
In Silico Drug Repurposing of FDA-Approved Drugs Highlighting Promacta as a Potential Inhibitor of H7N9 Influenza Virus


Abstract
:1. Introduction
2. Results and Discussion
2.1. Molecular Docking Analysis
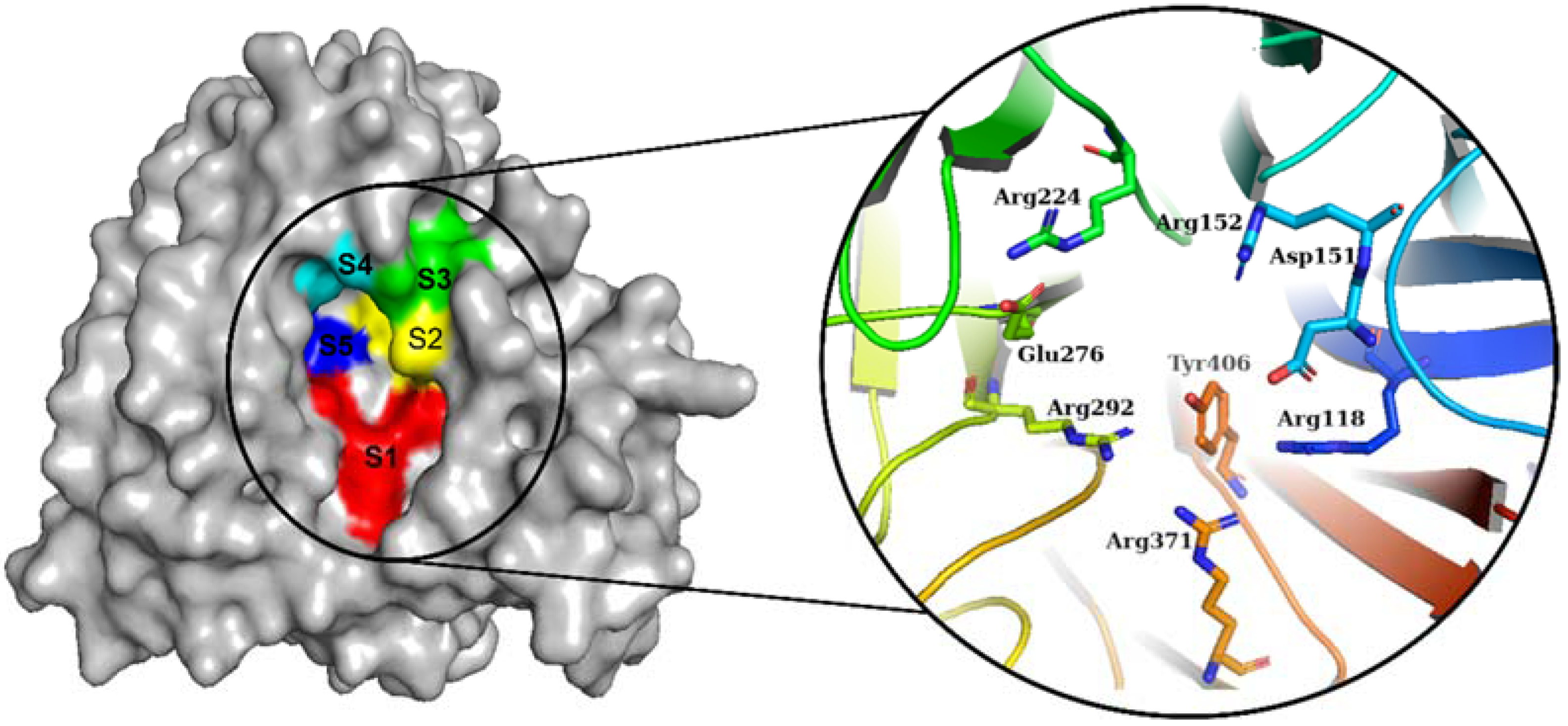
2.2. Binding Pose Analysis
2.3. Molecular Dynamics Trajectory Analysis
