, 275, 18557–18565. [Google Scholar] [CrossRef]
- Zaiss, D.M.; Standera, S.; Holzhütter, H.; Kloetzel, P.; Sijts, A.J. The proteasome inhibitor PI31 competes with PA28 for binding to 20S proteasomes. FEBS Lett. 1999, 457, 333–338. [Google Scholar] [CrossRef]
- Cho-Park, P.F.; Steller, H. Proteasome regulation by ADP-ribosylation. Cell 2013, 153, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tang, D.; Zhao, Y.C.; Liu, H.; Luo, S.; Stinchcombe, T.E.; Glass, C.; Su, L.; Shen, S.; Christiani, D.C.; et al. Potentially functional variants of ERAP1, PSMF1 and NCF2 in the MHC-I-related pathway predict non-small cell lung cancer survival. Cancer Immunol. Immunother. CII 2021, 70, 2819–2833. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, K.; Mimori, K.; Tanaka, F.; Kamohara, Y.; Inoue, H.; Sawada, T.; Hirakawa, K.; Mori, M. Prognostic relevance of Tensin4 expression in human gastric cancer. Ann. Surg. Oncol. 2008, 15, 2606–2613. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.Z.; Wang, W.J.; Chen, Y.X.; Fan, Z.W.; Xie, X.F.; Yang, L.Y.; Chang, C.; Cai, Y.; Hao, J.J.; Wang, M.R.; et al. The miR-1224-5p/TNS4/EGFR axis inhibits tumour progression in oesophageal squamous cell carcinoma. Cell Death Dis. 2020, 11, 597. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, D.; Zhao, D.; Hu, D. Aberrant Methylation and Differential Expression of SLC2A1, TNS4, GAPDH, ATP8A2, and CASZ1 Are Associated with the Prognosis of Lung Adenocarcinoma. BioMed Res. Int. 2020, 2020, 1807089. [Google Scholar] [CrossRef]
- Xie, J.J.; Jiang, Y.Y.; Jiang, Y.; Li, C.Q.; Lim, M.C.; An, O.; Mayakonda, A.; Ding, L.W.; Long, L.; Sun, C.; et al. Super-Enhancer-Driven Long Non-Coding RNA LINC01503, Regulated by TP63, Is Over-Expressed and Oncogenic in Squamous Cell Carcinoma. Gastroenterology 2018, 154, 2137–2151.e2131. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Han, B.; Sun, Y.; Yang, D.; Zhang, H.; Mo, S.; Chen, X.; Lu, H.; Mao, X.; Hu, J. USP22 promotes development of lung adenocarcinoma through ubiquitination and immunosuppression. Aging 2020, 12, 6990–7005. [Google Scholar] [CrossRef]
- Kong, W.; Chen, Y.; Zhao, Z.; Zhang, L.; Lin, X.; Luo, X.; Wang, S.; Song, Z.; Lin, X.; Lai, G.; et al. EXT1 methylation promotes proliferation and migration and predicts the clinical outcome of non-small cell lung carcinoma via WNT signalling pathway. J. Cell. Mol. Med. 2021, 25, 2609–2620. [Google Scholar] [CrossRef]
- Takanaga, H.; Mackenzie, B.; Hediger, M.A. Sodium-dependent ascorbic acid transporter family SLC23. Pflug. Arch. Eur. J. Physiol. 2004, 447, 677–682. [Google Scholar] [CrossRef]
- Gillberg, L.; Ørskov, A.D.; Liu, M.; Harsløf, L.B.S.; Jones, P.A.; Grønbæk, K. Vitamin C—A new player in regulation of the cancer epigenome. Semin. Cancer Biol. 2018, 51, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Gunbin, K.V.; Ruvinsky, A. Evolution of general transcription factors. J. Mol. Evol. 2013, 76, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Candia, J.; Bayarsaikhan, E.; Tandon, M.; Budhu, A.; Forgues, M.; Tovuu, L.O.; Tudev, U.; Lack, J.; Chao, A.; Chinburen, J.; et al. The genomic landscape of Mongolian hepatocellular carcinoma. Nat. Commun. 2020, 11, 4383. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wu, X.; Jiang, Q.; Sun, Y.; Liu, H.; Chen, R.; Wu, S. Downregulation of microRNA-1 and microRNA-145 contributes synergistically to the development of colon cancer. Int. J. Mol. Med. 2015, 36, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.A.; Buschhaus, J.M.; Chen, Y.C.; Haley, H.R.; Qyli, T.; Chiang, B.; Shen, N.; Rajendran, S.; Cutter, A.; Cheng, Y.H.; et al. Plasminogen Activator Inhibitor 1 (PAI1) Promotes Actin Cytoskeleton Reorganization and Glycolytic Metabolism in Triple-Negative Breast Cancer. Mol. Cancer Res. 2019, 17, 1142–1154. [Google Scholar] [CrossRef]
- Beckner, M.E.; Stracke, M.L.; Liotta, L.A.; Schiffmann, E. Glycolysis as primary energy source in tumor cell chemotaxis. J. Natl. Cancer Inst. 1990, 82, 1836–1840. [Google Scholar] [CrossRef]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef]
