
Novel Galactopyranoside Esters: Synthesis, Mechanism, In Vitro Antimicrobial Evaluation and Molecular Docking Studies

 ,
, 
 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
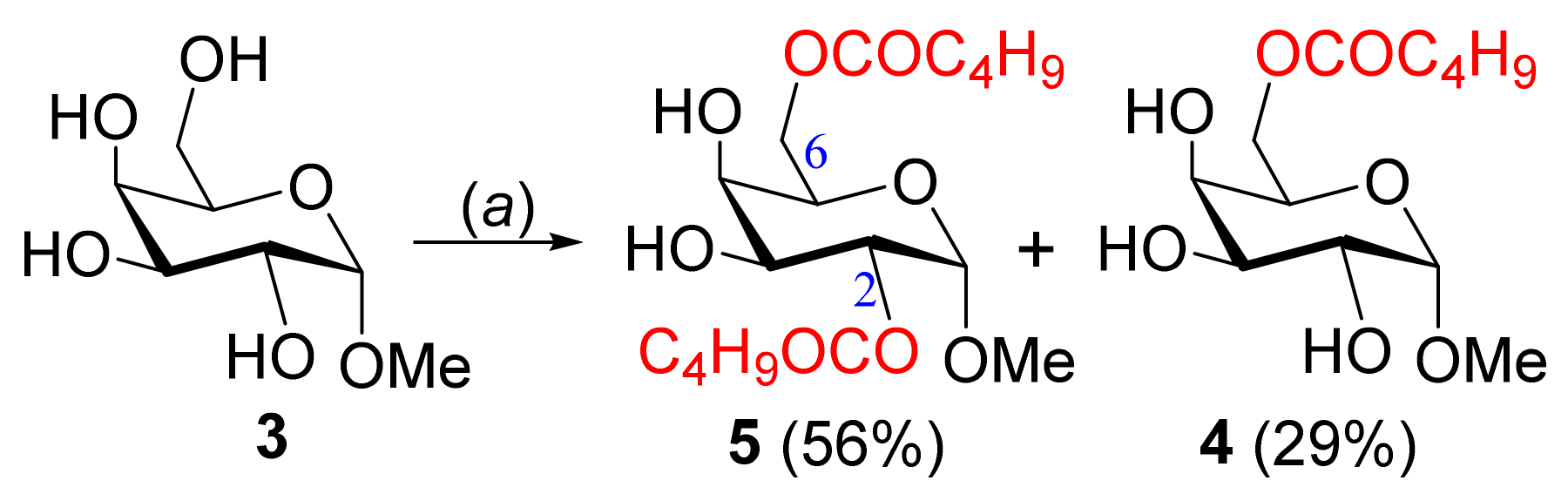
2.1. Selective 6-O-Valeroylation of MDG
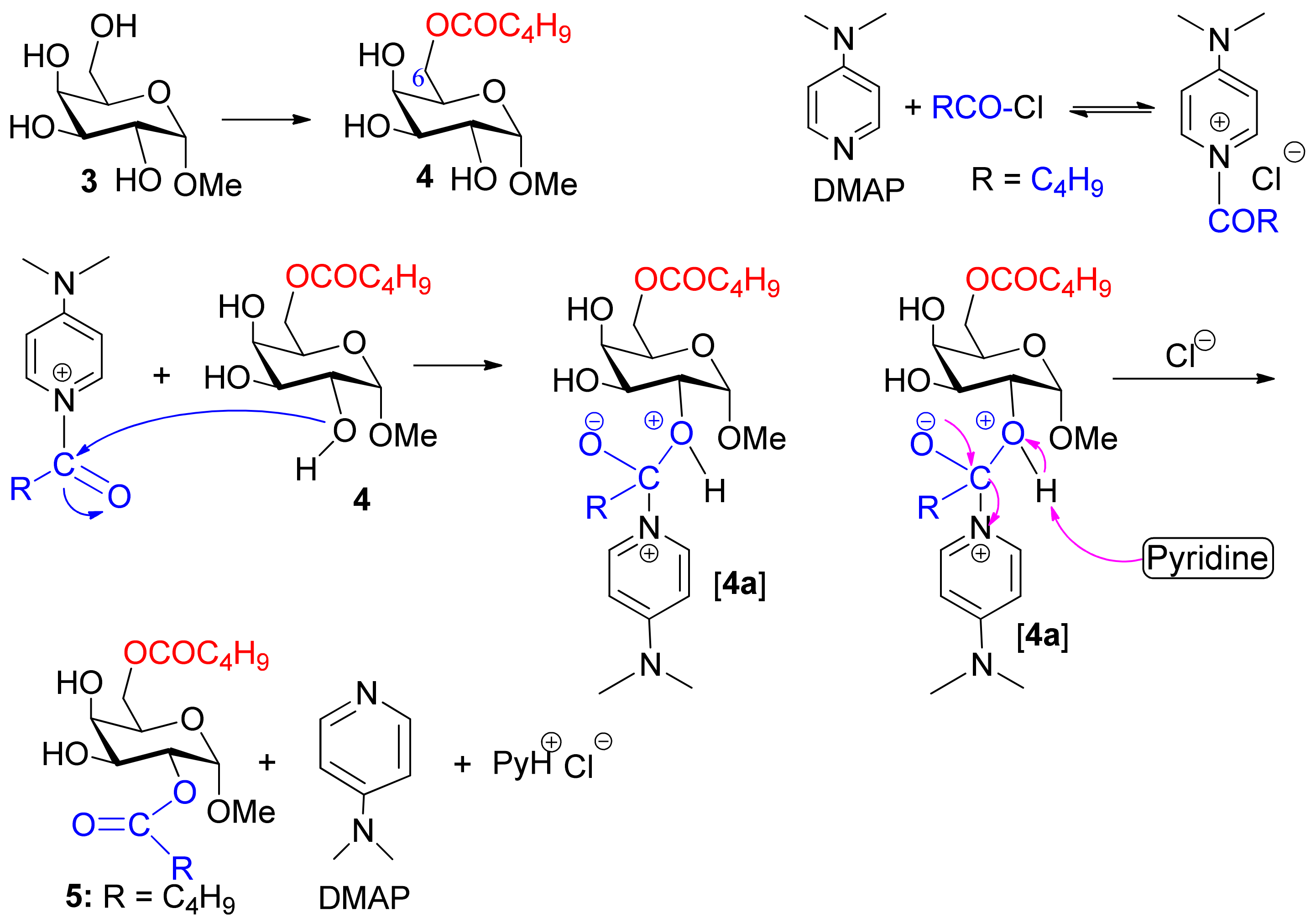
2.2. DMAP-Catalyzed Valeroylation, Reactivity Order of MDG, and Probable Mechanism
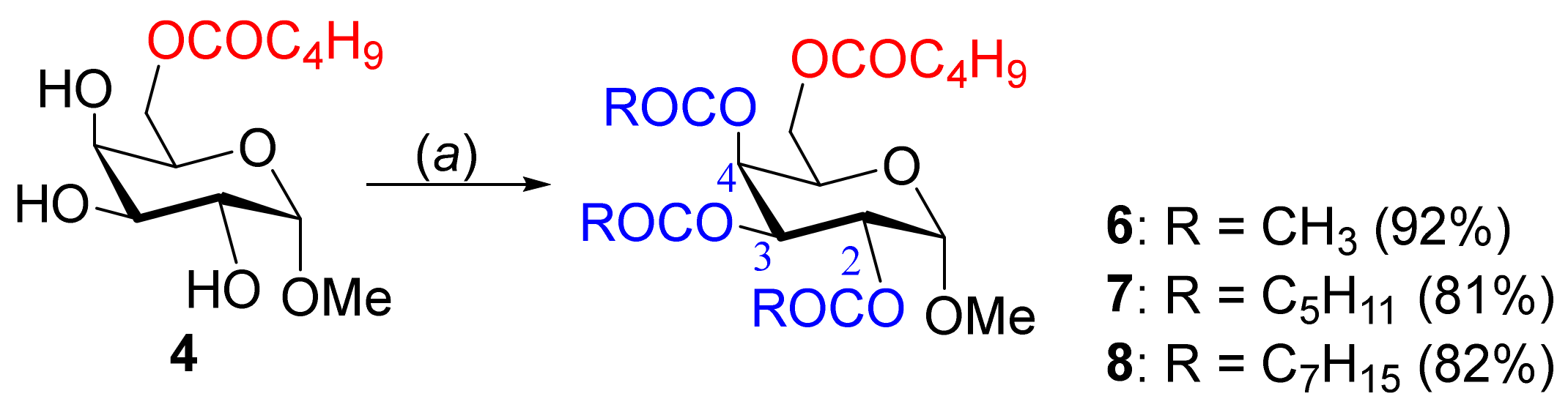
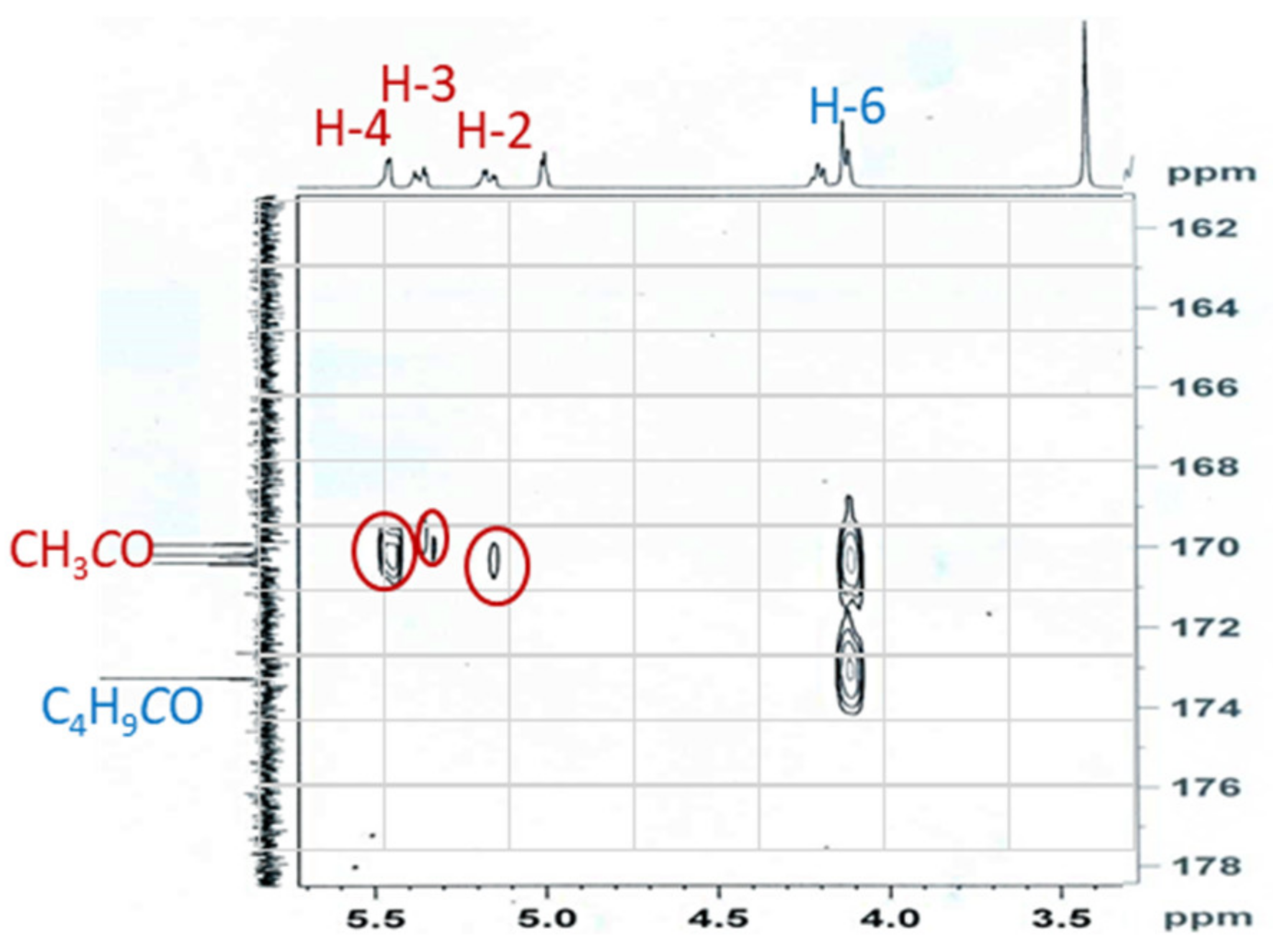
2.3. Synthesis of 2,3,4-Tri-O-acyl Esters from 6-O-Valeroate 4
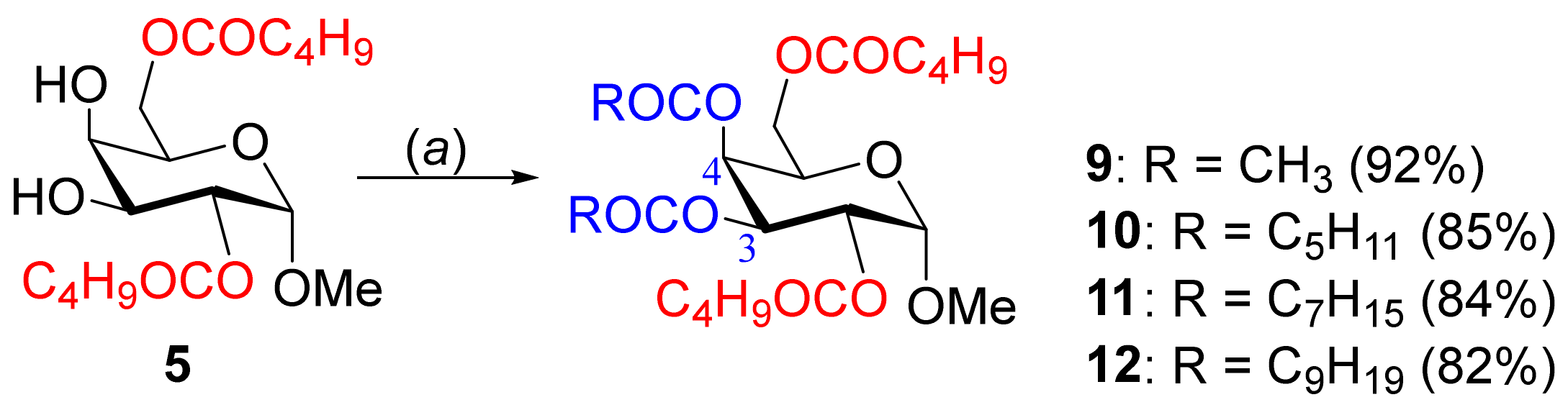
2.4. Synthesis of 3,4-Di-O-acyl Esters from 2,6-di-O-Valeroate 5
2.5. Prediction of Antimicrobial Activities of 3–12
2.6. In Vitro Antimicrobial Activities of MDG Esters 4–12
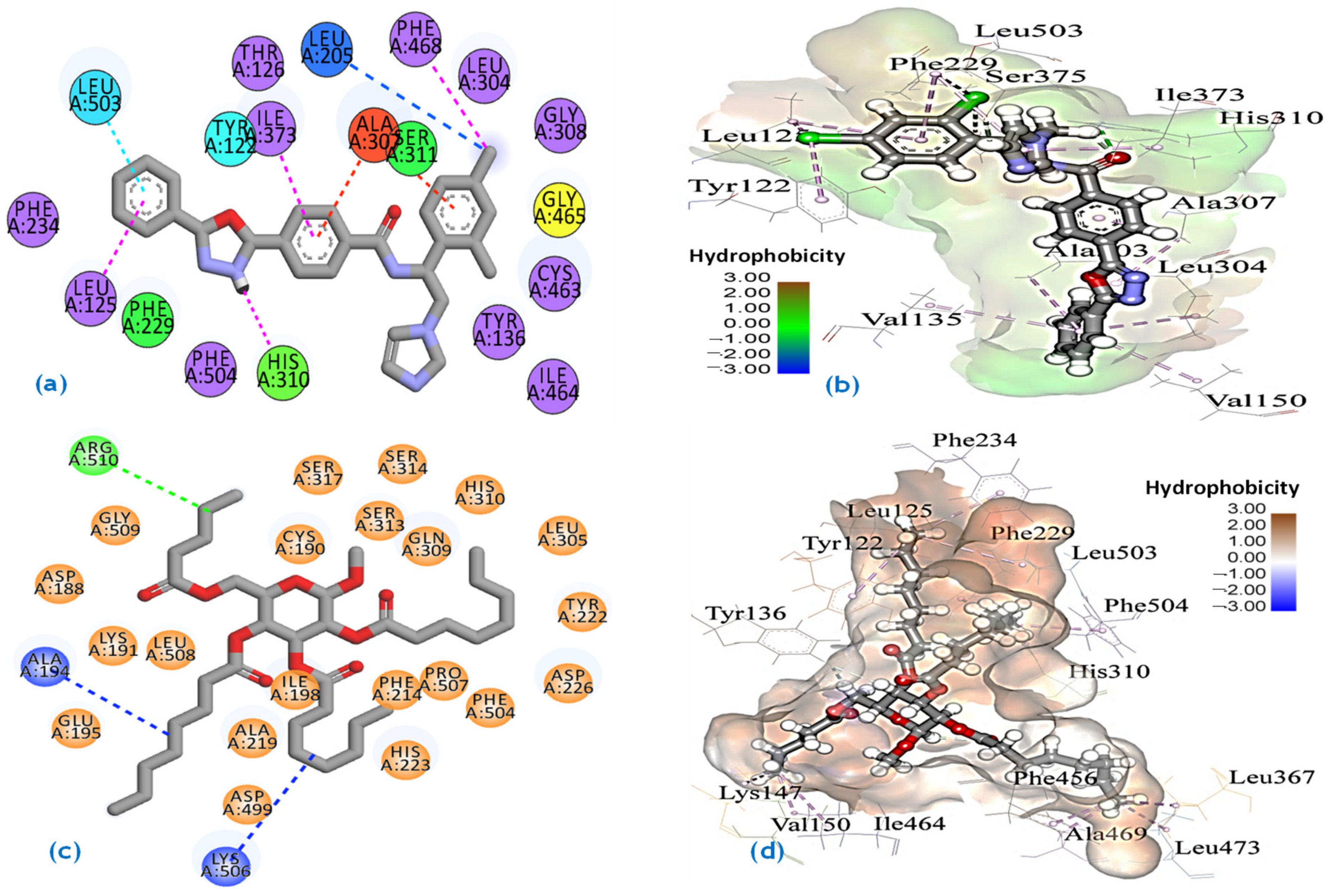
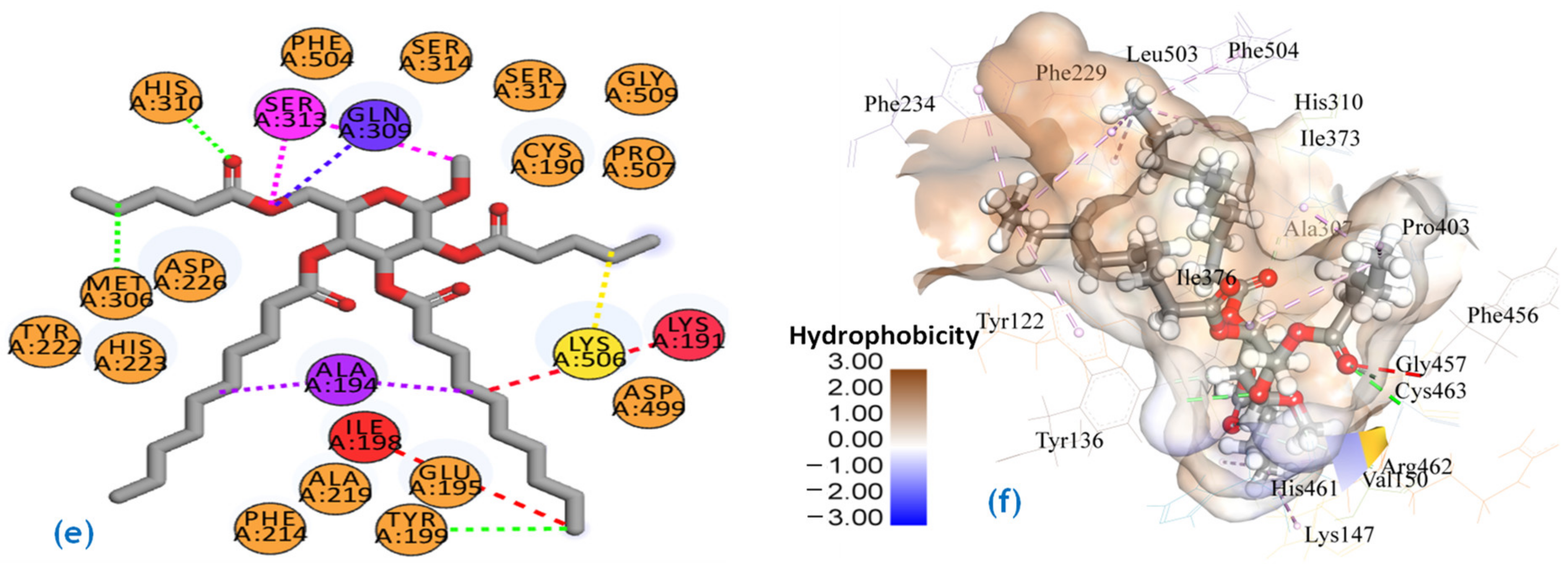
2.7. Molecular Docking Studies
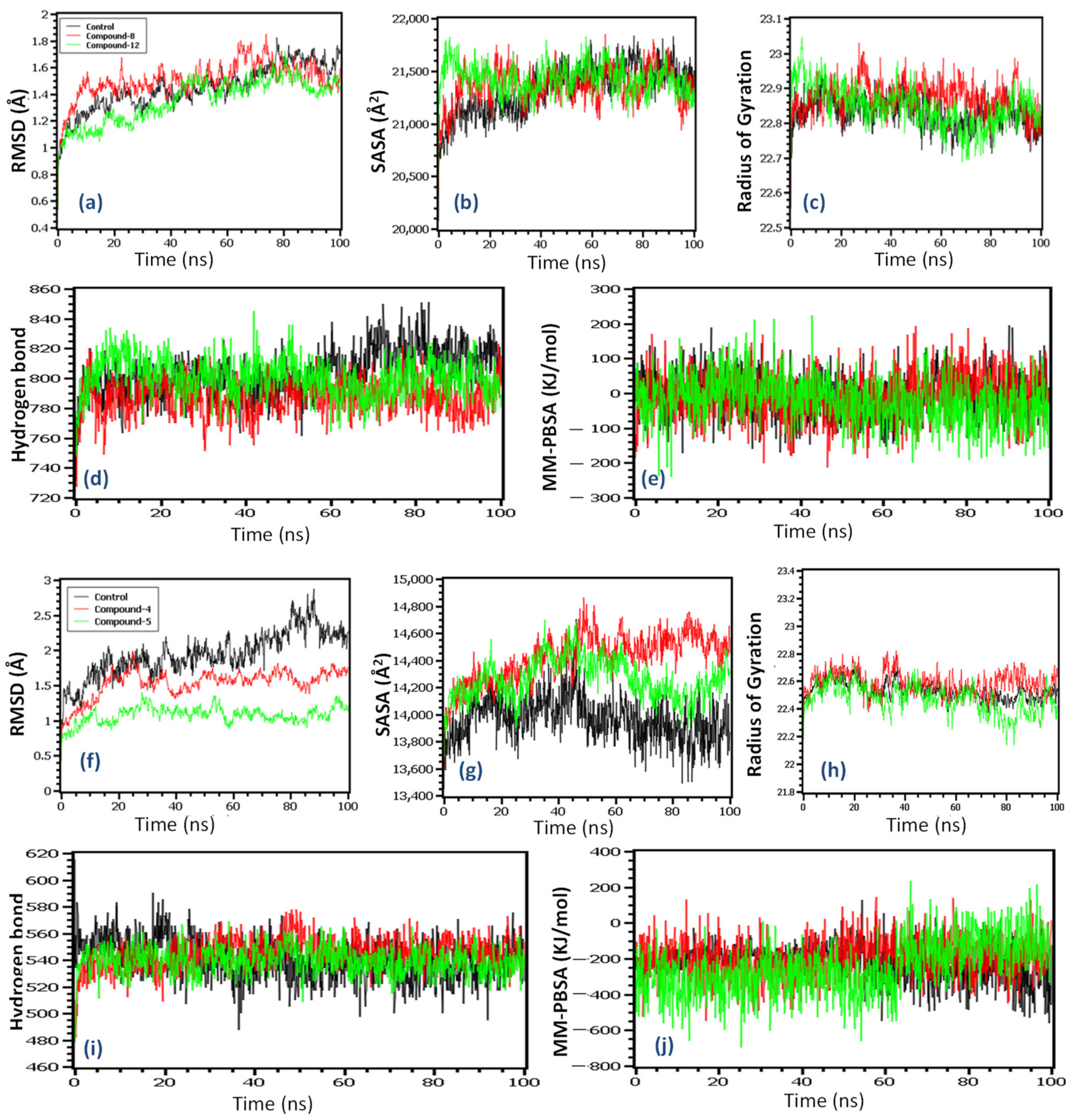
2.8. Molecular Dynamics Simulation
2.9. In Silico Inhibition Constant (Ki) Analysis
2.10. ADMET Studies
2.11. Drug-Likeness Results
3. Materials and Methods
3.1. General Methods and Instruments
3.2. Synthesis
3.3. Predication of Biological Activities
3.4. In Vitro Antimicrobial Potentiality Evaluation
3.5. Computational Methods
3.5.1. Molecular Docking
3.5.2. Molecular Dynamics Simulations
3.5.3. ADMET and Drug Friendliness Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Koester, D.C.; Holkenbrink, A.; Werz, D.B. Recent advances in the synthesis of carbohydrate mimics. Synthesis 2010, 2010, 3217–3242. [Google Scholar] [CrossRef]
- Bhat, S.H.; Ullah, M.F.; Abu-Duhier, F.M. Bioactive extract of artemisia judaica causes in vitro inhibition of dipeptidyl peptidase iv and pancreatic/intestinal enzymes of the carbohydrate absorption cascade: Implication for anti-diabetic new molecular entities (nmes). Orient. Pharm. Exp. Med. 2019, 19, 71–80. [Google Scholar] [CrossRef]
- Leibeling, M.; Werz, D.B. Carbohydrate-based synthetic chemistry in the context of drug design. In Carbohydrates as Drugs; Springer: Cham, Switzerland, 2014; Volume 12, pp. 1–21. [Google Scholar] [CrossRef]
- Dubbu, S.; Chennaiah, A.; Verma, A.K.; Vankar, Y.D. Stereoselective synthesis of 2-deoxy-β-C-aryl/alkyl glycosides using Prins cyclization: Application in the synthesis of C-disaccharides and differently protected C-aryl glycosides. Carbohydr. Res. 2018, 468, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Dhavale, D.D.; Matin, M.M. Selective sulfonylation of 4-C-hydroxymethyl-β-L-threo-pento-1,4-furanose: Synthesis of bicyclic diazasugars. Tetrahedron 2004, 60, 4275–4281. [Google Scholar] [CrossRef]
- Dhavale, D.D.; Matin, M.M. Piperidine homoazasugars: Natural occurrence, synthetic aspects and biological activity study. ARKIVOC 2005, 2005, 110–132. [Google Scholar] [CrossRef] [Green Version]
- Matin, M.M.; Ibrahim, M.; Anisa, T.R.; Rahman, M.R. Synthesis, characterization, in silico optimization, and conformational studies of methyl 4-O-palmitoyl-α-L-rhamnopyranosides. Malays. J. Sci. 2022, 41, 91–105. [Google Scholar] [CrossRef]