2.3.1. Root Mean Square Deviation (RMSD)
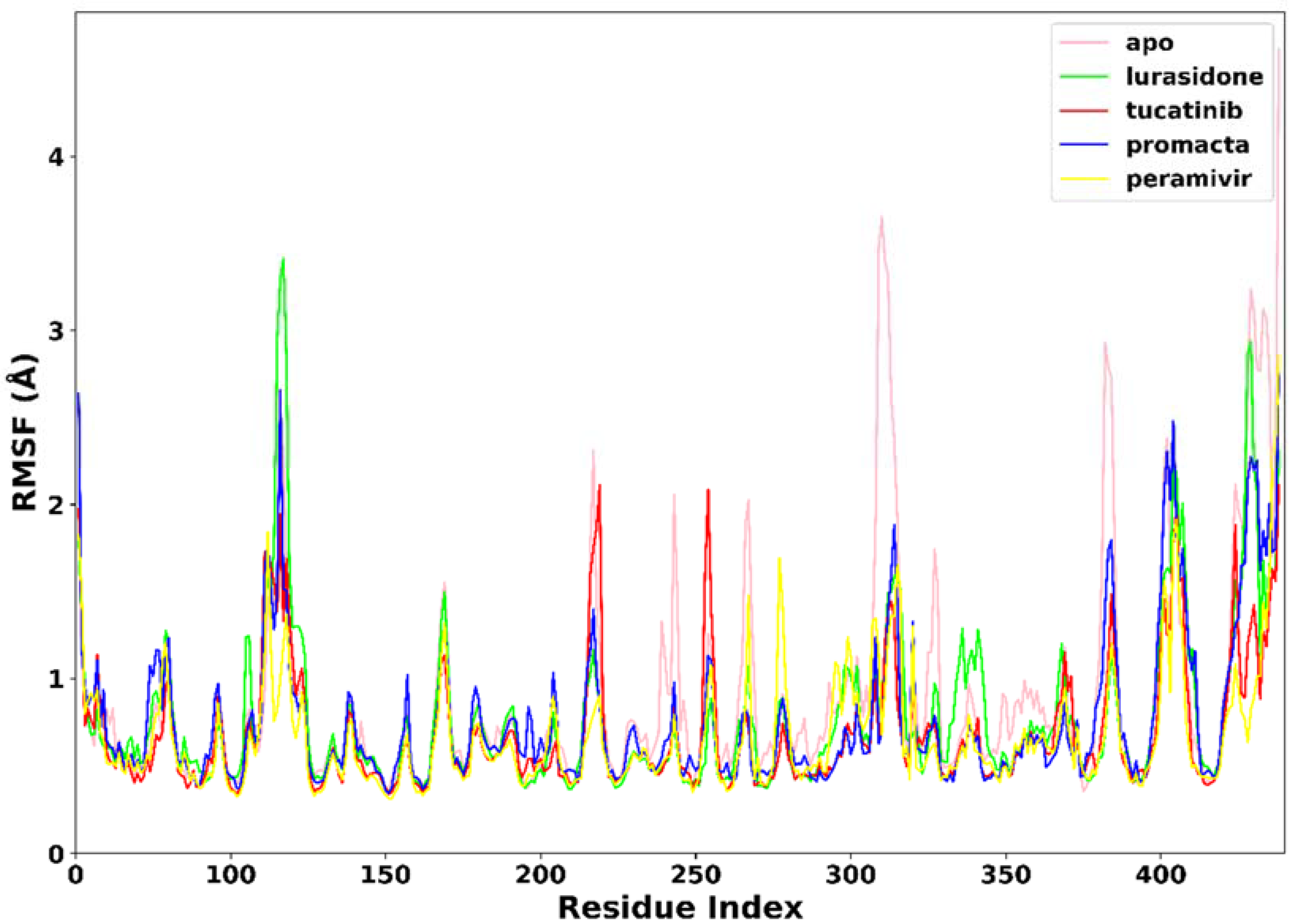
2.3.2. Root Mean Square Fluctuation (RMSF)
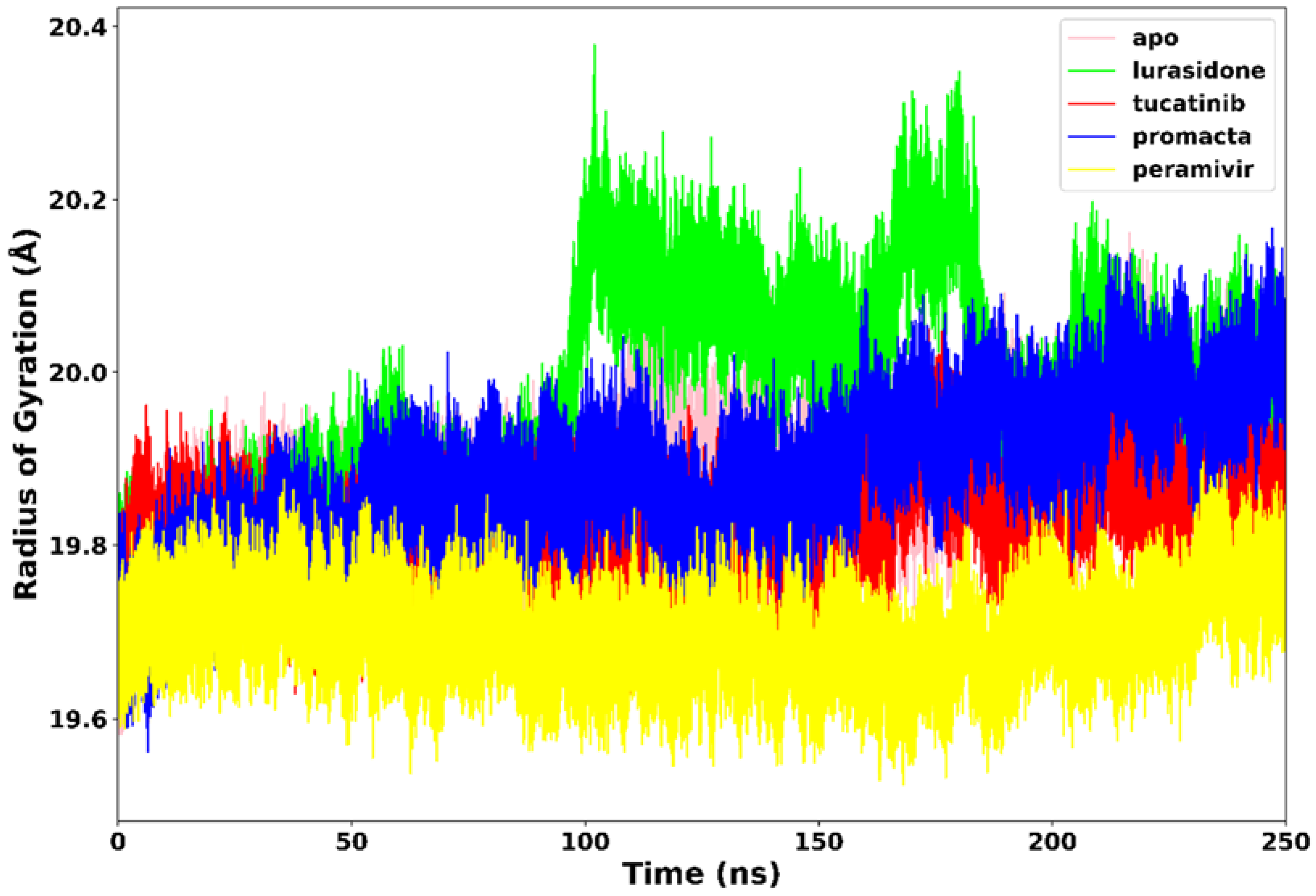
2.3.3. Radius of Gyration (RoG)
2.3.4. Hydrogen Bond Analysis
2.4. Binding Free Energy Analysis
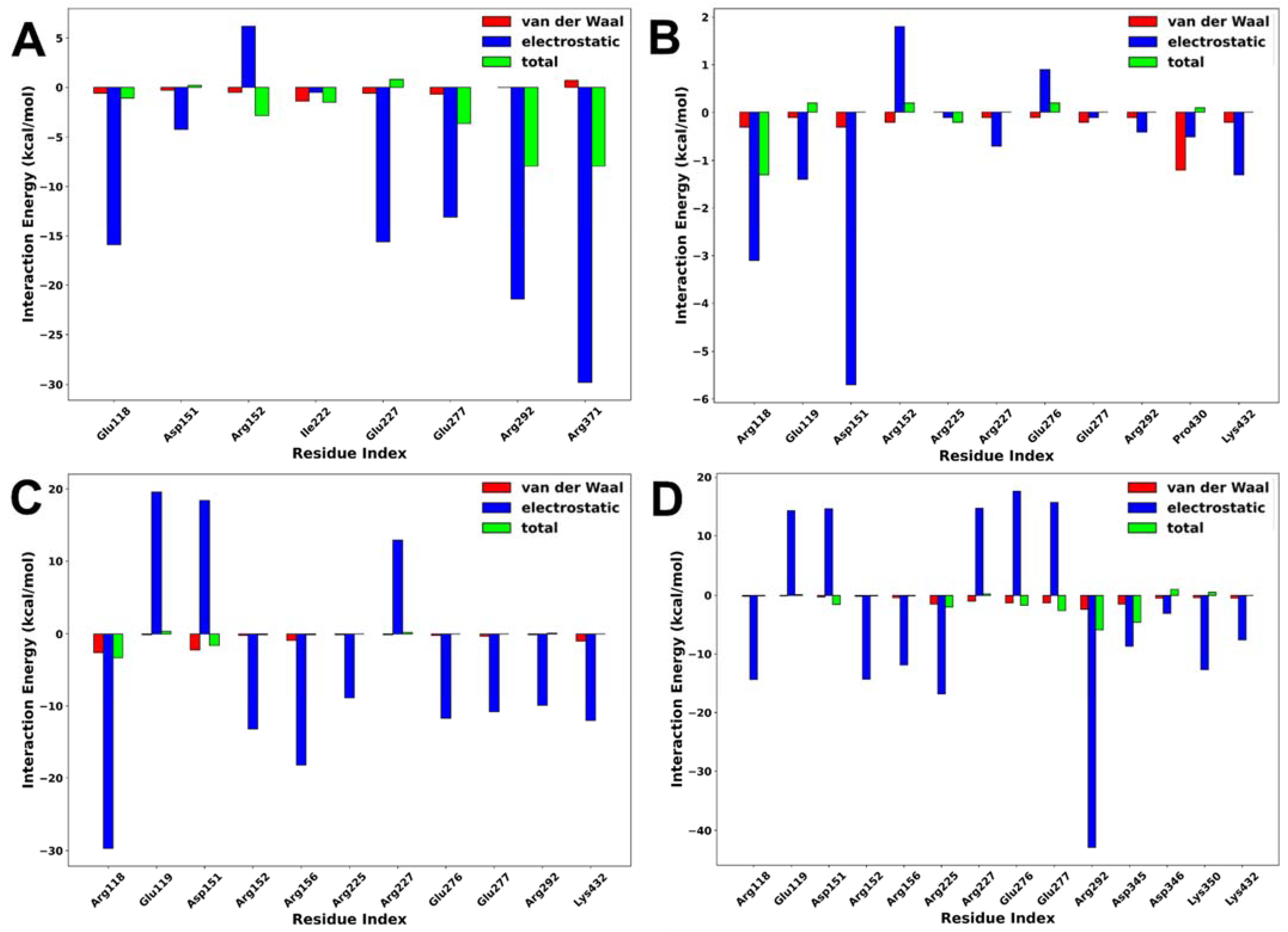
2.5. Interaction Energy Decomposition Analysis
2.6. Pharmacokinetic Analyses
2.7. Toxicological Analyses
3. Materials and Methods
3.1. Receptor and Ligand Preparation
3.2. Molecular Docking-Based Virtual Screening
3.3. Pharmacokinetic and Toxicological Predictions
3.4. Molecular Dynamics Simulations
3.5. Molecular Dynamics Trajectory Analyses
3.6. Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization (WHO). Analysis of Recent Scientific Information on Avian Influenza A(H7N9) Virus; World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/influenza/human_animal_interface/avian_influenza/riskassessment_AH7N9_201702/en/ (accessed on 29 March 2022).