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Huang, Y.; Gan, L.; Long, L.; Pu, A.; Xie, R. TFRC promotes epithelial ovarian cancer cell proliferation and metastasis via up-regulation of AXIN2 expression. Am. J. Cancer Res. 2020, 10, 131–147. [Google Scholar]
- Jiang, Y.; Zhao, J.; Zhang, Y.; Li, K.; Li, T.; Chen, X.; Zhao, S.; Zhao, S.; Liu, K.; Dong, Z. Establishment of lung cancer patient-derived xenograft models and primary cell lines for lung cancer study. J. Transl. Med. 2018, 16, 138. [Google Scholar] [CrossRef]
- Yatabe, Y.; Dacic, S.; Borczuk, A.C.; Warth, A.; Russell, P.A.; Lantuejoul, S.; Beasley, M.B.; Thunnissen, E.; Pelosi, G.; Rekhtman, N.; et al. Best Practices Recommendations for Diagnostic Immunohistochemistry in Lung Cancer. J. Thorac. Oncol. 2019, 14, 377–407. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, L.; Liu, D.; Li, W.M. Expression of TTF-1, NapsinA, P63, CK5/6 in Lung Cancer and Its Diagnostic Values for Histological Classification. Sichuan Da Xue Xue Bao. Yi Xue Ban J. Sichuan University. Med. Sci. Ed. 2017, 48, 336–341. [Google Scholar]
- Yin, P.; Ono, M.; Moravek, M.B.; Coon, J.S.T.; Navarro, A.; Monsivais, D.; Dyson, M.T.; Druschitz, S.A.; Malpani, S.S.; Serna, V.A.; et al. Human uterine leiomyoma stem/progenitor cells expressing CD34 and CD49b initiate tumors in vivo. J. Clin. Endocrinol. Metab. 2015, 100, E601-606. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. p53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Yasutis, K.M.; Kozminski, K.G. Cell cycle checkpoint regulators reach a zillion. Cell Cycle 2013, 12, 1501–1509. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qu, L.; Shou, C. Role and Characterization of Synuclein-γ Unconventional Protein Secretion in Cancer Cells. Methods Mol. Biol. 2016, 1459, 215–227. [Google Scholar] [CrossRef]
- Schulze, A.; Oshi, M.; Endo, I.; Takabe, K. MYC Targets Scores Are Associated with Cancer Aggressiveness and Poor Survival in ER-Positive Primary and Metastatic Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8127. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef]
- Candido, J.; Hagemann, T. Cancer-related inflammation. J. Clin. Immunol. 2013, 33 (Suppl 1), 79–84. [Google Scholar] [CrossRef]
- Zhong, R.; Chen, D.; Cao, S.; Li, J.; Han, B.; Zhong, H. Immune cell infiltration features and related marker genes in lung cancer based on single-cell RNA-seq. Clin. Transl. Oncol. 2021, 23, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Virk, H.; Arthur, G.; Bradding, P. Mast cells and their activation in lung disease. Transl. Res. J. Lab. Clin. Med. 2016, 174, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Cui, W.Q.; Wei, Y.; Cui, J.; Qiu, J.; Hu, L.L.; Gong, W.Y.; Dong, J.C.; Liu, B.J. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J. Exp. Clin. Cancer Res. 2018, 37, 207. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual Faces of IFNγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Obleukhova, I.; Kiryishina, N.; Falaleeva, S.; Lopatnikova, J.; Kurilin, V.; Kozlov, V.; Vitsin, A.; Cherkasov, A.; Kulikova, E.; Sennikov, S. Use of antigen-primed dendritic cells for inducing antitumor immune responses in vitro in patients with non-small cell lung cancer. Oncol. Lett. 2018, 15, 1297–1306. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Carlsen, L.; Schorl, C.; Huntington, K.; Hernandez-Borrero, L.; Jhaveri, A.; Zhang, S.; Zhou, L.; El-Deiry, W.S. Pan-drug and drug-specific mechanisms of 5-FU, irinotecan (CPT-11), oxaliplatin, and cisplatin identified by comparison of transcriptomic and cytokine responses of colorectal cancer cells. Oncotarget 2021, 12, 2006–2021. [Google Scholar] [CrossRef]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ortiz, J.A.; Taing, L.; Meyer, C.A.; Lee, B.; Zhang, Y.; Shin, H.; Wong, S.S.; Ma, J.; Lei, Y.; et al. Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 2011, 12, R83. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Lai, F.; Gardini, A.; Zhang, A.; Shiekhattar, R. Integrator mediates the biogenesis of enhancer RNAs. Nature 2015, 525, 399–403. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Meng, T.; Huang, R.; Zeng, Z.; Huang, Z.; Yin, H.; Jiao, C.; Yan, P.; Hu, P.; Zhu, X.; Li, Z.; et al. Identification of Prognostic and Metastatic Alternative Splicing Signatures in Kidney Renal Clear Cell Carcinoma. Front. Bioeng. Biotechnol. 2019, 7, 270. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.D.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e315. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Linderman, G.C.; Rachh, M.; Hoskins, J.G.; Steinerberger, S.; Kluger, Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 2019, 16, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G.; Wang, M.; Ajani, J.; Futreal, A.; Wang, L. iTALK: An R Package to Characterize and Illustrate Intercellular Communication. BioRxiv 2019, 507871. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281.e277. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Zheng, R.; Zang, C.; Zhu, M.; Wu, J.; Shi, X.; Taing, L.; et al. Cistrome Data Browser: A data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017, 45, D658–D662. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Reviews. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef]
- Yamane, A.; Robbiani, D.F.; Resch, W.; Bothmer, A.; Nakahashi, H.; Oliveira, T.; Rommel, P.C.; Brown, E.J.; Nussenzweig, A.; Nussenzweig, M.C.; et al. RPA accumulation during class switch recombination represents 5′-3′ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Rep. 2013, 3, 138–147. [Google Scholar] [CrossRef]
- Periyasamy, M.; Patel, H.; Lai, C.F.; Nguyen, V.T.M.; Nevedomskaya, E.; Harrod, A.; Russell, R.; Remenyi, J.; Ochocka, A.M.; Thomas, R.S.; et al. APOBEC3B-Mediated Cytidine Deamination Is Required for Estrogen Receptor Action in Breast Cancer. Cell Rep. 2015, 13, 108–121. [Google Scholar] [CrossRef]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Perona, A.; Hoang-Phou, S.; Leitner, M.C.; Okuniewska, M.; Meehan, S.; Schober, M. De Novo PITX1 Expression Controls Bi-Stable Transcriptional Circuits to Govern Self-Renewal and Differentiation in Squamous Cell Carcinoma. Cell Stem Cell 2019, 24, 390–404.e398. [Google Scholar] [CrossRef] [PubMed]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.J.; Bayles, I.; Funnell, A.P.W.; Miller, T.E.; Saiakhova, A.; Lizardo, M.M.; Bartels, C.F.; Kapteijn, M.Y.; Hung, S.; Mendoza, A.; et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat. Med. 2018, 24, 176–185. [Google Scholar] [CrossRef]
- Watanabe, H.; Ma, Q.; Peng, S.; Adelmant, G.; Swain, D.; Song, W.; Fox, C.; Francis, J.M.; Pedamallu, C.S.; DeLuca, D.S.; et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J. Clin. Investig. 2014, 124, 1636–1645. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Total Patients (N = 829) |
|---|---|
| Age, years | |
| 66.30 ± 9.35 |
| 68 (33–87) |
| Gender | |
| 301 (36.31%) |
| 528 (63.69%) |
| Stages | |
| 415 (50.06%) |
| 242 (29.19%) |
| 144 (17.37%) |
| 28 (3.38%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Jia, J.; Wang, S.; Zhang, J.; Xian, S.; Zheng, Z.; Deng, L.; Feng, Y.; Zhang, Y.; Zhang, J. Prognostic Ability of Enhancer RNAs in Metastasis of Non-Small Cell Lung Cancer. Molecules 2022, 27, 4108. https://doi.org/10.3390/molecules27134108
Liu J, Jia J, Wang S, Zhang J, Xian S, Zheng Z, Deng L, Feng Y, Zhang Y, Zhang J. Prognostic Ability of Enhancer RNAs in Metastasis of Non-Small Cell Lung Cancer. Molecules. 2022; 27(13):4108. https://doi.org/10.3390/molecules27134108
Chicago/Turabian StyleLiu, Jun, Jingyi Jia, Siqiao Wang, Junfang Zhang, Shuyuan Xian, Zixuan Zheng, Lin Deng, Yonghong Feng, Yuan Zhang, and Jie Zhang. 2022. "Prognostic Ability of Enhancer RNAs in Metastasis of Non-Small Cell Lung Cancer" Molecules 27, no. 13: 4108. https://doi.org/10.3390/molecules27134108
APA StyleLiu, J., Jia, J., Wang, S., Zhang, J., Xian, S., Zheng, Z., Deng, L., Feng, Y., Zhang, Y., & Zhang, J. (2022). Prognostic Ability of Enhancer RNAs in Metastasis of Non-Small Cell Lung Cancer. Molecules, 27(13), 4108. https://doi.org/10.3390/molecules27134108