
A Computational Approach Applied to the Study of Potential Allosteric Inhibitors Protease NS2B/NS3 from Dengue Virus

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
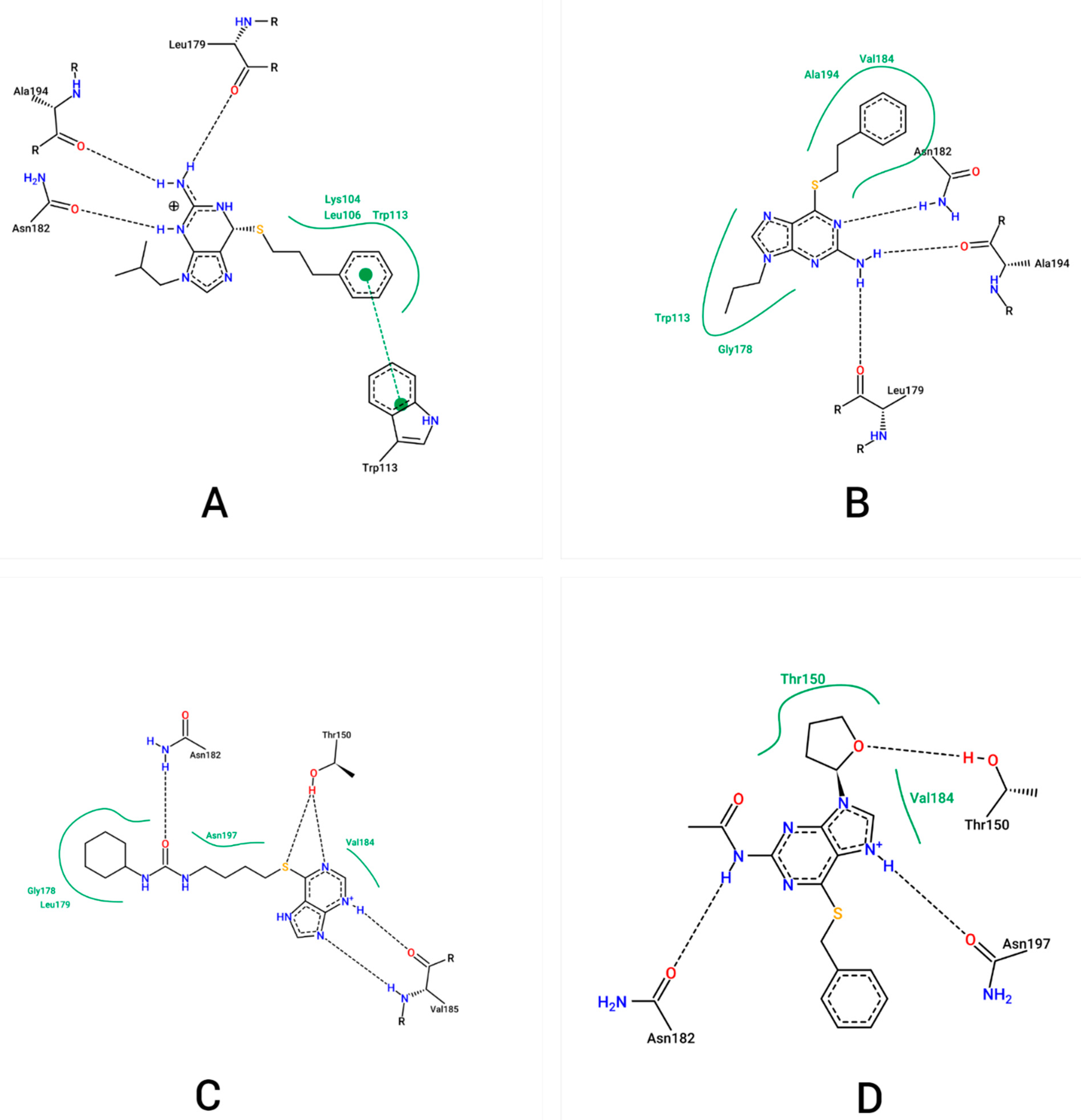
2.1. Molecular Docking
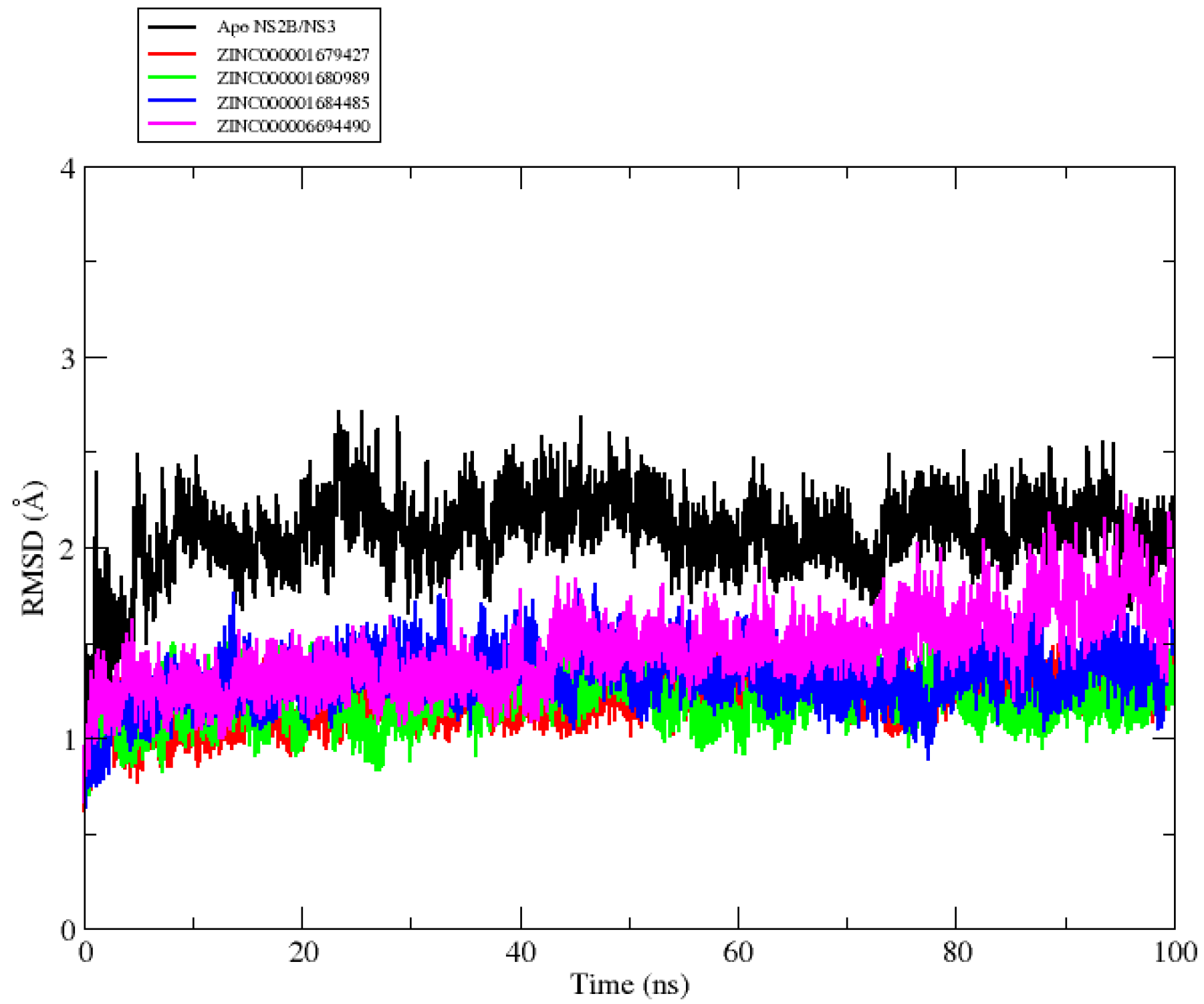
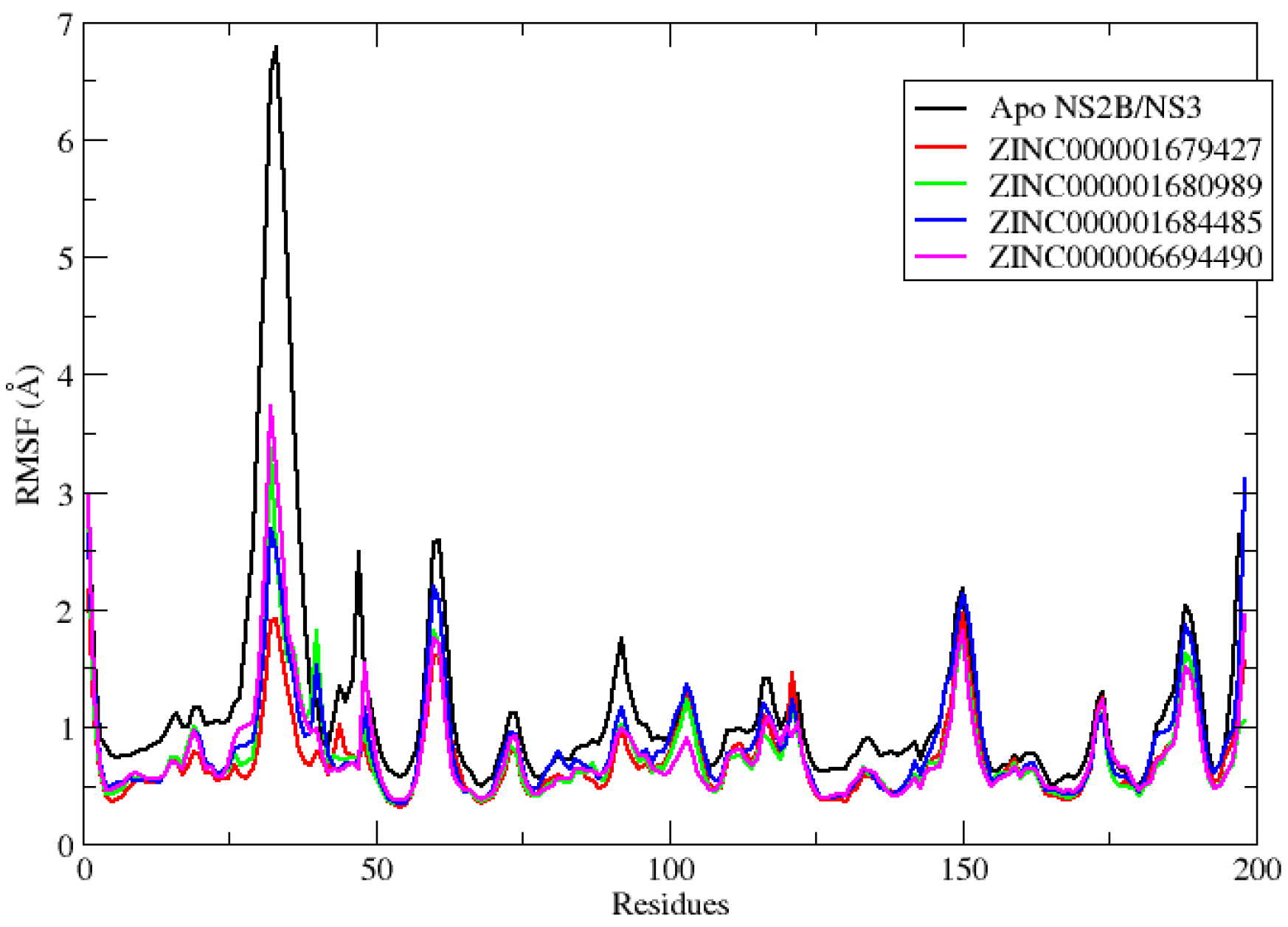
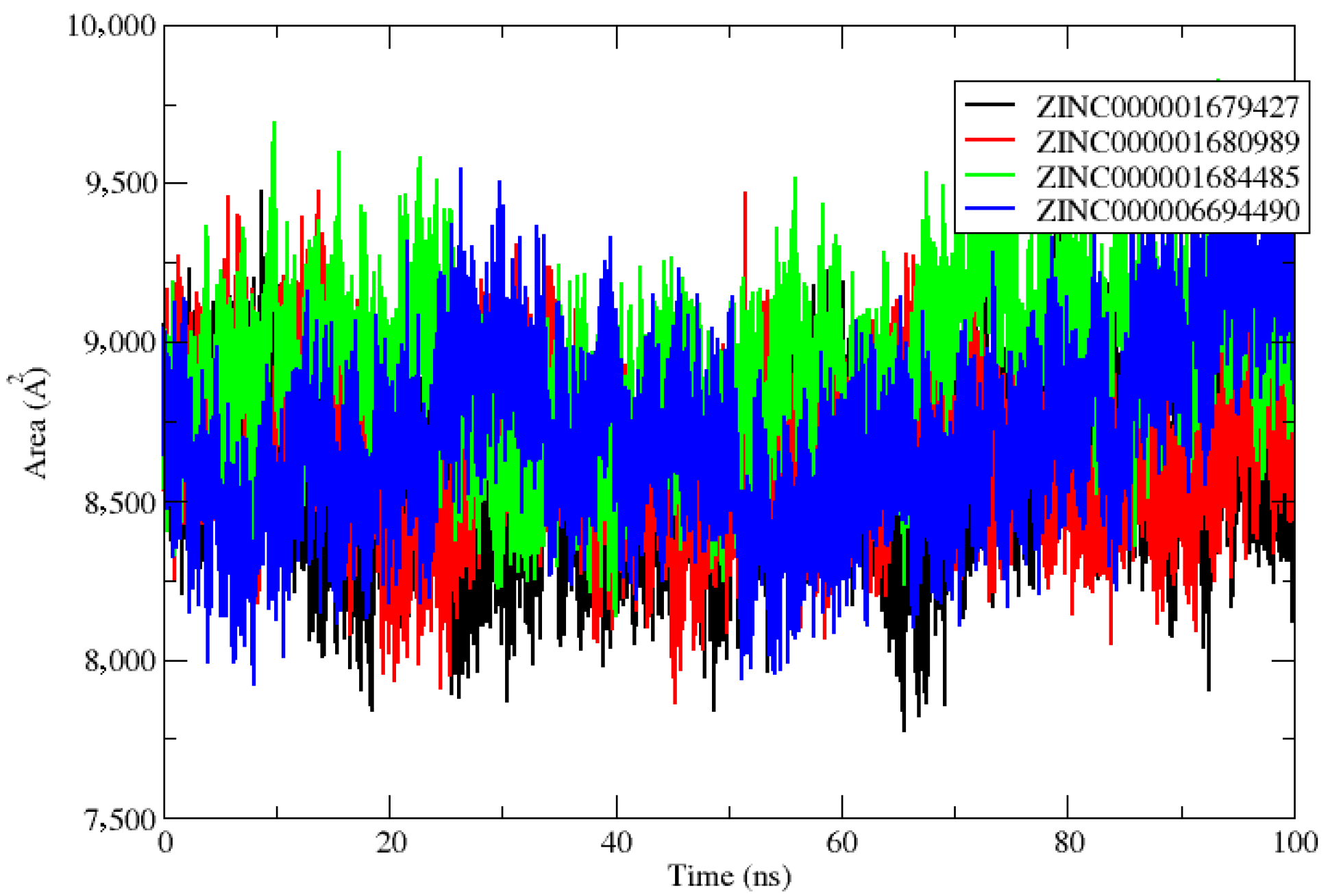
2.2. Molecular Dynamics
2.3. SwissADME Prediction of Selected Compounds
2.4. Toxicological Analysis by Derek NexusTM
2.5. Chromosome Damage: In Vivo Chromosome Aberration Test; In Vivo Micronucleus Test
3. Materials and Methods
3.1. Virtual Screening for Ligand Selection
3.2. Molecular Docking
3.3. Molecular Dynamics Simulation
3.4. Binding Free Energy Calculations MM/PBSA and MM/GBSA
3.5. Decomposition Energy by Residue
3.6. Pharmacokinetics Analysis by SwissADME
3.7. Toxicity Analysis by DerekTM
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clapham, H. Zika Virus Increases Risk of Dengue Disease. Science 2020, 369, 1055–1056. [Google Scholar] [CrossRef]
- Tovar, J.H.; Pinzón, M.A.; Rincón, D.; Jiménez-Canizalez, C.E.; Arrieta-Mendoza, M.E.; Mondragón-Cardona, Á. Consideraciones Anestésicas En El Paciente Con Enfermedad Por Virus Dengue. Rev. Chil. Anest. 2018, 47, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Vial, T.; Tan, W.-L.; Deharo, E.; Missé, D.; Marti, G.; Pompon, J. Mosquito Metabolomics Reveal That Dengue Virus Replication Requires Phospholipid Reconfiguration via the Remodeling Cycle. Proc. Natl. Acad. Sci. USA 2020, 117, 27627–27636. [Google Scholar] [CrossRef]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of Dengue Virus. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Green, A.M.; Beatty, P.R.; Hadjilaou, A.; Harris, E. Innate Immunity to Dengue Virus Infection and Subversion of Antiviral Responses. J. Mol. Biol. 2014, 426, 1148–1160. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Huang, Y.; Wu, Y.; Chen, H.; Wu, Y.; Hsu, C.; Hsu, Y.; Lee, J. MicroRNA-155 Inhibits Dengue Virus Replication by Inducing Heme Oxygenase-1-mediated Antiviral Interferon Responses. FASEB J. 2020, 34, 7283–7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecher, M.; Zhang, J.; Li, H. The Flavivirus Protease as a Target for Drug Discovery. Virol. Sin. 2013, 28, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Vasudevan, S.G.; Lescar, J. The Flavivirus NS2B–NS3 Protease–Helicase as a Target for Antiviral Drug Development. Antivir. Res. 2015, 118, 148–158. [Google Scholar] [CrossRef]