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.-N.; Lu, H.-Z.; Cao, B.; Du, B.; Shang, H.; Gan, J.-H.; Lu, S.-H.; Yang, Y.-D.; Fang, Q.; Shen, Y.-Z.; et al. Clinical Findings in 111 Cases of Influenza A (H7N9) Virus Infection. N. Engl. J. Med. 2013, 368, 2277–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, N.; Li, X.; Ren, R.; Wang, D.; Zhou, S.; Greene, C.M.; Song, Y.; Zhou, L.; Yang, L.; Davis, C.T.; et al. Assessing Change in Avian Influenza A(H7N9) Virus Infections During the Fourth Epidemic—China, September 2015–August 2016. MMWR. Morb. Mortal. Wkly. Rep. 2016, 65, 1390–1394. [Google Scholar] [CrossRef] [Green Version]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef]
- Mtambo, S.E.; Amoako, D.G.; Somboro, A.M.; Agoni, C.; Lawal, M.M.; Gumede, N.S.; Khan, R.B.; Kumalo, H.M. Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design. Molecules 2021, 26, 880. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kiso, M.; Fukuyama, S.; Nakajima, N.; Imai, M.; Yamada, S.; Murakami, S.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Sakoda, Y.; et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 2013, 501, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, D.; Gao, R.; Zhao, B.; Song, J.; Qi, X.; Zhang, Y.; Shi, Y.; Yang, L.; Zhu, W.; et al. Biological features of novel avian influenza A (H7N9) virus. Nature 2013, 499, 500–503. [Google Scholar] [CrossRef]
- Tharakaraman, K.; Jayaraman, A.; Raman, R.; Viswanathan, K.; Stebbins, N.W.; Johnson, D.; Shriver, Z.; Sasisekharan, V.; Sasisekharan, R. Glycan-Receptor Binding of the Influenza A Virus H7N9 Hemagglutinin. NIH Public Access 2013, 153, 1486–1493. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; McCauley, J.; Steinhauer, D. Receptor binding properties of the influenza virus hemagglutinin as a determinant of host range. Curr. Top. Microbiol. Immunol. 2014, 385, 63–91. [Google Scholar] [CrossRef]
- Bai, T.; Zhou, J.; Shu, Y. Serologic Study for Influenza A (H7N9) among High-Risk Groups in China. N. Engl. J. Med. 2013, 368, 2339–2340. [Google Scholar] [CrossRef]
- Aoki, F.Y. Antiviral Drugs for Influenza and Other Respiratory Virus Infections|Elsevier Enhanced Reader. Mand. Douglas Bennett’s Princ. Pract. Infect. Dis. 2015, 1, 531–545. [Google Scholar] [CrossRef]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Bi, Y.; Wang, J.; Wong, G.; Shi, W.; Hu, F.; Yang, Y.; Yang, L.; Deng, X.; Jiang, S.; et al. Human infections with recently-emerging highly pathogenic H7N9 avian influenza virus in China. J. Infect. 2017, 75, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, L.; Zhou, M.; Chen, Z.; Li, F.; Wu, H.; Xiang, N.; Chen, E.; Tang, F.; Wang, D.; et al. Epidemiology of Human Infections with Avian Influenza A(H7N9) Virus in China. N. Engl. J. Med. 2014, 370, 520–532. [Google Scholar] [CrossRef]
- Du, Z.; Nugent, C.; Galvani, A.P.; Krug, R.M.; Meyers, L.A. Modeling mitigation of influenza epidemics by baloxavir. Nat. Commun. 2020, 11, 2750. [Google Scholar] [CrossRef]
- Koshimichi, H.; Ishibashi, T.; Kawaguchi, N.; Sato, C.; Kawasaki, A.; Wajima, T. Safety, Tolerability, and Pharmacokinetics of the Novel Anti-influenza Agent Baloxavir Marboxil in Healthy Adults: Phase I Study Findings. Clin. Drug Investig. 2018, 38, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- RJ, R.; LF, H.; DJ, S.; PJ, C.; YP, L.; GM, B.; AJ, H.; SJ, G.; JJ, S. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar] [CrossRef]