- Matin, M.M.; Iqbal, M.Z. Methyl 4-O-(2-chlorobenzoyl)-α-L-rhamnopyranosides: Synthesis, characterization, and thermodynamic studies. Orbital Electron. J. Chem. 2021, 13, 19–27. [Google Scholar] [CrossRef]
- Matin, M.M. One step intramolecular cyclization of diol via mesylation: Efficient synthesis of sugar derived [1,4]oxazepanes. J. Bangladesh Chem. Soc. 2008, 21, 179–183. [Google Scholar]
- Marshall, D.L.; Bullermann, L.B. Antimicrobial properties of sucrose fatty acid esters. In Carbohydrate Polymers as Fat Substitutes; Akoh, C.C., Swanson, B.G., Eds.; Marcel Dekker: New York, NY, USA, 1994; pp. 149–167. [Google Scholar]
- Chang, S.W.; Shaw, J.F. Biocatalysis for the production of carbohydrate esters. New Biotechnol. 2009, 26, 109–116. [Google Scholar] [CrossRef]
- Okabe, S.; Suganuma, M.; Tada, Y.; Ochiai, Y.; Sueoka, E.; Kohya, H.; Shibata, A.; Takahashi, M.; Mizutani, M.; Matsuzaki, T.; et al. Disaccharide esters screened for inhibition of tumor necrosis factor-α release are new anti-cancer agents. Cancer Res. 1999, 90, 669–676. [Google Scholar] [CrossRef]
- Matin, M.M.; Bhattacharjee, S.C.; Chakraborty, P.; Alam, M.S. Synthesis, PASS predication, in vitro antimicrobial evaluation and pharmacokinetic study of novel n-octyl glucopyranoside esters. Carbohydr. Res. 2019, 485, 107812. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, Y.C. Neuroprotective phenylpropanoid esters of rhamnose isolated from roots of Scrophularia buergeriana. Phytochemistry 2000, 54, 503–509. [Google Scholar] [CrossRef]
- Elmaidomy, A.H.; Mohammed, R.; Owis, A.I.; Hetta, M.H.; AboulMagd, A.M.; Siddique, A.B.; Abdelmohsen, U.R.; Rateb, M.E.; Sayed, K.A.E.; Hassan, H.M. Triple-negative breast cancer suppressive activities, antioxidants and pharmacophore model of new acylated rhamnopyranoses from Premna odorata. RSC Adv. 2020, 10, 10584. [Google Scholar] [CrossRef] [Green Version]
- Lokhande, K.B.; Apte, G.R.; Shrivastava, A.; Singh, A.; Pal, J.K.; Swamy, K.V.; Gupta, R.K. Sensing the interactions between carbohydrate-binding agents and N-linked glycans of SARS-CoV-2 spike glycoprotein using molecular docking and simulation studies. J. Biomol. Struct. Dyn. 2020, 40, 3880–3898. [Google Scholar] [CrossRef]
- Chen, P.; Yang, J.-S. Flavonol galactoside caffeiate ester and homoisoflavones from Caesalpinia millettii Hook. Chem. Pharm. Bull. 2007, 55, 655–657. [Google Scholar] [CrossRef] [Green Version]
- Viana, P.A.; de Rezende, S.T.; Alves, A.D.A.; Manfrini, R.M.; Alves, R.J.; Bemquerer, M.P.; Santoro, M.M.; Guimarães, V.M. Activity of Debaryomyces hansenii UFV-1 α-galactosidases against α-D-galactopyranoside derivatives. Carbohydr. Res. 2011, 346, 602–605. [Google Scholar] [CrossRef]
- Kumar, V.; Ahmed, D.; Verma, A.; Anwar, F.; Ali, M.; Mujeeb, M. Umbelliferone β-D-galactopyranoside from Aegle marmelos (L.) corr. an ethnomedicinal plant with antidiabetic, antihyperlipidemic and antioxidative activity. BMC Complement. Altern. Med. 2013, 13, 273. [Google Scholar] [CrossRef] [Green Version]