- Merdanovic, M.; Mönig, T.; Ehrmann, M.; Kaiser, M. Diversity of Allosteric Regulation in Proteases. ACS Chem. Biol. 2013, 8, 19–26. [Google Scholar] [CrossRef]
- Hauske, P.; Ottmann, C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Allosteric Regulation of Proteases. ChemBioChem 2008, 9, 2920–2928. [Google Scholar] [CrossRef]
- Mukhametov, A.; Newhouse, E.I.; Aziz, N.A.; Saito, J.A.; Alam, M. Allosteric Pocket of the Dengue Virus (Serotype 2) NS2B/NS3 Protease: In Silico Ligand Screening and Molecular Dynamics Studies of Inhibition. J. Mol. Graph. Model. 2014, 52, 103–113. [Google Scholar] [CrossRef]
- Millies, B.; von Hammerstein, F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-Based Allosteric Inhibitors of Zika and Dengue Virus NS2B/NS3 Proteases. J. Med. Chem. 2019, 62, 11359–11382. [Google Scholar] [CrossRef]
- Lim, L.; Dang, M.; Roy, A.; Kang, J.; Song, J. Curcumin Allosterically Inhibits the Dengue NS2B-NS3 Protease by Disrupting Its Active Conformation. ACS Omega 2020, 5, 25677–25686. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, M.; Ghosh, S.; Bell, J.A.; Sherman, W.; Hardy, J.A. Allosteric Inhibition of the NS2B-NS3 Protease from Dengue Virus. ACS Chem. Biol. 2013, 8, 2744–2752. [Google Scholar] [CrossRef] [Green Version]
- Uday, R.V.S.; Misra, R.; Harika, A.; Dolui, S.; Saha, A.; Pal, U.; Ravichandiran, V.; Maiti, N.C. Dabrafenib, Idelalisib and Nintedanib Act as Significant Allosteric Modulator for Dengue NS3 Protease. PLoS ONE 2021, 16, e0257206. [Google Scholar] [CrossRef]
- Brecher, M.; Li, Z.; Liu, B.; Zhang, J.; Koetzner, C.A.; Alifarag, A.; Jones, S.A.; Lin, Q.; Kramer, L.D.; Li, H. A Conformational Switch High-Throughput Screening Assay and Allosteric Inhibition of the Flavivirus NS2B-NS3 Protease. PLoS Pathog. 2017, 13, e1006411. [Google Scholar] [CrossRef]
- Hariono, M.; Choi, S.B.; Roslim, R.F.; Nawi, M.S.; Tan, M.L.; Kamarulzaman, E.E.; Mohamed, N.; Yusof, R.; Othman, S.; Abd Rahman, N.; et al. Thioguanine-Based DENV-2 NS2B/NS3 Protease Inhibitors: Virtual Screening, Synthesis, Biological Evaluation and Molecular Modelling. PLoS ONE 2019, 14, e0210869. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural Basis for the Activation of Flaviviral NS3 Proteases from Dengue and West Nile Virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- da Costa, R.A.; da Rocha, J.A.P.; Pinheiro, A.S.; da Costa, A.S.S.; da Rocha, E.C.M.; Josino, L.P.C.; da Gonçalves, A.S.; Lima, A.H.L.; Brasil, D.S.B. In Silico Identification of Novel Allosteric Inhibitors of Dengue Virus NS2B/NS3 Serine Protease. J. Serb. Chem. Soc. 2022, 11. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure Based Pharmacophore Modeling, Virtual Screening, Molecular Docking and ADMET Approaches for Identification of Natural Anti-Cancer Agents Targeting XIAP Protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Kato, K.; Nakayoshi, T.; Fukuyoshi, S.; Kurimoto, E.; Oda, A. Validation of Molecular Dynamics Simulations for Prediction of Three-Dimensional Structures of Small Proteins. Molecules 2017, 22, 1716. [Google Scholar] [CrossRef] [Green Version]
- Pal, U.; Sen, S.; Maiti, N.C. Cα–H Carries Information of a Hydrogen Bond Involving the Geminal Hydroxyl Group: A Case Study with a Hydrogen-Bonded Complex of 1,1,1,3,3,3-Hexafluoro-2-Propanol and Tertiary Amines. J. Phys. Chem. A 2014, 118, 1024–1030. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Shahroz, M.M.; Sharma, H.K.; Altamimi, A.S.A.; Alamri, M.A.; Ali, A.; Ali, A.; Alqahtani, S.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; et al. Novel and Potential Small Molecule Scaffolds as DYRK1A Inhibitors by Integrated Molecular Docking-Based Virtual Screening and Dynamics Simulation Study. Molecules 2022, 27, 1159. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 7. Entropy Effects on the Performance of End-Point Binding Free Energy Calculation Approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Wu, H.; Bock, S.; Snitko, M.; Berger, T.; Weidner, T.; Holloway, S.; Kanitz, M.; Diederich, W.E.; Steuber, H.; Walter, C.; et al. Novel Dengue Virus NS2B/NS3 Protease Inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1100–1109. [Google Scholar] [CrossRef] [Green Version]
- Othman, R.; Kiat, T.S.; Khalid, N.; Yusof, R.; Irene Newhouse, E.; Newhouse, J.S.; Alam, M.; Rahman, N.A. Docking of Noncompetitive Inhibitors into Dengue Virus Type 2 Protease: Understanding the Interactions with Allosteric Binding Sites. J. Chem. Inf. Model. 2008, 48, 1582–1591. [Google Scholar] [CrossRef]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of LogP Methods on More than 96,000 Compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The Influence of Lipophilicity in Drug Discovery and Design. Expert Opin. Drug Dis. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Ji, D.; Xu, M.; Udenigwe, C.C.; Agyei, D. Physicochemical Characterisation, Molecular Docking, and Drug-Likeness Evaluation of Hypotensive Peptides Encrypted in Flaxseed Proteome. Curr. Res. Food Sci. 2020, 3, 41–50. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.C.; Freitas, H.F.; Campos, J.M.; Kimani, N.M.; Silva, C.H.T.P.; Borges, R.S.; Pita, S.S.R.; Santos, C.B.R. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. Int. J. Mol. Sci. 2021, 22, 11739. [Google Scholar] [CrossRef]
- Silva, R.; Poiani, J.; Ramos, R.; Costa, J.; Silva, C.; Brasil, D.; Santos, C. Ligand and Structure-Based Virtual Screening from 16-((Diisobutylamino)Methyl)-6α-Hydroxyivouacapane-7β,17β-Lactone a Compound with Potential Anti-Prostate Cancer Activity. J. Serb. Chem. Soc. 2019, 84, 153–174. [Google Scholar] [CrossRef] [Green Version]
- Kontorinis, N.; Agarwal, K.; Gondolesi, G.; Fiel, M.I.; O’Rourke, M.; Schiano, T.D. Diagnosis of 6 Mercaptopurine Hepatotoxicity Post Liver Transplantation Utilizing Metabolite Assays. Am. J. Transp. 2004, 4, 1539–1542. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.A.; Philips, F.S.; Sternberg, S.S.; Stock, C.C.; Elion, G.B.; Hitchings, G.H. 6-Mercaptopurine: Effects in Mouse Sarcoma 180 and in Normal Animals. Cancer Res. 1953, 13, 593–604. [Google Scholar] [PubMed]
- Gisbert, J.P.; González-Lama, Y.; Maté, J. Thiopurine-Induced Liver Injury in Patients with Inflammatory Bowel Disease: A Systematic Review. Am. J. Gastroenterol. 2007, 102, 1518–1527. [Google Scholar] [CrossRef]
- Bissuel, F.; Bruneel, F.; Habersetzer, F.; Chassard, D.; Cotte, L.; Chevallier, M.; Bernuau, J.; Lucet, J.-C.; Trepo, C. Fulminant Hepatitis with Severe Lactate Acidosis in HIV-Infected Patients on Didanosine Therapy. J. Int. Med. 1994, 235, 367–372. [Google Scholar] [CrossRef]
- Maida, I.; Núñez, M.; Ríos, M.J.; Martín-Carbonero, L.; Sotgiu, G.; Toro, C.; Rivas, P.; Barreiro, P.; Mura, M.S.; Babudieri, S.; et al. Severe Liver Disease Associated with Prolonged Exposure to Antiretroviral Drugs. JAIDS J. Acquir. Immune Defic. Syndr. 2006, 42, 177–182. [Google Scholar] [CrossRef]
- Hu, B.; French, S.W. 2′,3′-Dideoxyinosine–Induced Mallory Bodies in Patients With HIV. Am. J. Clin. Pathol. 1997, 108, 280–283. [Google Scholar] [CrossRef] [Green Version]