- Varghese, J.N.; Colman, P.M. Three-dimensional Structure of the Negraminidase of Influenza Virus A/Tokyo/3/67 at 2-2 A Resolution receptor. J. Mol. Biol. 1991, 221, 473–486. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Honarparvar, B.; Govender, T.; Maguire, G.E.M.; Soliman, M.E.S.; Kruger, H.G. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2014, 114, 493–537. [Google Scholar] [CrossRef]
- Usha, T.; Shanmugarajan, D.; Goyal, A.K.; Kumar, C.S.; Middha, S.K. Recent Updates on Computer-aided Drug Discovery: Time for a Paradigm Shift. Curr. Top. Med. Chem. 2017, 17, 3296–3307. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Soto, C.S.; Honig, B. Evaluating conformational free energies: The colony energy and its application to the problem of loop prediction. Proc. Natl. Acad. Sci. USA 2002, 99, 7432–7437. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, E.; Akimoto, T.; Mitsutake, A.; Metzler, R. Universal Relation between Instantaneous Diffusivity and Radius of Gyration of Proteins in Aqueous Solution. Phys. Rev. Lett. 2021, 126, 128101. [Google Scholar] [CrossRef]
- Gomes, D.E.B.; Lins, R.D.; Pascutti, P.G.; Lei, C.; Soares, T.A. The Role of Non-Bonded Interactions in the Conformational Dynamics of Organophosphorous Hydrolase Adsorbed onto Functionalized Mesoporous Silica Surfaces. J. Phys. Chem. B 2010, 114, 531. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959. [Google Scholar] [CrossRef]
- Gupta, P.; Garg, T.; Tanmay, M.; Arora, S. Polymeric Drug-Delivery Systems: Role in P-gp Efflux System Inhibition. Crit. Rev. Ther. Drug Carrier Syst. 2015, 32, 247–275. [Google Scholar] [CrossRef]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 30, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment, Release 2020; Dassault Systems: San Diego, CA, USA, 2016; Available online: https://www.3ds.com/ (accessed on 21 April 2022).
- Schrödinger, L.; DeLano, W. PyMol 2021. Available online: https://pymol.org/pymol (accessed on 25 April 2022).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257. [Google Scholar] [CrossRef] [Green Version]
- Braga, R.C.; Alves, V.M.; Silva, M.F.B.; Muratov, E.; Fourches, D.; Lião, L.M.; Tropsha, A.; Andrade, C.H. Pred-hERG: A Novel web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform. 2015, 34, 698–701. [Google Scholar] [CrossRef] [Green Version]
- Ben-shalom, I.Y.; Lin, C.; Radak, B.K.; Sherman, W.; Gilson, M.K. Fast Equilibration of Water between Buried Sites and Bulk by MD with Parallel Monte Carlo Water Moves on GPUs. J. Chem. Theory. Comput. 2021, 7366–7372. [Google Scholar] [CrossRef]
- Du, Q.; Qian, Y.; Xue, W. Cross-reactivity of two human IL-6 family cytokines OSM and LIF explored by protein-protein docking and molecular dynamics simulation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129907. [Google Scholar] [CrossRef]
- Fakhar, Z.; Hejazi, L.; Tabatabai, S.A.; Munro, O.Q. Discovery of novel heterocyclic amide-based inhibitors: An integrative in-silico approach to targeting soluble epoxide hydrolase. J. Biomol. Struct. Dyn. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.; Xu, T.; Pan, P.; Yu, Q.; Xu, L.; Xiong, X.; Hou, T.; Cui, S.; Sun, Y. Discovery of a small molecule inhibitor of cullin neddylation that triggers ER stress to induce autophagy. Acta Pharm. Sin. B 2021, 11, 3567–3584. [Google Scholar] [CrossRef]
- Wolf, S.; Sohmen, B.; Hellenkamp, B.; Thurn, J.; Stock, G.; Hugel, T. Hierarchical dynamics in allostery following ATP hydrolysis monitored by single molecule FRET measurements and MD simulations. Chem. Sci. 2021, 12, 3350–3359. [Google Scholar] [CrossRef] [PubMed]