- Matin, M.M. Synthesis of some protected 6-O-acyl-galactopyranose derivatives for antibacterial evaluation. Chittagong Univ. J. Sci. 2006, 30, 59–65. [Google Scholar]
- Matin, M.M.; Azad, A.K.M.S. Synthesis of some protected 6-O-acyl-galactopyranose derivatives. J. Appl. Sci. Res. 2006, 2, 1199–1202. [Google Scholar]
- Abe, Y.; Fujiwara, M.; Ohbu, K.; Harata, K. Crystal structures of methyl 6-O-acyl-α-D-galactopyranosides. Carbohydr. Res. 1995, 275, 9–16. [Google Scholar] [CrossRef]
- Myszka, H.; Sokołowska, P.; Cieślińska, A.; Nowacki, A.; Jaśkiewicz, M.; Kamysz, W.; Liberek, B. Diosgenyl 2-amino-2-deoxy-β-D-galactopyranoside: Synthesis, derivatives and antimicrobial activity. Beilstein J. Org. Chem. 2017, 13, 2310–2315. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Bayata, S.; Rahmana, M.B.A.; Ibrahima, Z.; Abdulmalek, E. Synthesis and in-vitro bioactivity evaluation of new galactose and fructose ester derivatives of 5-aminosalicylic acid. Chem. Biodivers. 2017, 14, e1600362. [Google Scholar] [CrossRef]
- Lipták, A.; Balla, E.; Jánossy, L.; Sajtos, F.; Szilágyi, L. The first synthesis of secondary sugar sulfonic acids by nucleophilic displacement reactions. Tetrahedron Lett. 2004, 45, 839–842. [Google Scholar] [CrossRef]
- Dimakos, V.; Taylor, M.S. Site-selective functionalization of hydroxyl groups in carbohydrate derivatives. Chem. Rev. 2018, 118, 11457–11517. [Google Scholar] [CrossRef]
- Rahman, M.A.; Matin, M.M.; Kumer, A.; Chakma, U.; Rahman, M.R. Modified D-glucofuranoses as new black fungus protease inhibitors: Computational screening, docking, dynamics, and QSAR study. Phys. Chem. Res. 2022, 10, 189–203. [Google Scholar] [CrossRef]
- Matin, M.M.; Islam, N.; Siddika, A.; Bhattacharjee, S.C. Regioselective synthesis of some rhamnopyranoside esters for PASS predication, and ADMET studies. J. Turk. Chem. Soc. Sect. A Chem. 2021, 8, 363–374. [Google Scholar] [CrossRef]
- Matin, M.M. Synthesis and antimicrobial study of some methyl 4-O-palmitoyl-α-L-rhamnopyranoside derivatives. Orbital Electron. J. Chem. 2014, 6, 20–28. [Google Scholar] [CrossRef]
- Matin, M.M.; Chakraborty, P. Synthesis, spectral and DFT characterization, PASS predication, antimicrobial, and ADMET studies of some novel mannopyranoside esters. J. Appl. Sci. Process Eng. 2020, 7, 572–586. [Google Scholar] [CrossRef]
- Lawandi, J.; Rocheleau, S.; Moitessier, N. Regioselective acylation, alkylation, silylation and glycosylation of monosaccharides. Tetrahedron 2016, 72, 6283–6319. [Google Scholar] [CrossRef]
- Matin, M.M.; Bhuiyan, M.M.H.; Kabir, E.; Sanaullah, A.F.M.; Rahman, M.A.; Hossain, M.E.; Uzzaman, M. Synthesis, characterization, ADMET, PASS predication, and antimicrobial study of 6-O-lauroyl mannopyranosides. J. Mol. Struct. 2019, 1195, 189–197. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Wang, C.-C.; Sabbavarapu, N.M.; Podilapu, A.R.; Liao, P.-H.; Hung, S.-C. “One-Pot” protection, glycosylation, and protection–glycosylation strategies of carbohydrates. Chem. Rev. 2018, 118, 8025–8104. [Google Scholar] [CrossRef] [PubMed]