- Lacaille, F.; Ortigao, M.B.; Debré, M.; Rouzioux, C.; Brousse, N.; Blanche, S. Hepatic Toxicity Associated with 2′-3ʼDideoxyinosine in Children with AIDS. J. Pediatr. Gastroenterol. Nutr. 1995, 20, 287–290. [Google Scholar] [CrossRef]
- Bastida, G.; Nos, P.; Aguas, M.; Beltran, B.; Rubin, A.; Dasi, F.; Ponce, J. Incidence, Risk Factors and Clinical Course of Thiopurine-Induced Liver Injury in Patients with Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2005, 22, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Eddleston, A.L.; Williams, R. Hypersensitivity and Jaundice Due to Azathioprine. Postgrad. Med. J. 1980, 56, 274–275. [Google Scholar] [CrossRef]
- Jeurissen, M.E.; Boerbooms, A.M.; van de Putte, L.B.; Kruijsen, M.W. Azathioprine Induced Fever, Chills, Rash, and Hepatotoxicity in Rheumatoid Arthritis. Ann. Rheum. Dis. 1990, 49, 25–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, P.; Lambert, T.R.; de G. Mitchell, I. Comparative In-Vivo Genotoxicity of Antiviral Nucleoside Analogues; Penciclovir, Acyclovir, Ganciclovir and the Xanthine Analogue, Caffeine, in the Mouse Bone Marrow Micronucleus Assay. Mutat. Res. Genet. Toxicol. 1996, 369, 65–74. [Google Scholar] [CrossRef]
- Hara, T.; Makita, T.; Horiya, N.; Ozawa, S.; Ohba, M.; Naito, J.; Shibuya, T. Micronucleus Test with 6-Mercaptopurine Monohydrate Administered Intraperitoneally and Orally. Mutat. Res. Genet. Toxicol. 1989, 223, 349–352. [Google Scholar] [CrossRef]
- Holden, H.E.; Ray, V.A.; Wahrenburg, M.G.; Zelenski, J.D. Mutagenicity Studies with 6-Mercaptopurine: I. Cytogenetic Activity in Vivo. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1973, 20, 257–263. [Google Scholar] [CrossRef]
- Phillips, M.D.; Nascimbeni, B.; Tice, R.R.; Shelby, M.D.; van Zeeland, A.A. Induction of Micronuclei in Mouse Bone Marrow Cells: An Evaluation of Nucleoside Analogues Used in the Treatment of AIDS. Environ. Mol. Mutagen. 1991, 18, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, R.; Mitsuya, H.; Thomas, R.V.; Pluda, J.M.; Hartman, N.R.; Perno, C.-F.; Marczyk, K.S.; Allain, J.-P.; Johns, D.G.; Broder, S. In Vivo Activity Against HIV and Favorable Toxicity Profile of 2′,3′-Dideoxyinosine. Science 1989, 245, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Wutzler, P.; Thust, R. Genetic Risks of Antiviral Nucleoside Analogues–a Survey. Antivir. Res. 2001, 49, 55–74. [Google Scholar] [CrossRef]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef]