- Panwar, A.; Kumar, A. In-silico Analysis and Molecular Dynamics Simulations of Lysozyme by GROMACS 2020.2. Ann. Rom. Soc. Cell Biol. 2021, 25, 9679–9685. [Google Scholar]
- Jin, T.; Patel, S.J.; Lehn, R.C. Van Molecular simulations of lipid membrane partitioning and translocation by bacterial quorum sensing modulators. PLoS ONE 2021, 16, e0246187. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DrugBank ID | Generic Name | Physicochemical Properties | Structures | Binding Residues | Binding Affinity (kcal/mol) | Function |
|---|---|---|---|---|---|---|
| DB06614 | Peramivir | Mw = 328.41 logP = 0.08 HBA = 5 HBD = 6 |  | Glu119, Asp151, Trp178, Ile222, Arg227, Glu227 Ala246, Glu277, Arg292, Tyr406 | −6.8 | Treatment of influenza |
| DB08815 | Lurasidone | Mw = 492.68 logP = 4.22 HBA = 4 HBD = 0 | 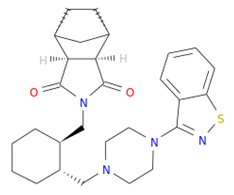 | Asp151, Ala246, Glu277, Arg292, Arg371, Trp403, Tyr406, Ile427, Ly432 | −9.9 | Treatment of schizophrenia |
| DB11652 | Tucatinib | Mw = 480.53 logP = 3.77 HBA = 7 HBD = 2 | 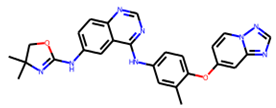 | Ile149, Asp151, Arg152, Arg224, Ala246, Arg292, Asp294, Arg371, Ile427, Lys432, Pro431 | −9.8 | Treatment of metastatic breast cancer |
| DB06210 | Promacta | Mw = 442.47 logP = 3.74 HBA = 6 HBD = 3 | 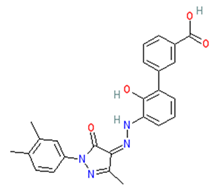 | Arg118, Asp151, Ser179, Arg224, Arg292, Arg371, Ile427, Pro431, Lys432 | −10.0 | Treatment of thrombocytopenia or aplastic anaemia |
| Complexes | ΔGbind | ΔEvdw | ΔEele | ΔGpol | ΔGnonpol |
|---|---|---|---|---|---|
| NA–lurasidone | −22.59 ± 0.14 | −28.20 ± 0.09 | −32.20 ± 0.51 | 41.27 ± 0.44 | −3.33 ± 0.01 |
| NA–tucatinib | −54.11 ± 0.11 | −57.95 ± 0.09 | −41.76 ± 0.25 | 51.50 ± 0.22 | −5.91 ± 0.02 |
| NA–Promacta | −56.20 ± 0.19 | −39.17 ± 0.12 | −76.47 ± 0.43 | 65.07 ± 0.30 | −5.66 ± 0.01 |
| NA–peramivir | −49.09 ± 0.13 | −28.86 ± 0.08 | −128.21 ± 0.35 | 115.11 ± 0.26 | −15.12 ± 0.00 |
| Parameters | Lurasidone | Tucatinib | Promacta | Peramivir |
|---|---|---|---|---|
| GI absorption | High | High | High | Low |
| BBB permeant | No | No | No | No |
| P-gp substrate | No | Yes | No | Yes |
| CYP1A2 inhibitor | No | Yes | No | No |
| CYP2C19 inhibitor | Yes | Yes | No | No |
| CYP2C9 inhibitor | Yes | Yes | Yes | No |
| CYP2D6 inhibitor | No | Yes | No | No |
| CYP3A4 inhibitor | Yes | Yes | No | No |
| Parameters | Lurasidone | Tucatinib | Promacta | Peramivir |
|---|---|---|---|---|
| Carcinogenicity | No | Yes | No | No |
| Immunotoxicity | No | Yes | No | No |
| Mutagenicity | No | No | No | No |
| Cytotoxicity | No | No | No | No |
| LD50 (mg/kg) | 660 | 3160 | 5000 | 1430 |
| Class | 4 | 5 | 5 | 4 |
| hERG inhibition | Yes (weak) | Yes (weak) | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mtambo, S.E.; Kumalo, H.M. In Silico Drug Repurposing of FDA-Approved Drugs Highlighting Promacta as a Potential Inhibitor of H7N9 Influenza Virus. Molecules 2022, 27, 4515. https://doi.org/10.3390/molecules27144515
Mtambo SE, Kumalo HM. In Silico Drug Repurposing of FDA-Approved Drugs Highlighting Promacta as a Potential Inhibitor of H7N9 Influenza Virus. Molecules. 2022; 27(14):4515. https://doi.org/10.3390/molecules27144515
Chicago/Turabian StyleMtambo, Sphamandla E., and Hezekiel M. Kumalo. 2022. "In Silico Drug Repurposing of FDA-Approved Drugs Highlighting Promacta as a Potential Inhibitor of H7N9 Influenza Virus" Molecules 27, no. 14: 4515. https://doi.org/10.3390/molecules27144515