- Matin, P.; Rahman, M.R.; Huda, D.; Bakri, M.K.B.; Uddin, J.; Yurkin, Y.; Burko, A.; Kuok, K.K.; Matin, M.M. Application of synthetic acyl glucopyranosides for white-rot and brown-rot fungal decay resistance in aspen and pine wood. BioResources 2022, 17, 3025–3041. [Google Scholar] [CrossRef]
- AlFindee, M.N.; Zhang, Q.; Subedi, Y.P.; Shrestha, J.P.; Kawasaki, Y.; Grilley, M.; Taemoto, J.Y.; Chang, C.W.T. One-step synthesis of carbohydrate esters as antibacterial and antifungal agents. Bioorg. Med. Chem. 2018, 26, 765–774. [Google Scholar] [CrossRef]
- Matin, M.M.; Uzzaman, M.; Chowdhury, S.A.; Bhuiyan, M.M.H. In vitro antimicrobial, physicochemical, pharmacokinetics, and molecular docking studies of benzoyl uridine esters against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2022, 40, 3668–3680. [Google Scholar] [CrossRef]
- Rashid, A.; Mackie, W.; Colquhoun, I.J.; Lamba, D. Novel synthesis of monosulphated methyl α-D-galactopyranosides. Can. J. Chem. 1990, 68, 1122–1127. [Google Scholar] [CrossRef]
- Tsuda, Y.; Haque, M.E.; Yoshimoto, K. Regioselective monoacylation of some glycopyranosides via cyclic tin intermediates. Chem. Pharm. Bull. 1983, 31, 1612–1624. [Google Scholar] [CrossRef] [Green Version]
- Fersht, A.R.; Jencks, W.P. Acetylpyridinium ion intermediate in pyridine-catalyzed hydrolysis and acyl transfer reactions of acetic anhydride. Observation, kinetics, structure-reactivity correlations, and effects of concentrated salt solutions. J. Am. Chem. Soc. 1970, 92, 5432–5442. [Google Scholar] [CrossRef]
- Sanaullah, A.F.M.; Bhuiyan, M.M.H.; Matin, M.M. Stearoyl glucopyranosides: Selective synthesis, PASS analysis, in vitro antimicrobial, and SAR study. Egypt. J. Chem. 2022, 65, 329–338. [Google Scholar] [CrossRef]
- Matin, M.M.; Nath, A.R.; Saad, O.; Bhuiyan, M.M.H.; Kadir, F.A.; Hamid, S.B.A.; Alhadi, A.A.; Ali, M.E.; Yehye, W.A. Synthesis, PASS-predication and in vitro antimicrobial activity of benzyl 4-O-benzoyl-α-L-rhamnopyranoside derivatives. Int. J. Mol. Sci. 2016, 17, 1412. [Google Scholar] [CrossRef] [Green Version]
- Sanaullah, A.F.M.; Matin, M.M.; Rahman, M.R.; Nayeem, S.M.A. Acyl glucopyranosides: Synthesis, PASS predication, antifungal activities, and molecular docking. Org. Commun. 2022, 15, 32–43. [Google Scholar] [CrossRef]
- Damme, E.V.; Meyer, S.D.; Bojkova, D.; Ciesek, S.; Cinatl, J.; Jonghe, S.D.; Jochmans, D.; Leyssen, P.; Buyck, C.; Neyts, J.; et al. In vitro activity of itraconazole against SARS-CoV-2. J. Med. Virol. 2021, 93, 4454–4460. [Google Scholar] [CrossRef]
- Srinivasan, S.; Sadasivam, S.K.; Gunalan, S.; Shanmugam, G.; Kothandan, G. Application of docking and active site analysis for enzyme linked biodegradation of textile dyes. Environ. Pollut. 2019, 248, 599–608. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, E.; Rajasekaran, R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val). RSC Adv. 2016, 6, 102744–102753. [Google Scholar] [CrossRef]