- Majumder, R.; Mandal, M. Screening of Plant-Based Natural Compounds as a Potential COVID-19 Main Protease Inhibitor: An in Silico Docking and Molecular Dynamics Simulation Approach. J. Biomol. Struct. Dyn. 2020, 40, 696–711. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.; Willem, M.; Nissink, J.; Taylor, R. Protein-Ligand Docking Virtual Screening with GOLD. In Virtual Screening in Drug Discovery; Taylor & Francis CRC Press: Boca Raton, FL, USA, 2005; pp. 379–415. [Google Scholar]
- Werner, T.; Morris, M.B.; Dastmalchi, S.; Church, W.B. Structural Modelling and Dynamics of Proteins for Insights into Drug Interactions. Adv. Drug Deliv. Rev. 2012, 64, 323–343. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Song, L.F.; Lee, T.-S.; Zhu, C.; York, D.M.; Merz, K.M. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 59, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09; Revision d. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Volume 201. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.A.; Cruz, J.N.; Nascimento, F.C.A.; Silva, S.G.; Silva, S.O.; Martelli, M.C.; Carvalho, S.M.L.; Santos, C.B.R.; Neto, A.M.J.C.; Brasil, D.S.B. Studies of NMR, Molecular Docking, and Molecular Dynamics Simulation of New Promising Inhibitors of Cruzaine from the Parasite Trypanosoma Cruzi. Med. Chem. Res. 2019, 28, 246–259. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME–Tox). Drug Dis. Today 2008, 13, 379–393. [Google Scholar] [CrossRef]
- Costa, E.B.; Silva, R.C.; Espejo-Román, J.M.; Neto, M.F.d.A.; Cruz, J.N.; Leite, F.H.A.; Silva, C.H.T.P.; Pinheiro, J.C.; Macêdo, W.J.C.; Santos, C.B.R. Chemometric Methods in Antimalarial Drug Design from 1,2,4,5-Tetraoxanes Analogues. SAR QSAR Environ. Res. 2020, 31, 677–695. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting Drug Metabolism: Experiment and/or Computation? Nat. Rev. Drug Dis. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, N.; Judson, P.N.; Langowski, J.J.; Marchant, C.A. Knowledge-Based Expert Systems for Toxicity and Metabolism Prediction: DEREK, StAR and METEOR. SAR QSAR Environ. Res. 1999, 10, 299–314. [Google Scholar] [CrossRef]
- Braggio, S.; Corsi, M.; Feriani, A.; Fontana, S.; Marocchio, L.; Virginio, C. CHAPTER 15. Discovery Toxicology in Lead Optimisation. In The Handbook of Medicinal Chemistry; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 364–412. [Google Scholar]