- Madeswaran, A.; Umamaheswari, M.; Asokkumar, K.; Sivashanmugam, T.; Subhadradevi, V.; Jagannath, P. In silico docking studies of phosphodiesterase inhibitory activity of commercially available flavonoids. Orient. Pharm. Expt. Med. 2012, 12, 301–306. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Matin, M.M.; Chakraborty, P.; Alam, M.S.; Islam, M.M.; Hanee, U. Novel mannopyranoside esters as sterol 14α-demethylase inhibitors: Synthesis, PASS predication, molecular docking, and pharmacokinetic studies. Carbohydr. Res. 2020, 496, 108130. [Google Scholar] [CrossRef]
- Tzvetkov, M.; Vormfelde, S.; Balen, D.; Meineke, I.; Schmidt, T.; Sehrt, D.; Sabolić, I.; Koepsell, H.; Brockmöller, J. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin. Pharmacol. Ther. 2009, 86, 299–306. [Google Scholar] [CrossRef]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincen, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matin, M.M.; Hasan, M.S.; Uzzaman, M.; Bhuiyan, M.M.H.; Kibria, S.M.; Hossain, M.E.; Roshid, M.H.O. Synthesis, spectroscopic characterization, molecular docking, and ADMET studies of mannopyranoside esters as antimicrobial agents. J. Mol. Struct. 2020, 1222, 128821. [Google Scholar] [CrossRef]
- Qureshi, A.; Kaur, G.; Kumar, M. AVCpred: An integrated web server for prediction and design of antiviral compounds. Chem. Biol. Drug Des. 2017, 89, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Matin, M.M.; Bhuiyan, M.M.H.; Azad, A.K.M.S.; Akther, N. Design and synthesis of benzyl 4-O-lauroyl-α-L-rhamnopyranoside derivatives as antimicrobial agents. Curr. Chem. Lett. 2017, 6, 31–40. [Google Scholar] [CrossRef]
- Matin, M.M.; Bhuiyan, M.H.; Hossain, M.M.; Roshid, M.H.O. Comparative antibacterial activities of some monosaccharide and disaccharide benzoates. Orbital Electron. J. Chem. 2015, 7, 160–167. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Costanzo, L.D.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Mitra, S.; Dash, R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP Bromodomain inhibitors. J. Mol. Graph. Model. 2018, 83, 42–52. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Antibacterial | Antifungal | ||
|---|---|---|---|---|
| Pa | Pi | Pa | Pi | |
| 3 | 0.541 | 0.013 | 0.628 | 0.016 |
| 4 | 0.528 | 0.014 | 0.669 | 0.012 |
| 5 | 0.568 | 0.011 | 0.695 | 0.010 |
| 6 | 0.558 | 0.012 | 0.675 | 0.011 |
| 7 | 0.551 | 0.012 | 0.673 | 0.011 |
| 8 | 0.551 | 0.012 | 0.673 | 0.011 |
| 9 | 0.574 | 0.010 | 0.692 | 0.010 |
| 10 | 0.551 | 0.012 | 0.673 | 0.011 |
| 11 | 0.551 | 0.012 | 0.673 | 0.011 |
| 12 | 0.551 | 0.012 | 0.673 | 0.011 |
| TC | 0.694 | 0.005 | 0.523 | 0.027 |
| FZ | - | - | 0.726 | 0.008 |
| Drug | General | HBV | HCV | HHV | HIV |
|---|---|---|---|---|---|
| 3 | 64.331 | 16.25 | 8.535 | 72.663 | 41.557 |
| 4 | 45.78 | 21.737 | 16.24 | 25.566 | 57.575 |
| 5 | 38.776 | 19.899 | 51.185 | 91.166 | 59.266 |
| 6 | 57.884 | 12.57 | 1.08 | 50.853 | 63.981 |
| 7 | 47.04 | 17.494 | −0.041 | 48.407 | 58.789 |
| 8 | 52.5 | 16.791 | 4.554 | 48.84 | 60.162 |
| 9 | 42.478 | 14.127 | 0.958 | 50.592 | 63.99 |
| 10 | 46.638 | 22.318 | 22.465 | 50.106 | 65.089 |
| 11 | 41.704 | 22.414 | 22.453 | 49.839 | 63.958 |
| 12 | 27.919 | 22.478 | 21.176 | 49.709 | 63.022 |
| Retrovir | 86.484 | 19.619 | 24.962 | 28.728 | 92.855 |
| Remdesivir | 55.91 | 13.244 | 57.744 | 34.473 | 63.377 |
| Diameter of Zone of Inhibition in mm (25 μg.dw/disc) | |||
|---|---|---|---|
| Drug | S. aureus | E. coli | P. aeruginosa |
| 3 | 07 ± 0.50 | NI | NI |
| 4 | 07 ± 0.66 | NI | NI |
| 5 | 10 ± 0.84 | NI | NI |
| 6 | 08 ± 0.33 | NI | NI |
| 7 | 09 ± 0.48 | NI | NI |
| 8 | 11 ± 0.83 | NI | NI |
| 9 | 08 ± 0.35 | NI | NI |
| 10 | 09 ± 0.74 | NI | NI |
| 11 | 10 ± 0.58 | NI | NI |
| 12 | 07 ± 0.59 | NI | NI |
| TC[b] | * 23 ± 0.58 | * 25 ± 0.50 | * 22 ± 0.41 |
| Percentage of Zone of Inhibition | |||
|---|---|---|---|
| Drug | wt.μg/mL PDA | A. fumigatus | A. niger |
| 3 | 100 | 55.6 ± 0.64 | 30.5 ± 0.64 |
| 4 | 100 | * 70.1 ± 0.68 | 54.5 ± 0.73 |
| 5 | 100 | * 71.0 ± 0.44 | 54.2 ± 0.40 |
| 6 | 100 | 56.7 ± 0.78 | 27.3 ± 0.29 |
| 7 | 100 | * 71.6 ± 0.54 | 54.5 ± 0.73 |
| 8 | 100 | * 76.0 ± 0.78 | * 66.7 ± 0.84 |
| 9 | 100 | * 61.7 ± 0.64 | 48.5 ± 0.29 |
| 10 | 100 | * 68.3 ± 0.68 | 51.5 ± 0.50 |
| 11 | 100 | * 78.3 ± 0.64 | 45.5 ± 0.50 |
| 12 | 100 | * 70.0 ± 0.58 | 39.4 ± 0.29 |
| FZ[a] | 12.5 | * 65.0 ± 0.25 | 37.1 ± 0.17 |
| Drug | XP Docking Score (kcal/mol) | Glide Ligand Efficiency (kcal/mol) | Glide Energy (kcal/mol) | Glide Emodel (kcal/mol) |
|---|---|---|---|---|
| 8 | −10.551 | −0.229 | −60.69 | −83.022 |
| 12 | −10.532 | −0.224 | −63.084 | −91.061 |
| VNI | −10.249 | −0.278 | −57.187 | −83.708 |
| 11 | −9.79 | −0.228 | −55.28 | −91.442 |
| 7 | −9.651 | −0.241 | −61.729 | −86.997 |
| 10 | −8.727 | −0.224 | −55.244 | −79.947 |
| 9 | −7.502 | −0.242 | −54.585 | −75.466 |
| Fluconazole | −7.147 | −0.325 | −36.927 | −49.09 |
| 4 | −6.889 | −0.363 | −31.935 | −37.695 |
| 6 | −6.825 | −0.244 | −50.208 | −60.244 |
| 5 | −6.605 | −0.264 | −42.718 | −54.36 |
| Tetracycline | −6.384 | −0.199 | −40.925 | −56.193 |
| 3 | −5.859 | −0.451 | −22.873 | −24.96 |
| Valeric acid | −3.656 | −0.522 | −14.299 | −13.335 |
| Caproic acid | −3.974 | −0.496 | −16.528 | −16.282 |
| Caprylic acid | −4.648 | −0.464 | −19.635 | −19.357 |
| Capric acid | −3.854 | −0.321 | −23.177 | −26.672 |
| Drug | XP Docking Score (kcal/mol) | Glide Ligand Efficiency (kcal/mol) | Glide Energy (kcal/mol) | Glide Emodel (kcal/mol) |
|---|---|---|---|---|
| 4 | −8.976 | −0.472 | −36.901 | −45.516 |
| 5 | −8.464 | −0.339 | −40.331 | −49.196 |
| N3 | −8.151 | −0.166 | −72.606 | −103.33 |
| 3 | −6.489 | −0.499 | −28.094 | −34.283 |
| 9 | −6.379 | −0.206 | −45.505 | −55.634 |
| 6 | −5.908 | −0.211 | −42.797 | −49.515 |
| 8 | −5.774 | −0.126 | −63.605 | −83.924 |
| 11 | −5.745 | −0.134 | −55.857 | −74.402 |
| 12 | −5.517 | −0.117 | −58.997 | −76.72 |
| 10 | −5.473 | −0.14 | −55.326 | −73.864 |
| Fluconazole | −4.65 | −0.211 | −37.181 | −46.454 |
| 7 | −4.212 | −0.105 | −53.714 | −78.741 |
| Tetracycline | −5.725 | −0.107 | −34.956 | −44.252 |
| Receptor Ligand | Estimated Inhibition Constant |
|---|---|
| 8-4UYL | 313.40 µM |
| 12-4UYL | 1260 µM |
| VNI-4UYL | 101.17 nM |