- Sutter, A.; Amberg, A.; Boyer, S.; Brigo, A.; Contrera, J.F.; Custer, L.L.; Dobo, K.L.; Gervais, V.; Glowienke, S.; van Gompel, J.; et al. Use of in Silico Systems and Expert Knowledge for Structure-Based Assessment of Potentially Mutagenic Impurities. Regul. Toxicol. Pharmacol. 2013, 67, 39–52. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | PLPchem | Structures |
|---|---|---|
| ZINC000001680989 | 62.05 | 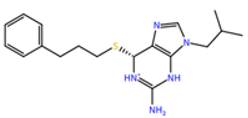 |
| ZINC000001679427 | 59.69 | 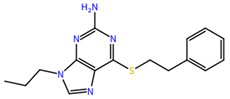 |
| ZINC000006694490 | 58.41 | 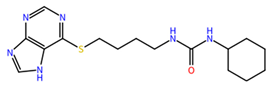 |
| ZINC000001684485 | 58.36 | 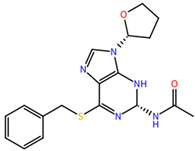 |
| Ligand ID | Terms | |||||
|---|---|---|---|---|---|---|
| ΔEvdW | ΔEele | ΔGGB | ΔGnonpol | ΔGMMGBSA | ΔGMMPBSA | |
| ZINC000001680989 | −44.83 ± 0.10 | −21.84 ± 0.11 | 31.28 ± 0.09 | −5.15 ± 0.01 | −40.65 ± 0.09 | −39.43 ± 0.12 |
| ZINC000001679427 | −42.58 ± 0.10 | −21.53 ± 0.19 | 33.98 ± 0.16 | −5.40 ± 0.01 | −35.53 ± 0.11 | −25.72 ± 0.13 |
| ZINC000006694490 | −38.22 ± 0.09 | −33.08 ± 0.25 | 48.50 ± 0.20 | −4.96 ± 0.01 | −27.76 ± 0.09 | −30.30 ± 0.12 |
| ZINC000001684485 | −22.14 ± 0.07 | −31 11± 0.15 | 45.84 ± 0.35 | −4.31 ± 0.01 | −11.71 ± 0.14 | −19.67 ± 0.22 |
| Complex | Hydrogen Bond Formation | Distance (Å) | Occupancy (%) |
|---|---|---|---|
| NS2B/NS3pro-ZINC000001680989 | LEU179@O-LIG@H16-LIG@N3 | 2.82 | 86.39 |
| ALA194@O-LIG@H15-LIG@N3 | 2.83 | 82.74 | |
| LIG@N2-ASN182@HD21-ASN_182@ND2 | 2.91 | 20.09 | |
| ALA194@O-LIG@H16-LIG@N3 | 2.82 | 16.96 | |
| LEU179@O-LIG@H15-LIG@N3 | 2.82 | 16.85 | |
| LIG@N4-LEU179@H-LEU179@N | 2.94 | 8.91 | |
| LIG@N3-ASN182@HD21-ASN182@ND2 | 2.93 | 2.49 | |
| NS2B/NS3pro-ZINC000001679427 | ALA194@O-LIG@H15-LIG@N3 | 2.83 | 46.96 |
| LEU179@O-LIG@H14-LIG@N3 | 2.83 | 32.68 | |
| ALA194@O-LIG@H14-LIG@N3 | 2.83 | 26.39 | |
| LEU179@O-LIG@H15-LIG@N3 | 2.82 | 15.65 | |
| LIG@N3-ASN182@HD21-ASN182@ND2 | 2.92 | 11.13 | |
| LIG@N4-ASN182@HD21- ASN182@ND2 | 2.93 | 3.37 | |
| LIG@N2-LEU179@H-LEU179@N | 2.94 | 2.58 | |
| LYS104@O-LIG@H14-LIG@N3 | 2.82 | 2.37 | |
| NS2B/NS3pro-ZINC000006694490 | ASN182@OD1- LIG@H23-LIG@N4 | 2.82 | 62.53 |
| LYS104@O-LIG@H23-LIG@N4 | 2.83 | 8.58 | |
| ILE195@O-LIG@H11-LIG@N5 | 2.88 | 6.83 | |
| ILE_195@O-LIG@H-LIG@N | 2.88 | 5.58 | |