| 4-6LU7 | 281.23 µM |
| 5-6LU7 | 727.23 µM |
| N3-6LU7 | 840.87 µM |
| Property | Model Name | Unit | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | TC | FC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Absorption | Water solubility | Numeric (log mol/L) | −0.678 | −1.326 | −2.176 | −2.75 | −4.37 | −3.942 | −3.506 | −4.398 | −4.258 | −3.884 | −2.418 | −3.293 |
| Absorption | Caco2 permeability | Numeric (log Papp in 10−6 cm/s) | −0.247 | −0.141 | −0.041 | 0.302 | 0.783 | 0.719 | 0.881 | 0.783 | 0.741 | 0.692 | 0.161 | 0.905 |
| Absorption | Intestinal absorption (human) | Numeric (% Absorbed) | 33.429 | 61.533 | 68.549 | 51.258 | 64.016 | 68.424 | 57.818 | 66.143 | 69.082 | 71.983 | 45.19 | 94.964 |
| Absorption | Skin permeability | Numeric (log Kp) | −3.442 | −3.336 | −2.477 | −2.57 | −2.719 | −2.732 | −2.503 | −2.713 | −2.727 | −2.733 | −2.735 | −2.8 |
| Absorption | P-glycoprotein substrate | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | Yes | No |
| Absorption | P-glycoprotein I inhibitor | Categorical (Yes/No) | No | No | No | No | Yes | Yes | Yes | Yes | Yes | Yes | No | No |
| Absorption | P-glycoprotein II inhibitor | Categorical (Yes/No) | No | No | No | No | Yes | Yes | No | Yes | Yes | Yes | No | No |
| Distribution | VDss (human) | Numeric (log L/kg) | −0.331 | −0.464 | −0.523 | −0.128 | −0.239 | −0.457 | −0.129 | −0.224 | −0.34 | −0.549 | 1.194 | −0.441 |
| Distribution | Fraction unbound (human) | Numeric (Fu) | 0.915 | 0.758 | 0.523 | 0.483 | 0.089 | 0.044 | 0.366 | 0.109 | 0.049 | 0.048 | 0.535 | 0.381 |
| Distribution | BBB permeability | Numeric (log BB) | −0.992 | −1.076 | −1.084 | −1.597 | −1.845 | −1.977 | −1.664 | −1.827 | −1.914 | −2.002 | −0.841 | −1.067 |
| Distribution | CNS permeability | Numeric (log PS) | −3.622 | −3.237 | −3.144 | −3.081 | −2.975 | −2.676 | −3.016 | −3.054 | −2.859 | −2.783 | −3.934 | −3.185 |
| Metabolism | CYP2D6 substrate | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Metabolism | CYP3A4 substrate | Categorical (Yes/No) | No | No | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No |
| Metabolism | CYP1A2 inhibitior | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | Yes |
| Metabolism | CYP2C19 inhibitior | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Metabolism | CYP2C9 inhibitior | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Metabolism | CYP2D6 inhibitior | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Metabolism | CYP3A4 inhibitior | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Excretion | Total Clearance | Numeric (log ml/min/kg) | 0.671 | 1.557 | 1.659 | 1.565 | 1.685 | 1.791 | 1.666 | 1.667 | 1.737 | 1.804 | 0.291 | 0.29 |
| Excretion | Renal OCT2 substrate | Categorical (Yes/No) | No | No | No | No | Yes | No | No | Yes | Yes | No | No | No |
| Toxicity | AMES toxicity | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Toxicity | Hepatotoxicity | Categorical (Yes/No) | No | No | No | Yes | No | No | No | No | No | No | No | Yes |
| Toxicity | Skin sensitization | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Toxicity | hERG I inhibitor | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Toxicity | hERG II inhibitor | Categorical (Yes/No) | No | No | No | No | No | No | No | No | No | No | No | No |
| Toxicity | Oral Rat Acute Toxicity (LD50) | Numeric (mol/kg) | 1.157 | 1.62 | 2.232 | 2.58 | 1.206 | 1.475 | 2.281 | 1.212 | 1.212 | 1.484 | 2.214 | 2.328 |
| Toxicity | Oral Rat Chronic Toxicity (LOAEL) | Numeric (log mg/kg_bw/day) | 2.797 | 2.15 | 0.94 | 1.275 | −0.17 | −0.366 | 1.064 | −0.096 | −0.163 | −0.203 | 3.038 | 1.033 |
| Toxicity | Max. tolerated dose (human) | Numeric (log mg/kg/day) | 1.552 | 1.095 | 0.473 | 0.685 | 0.623 | 0.748 | 0.53 | 0.593 | 0.734 | 0.767 | 0.281 | 0.114 |
| Molecule | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | TC | FC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heavy atoms | 13 | 19 | 25 | 28 | 40 | 46 | 31 | 39 | 43 | 47 | 32 | 22 |
| Aromatic heavy atoms | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 16 |
| Fraction Csp3 | 1 | 0.92 | 0.88 | 0.78 | 0.87 | 0.89 | 0.81 | 0.86 | 0.88 | 0.89 | 0.41 | 0.23 |
| Rotatable bonds | 2 | 7 | 12 | 13 | 25 | 31 | 16 | 24 | 28 | 32 | 2 | 5 |
| H-bond acceptors | 6 | 7 | 8 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 9 | 7 |
| H-bond donors | 4 | 3 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 1 |
| Molar refractivity | 40.47 | 64.62 | 88.78 | 93.84 | 151.52 | 180.36 | 108.26 | 146.71 | 165.94 | 185.17 | 110.79 | 70.71 |
| TPSA | 99.38 | 105.45 | 111.52 | 123.66 | 123.66 | 123.66 | 123.66 | 123.66 | 123.66 | 123.66 | 181.62 | 81.65 |
| Lipinski violations | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 0 |
| Ghose violations | 1 | 1 | 0 | 0 | 3 | 4 | 0 | 3 | 4 | 4 | 1 | 0 |
| Veber violations | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 |
| Egan violations | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 |
| Muegge violations | 2 | 0 | 0 | 0 | 2 | 3 | 1 | 2 | 3 | 3 | 2 | 0 |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.11 | 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matin, P.; Hanee, U.; Alam, M.S.; Jeong, J.E.; Matin, M.M.; Rahman, M.R.; Mahmud, S.; Alshahrani, M.M.; Kim, B. Novel Galactopyranoside Esters: Synthesis, Mechanism, In Vitro Antimicrobial Evaluation and Molecular Docking Studies. Molecules 2022, 27, 4125. https://doi.org/10.3390/molecules27134125
Matin P, Hanee U, Alam MS, Jeong JE, Matin MM, Rahman MR, Mahmud S, Alshahrani MM, Kim B. Novel Galactopyranoside Esters: Synthesis, Mechanism, In Vitro Antimicrobial Evaluation and Molecular Docking Studies. Molecules. 2022; 27(13):4125. https://doi.org/10.3390/molecules27134125
Chicago/Turabian StyleMatin, Priyanka, Umme Hanee, Muhammad Shaiful Alam, Jae Eon Jeong, Mohammed Mahbubul Matin, Md. Rezaur Rahman, Shafi Mahmud, Mohammed Merae Alshahrani, and Bonglee Kim. 2022. "Novel Galactopyranoside Esters: Synthesis, Mechanism, In Vitro Antimicrobial Evaluation and Molecular Docking Studies" Molecules 27, no. 13: 4125. https://doi.org/10.3390/molecules27134125