| LIG@N1-ASN197@HD21-ASN197@ND2 | 2.91 | 4.47 | |
| LIG@O-LYS104@HZ1-LYS104@NZ | 2.82 | 3.27 | |
| LIG@OLYS104@HZ2-LYS104@NZ | 2.81 | 3.19 | |
| LIG@O-LYS104@HZ3-LYS104@NZ | 2.81 | 2.86 | |
| NS2B/NS3pro-ZINC000001684485 | LYS104@NZ-LIG@H-LIG@N | 2.80 | 95.48 |
| LIG@N-LYS104@HZ1-LYS104@NZ | 2.80 | 55.70 | |
| LIG@N-LYS104@HZ3-LYS104@NZ | 2.80 | 20.84 | |
| LIG@N-LYS104@HZ2-LYS104@NZ | 2.79 | 18.93 | |
| LIG@N4-ASN197@HD21-ASN197@ND2 | 2.91 | 2.04 |
| Ligands | MR | TPSA (Å2) | Log Po/w | Log S | Log Kp (cm/s) | BS | SA |
|---|---|---|---|---|---|---|---|
| ZINC000001684485 | 100.5 | 107.23 | 2.33 | Moderately soluble | −6.91 | 0.55 | 3.69 |
| ZINC000006694490 | 95.55 | 120.89 | 2.40 | Moderately soluble | −6.27 | 0.55 | 3.19 |
| ZINC000001680989 | 101.23 | 94.92 | 3.50 | Poorly soluble | −5.18 | 0.55 | 3.06 |
| ZINC000001679427 | 91.62 | 94.92 | 2.87 | Moderately soluble | −5.17 | 0.55 | 2.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, R.A.d.; Rocha, J.A.P.d.; Pinheiro, A.S.; Costa, A.d.S.S.d.; Rocha, E.C.M.d.; Silva, R.C.; Gonçalves, A.d.S.; Santos, C.B.R.; Brasil, D.d.S.B. A Computational Approach Applied to the Study of Potential Allosteric Inhibitors Protease NS2B/NS3 from Dengue Virus. Molecules 2022, 27, 4118. https://doi.org/10.3390/molecules27134118
Costa RAd, Rocha JAPd, Pinheiro AS, Costa AdSSd, Rocha ECMd, Silva RC, Gonçalves AdS, Santos CBR, Brasil DdSB. A Computational Approach Applied to the Study of Potential Allosteric Inhibitors Protease NS2B/NS3 from Dengue Virus. Molecules. 2022; 27(13):4118. https://doi.org/10.3390/molecules27134118
Chicago/Turabian StyleCosta, Renato A. da, João A. P. da Rocha, Alan S. Pinheiro, Andréia do S. S. da Costa, Elaine C. M. da Rocha, Rai. C. Silva, Arlan da S. Gonçalves, Cleydson B. R. Santos, and Davi do S. B. Brasil. 2022. "A Computational Approach Applied to the Study of Potential Allosteric Inhibitors Protease NS2B/NS3 from Dengue Virus" Molecules 27, no. 13: 4118. https://doi.org/10.3390/molecules27134118
APA StyleCosta, R. A. d., Rocha, J. A. P. d., Pinheiro, A. S., Costa, A. d. S. S. d., Rocha, E. C. M. d., Silva, R. C., Gonçalves, A. d. S., Santos, C. B. R., & Brasil, D. d. S. B. (2022). A Computational Approach Applied to the Study of Potential Allosteric Inhibitors Protease NS2B/NS3 from Dengue Virus. Molecules, 27(13), 4118. https://doi.org/10.3390/molecules27134118