
Harnessing Protein-Ligand Interaction Fingerprints to Predict New Scaffolds of RIPK1 Inhibitors

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
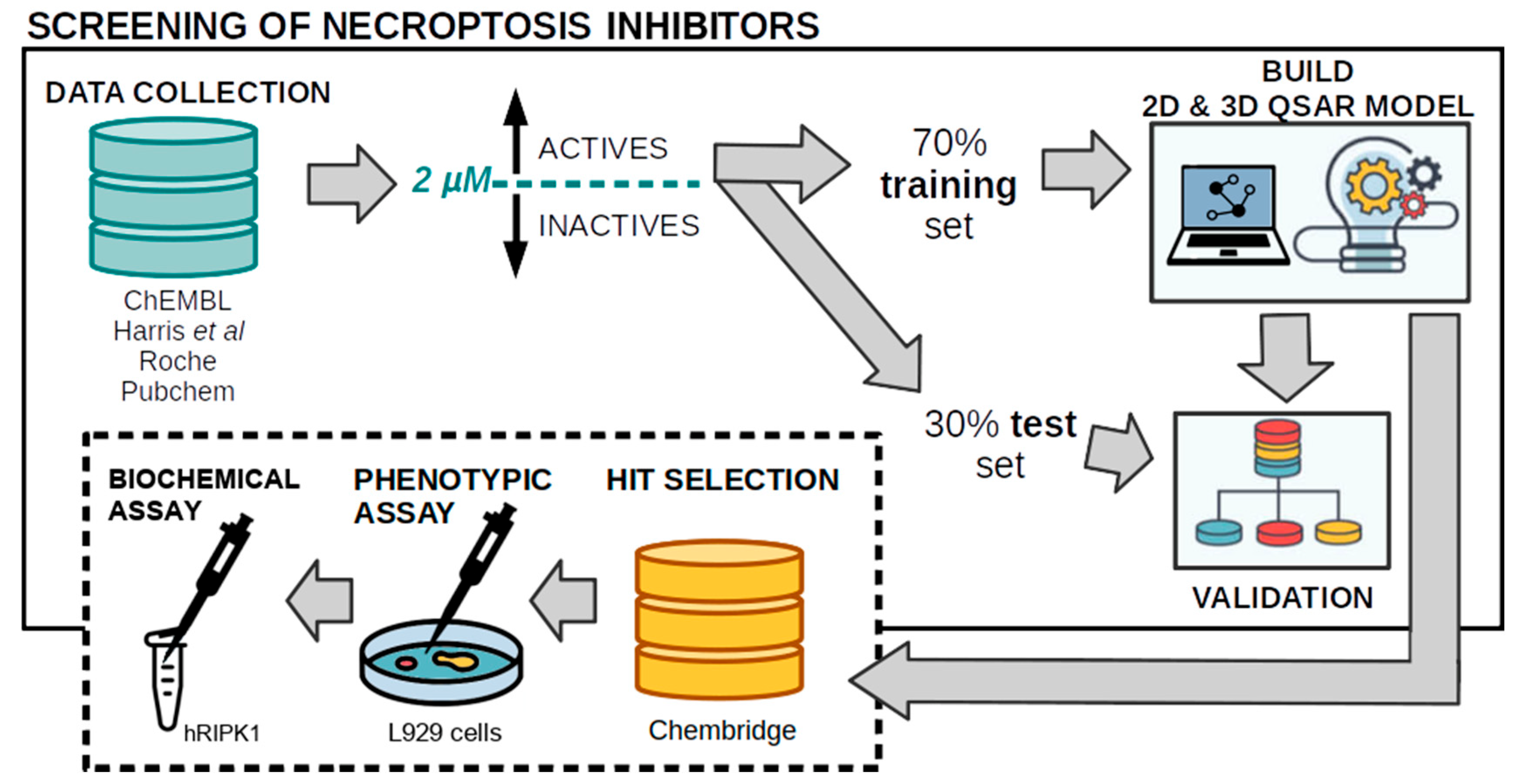
2. Methods
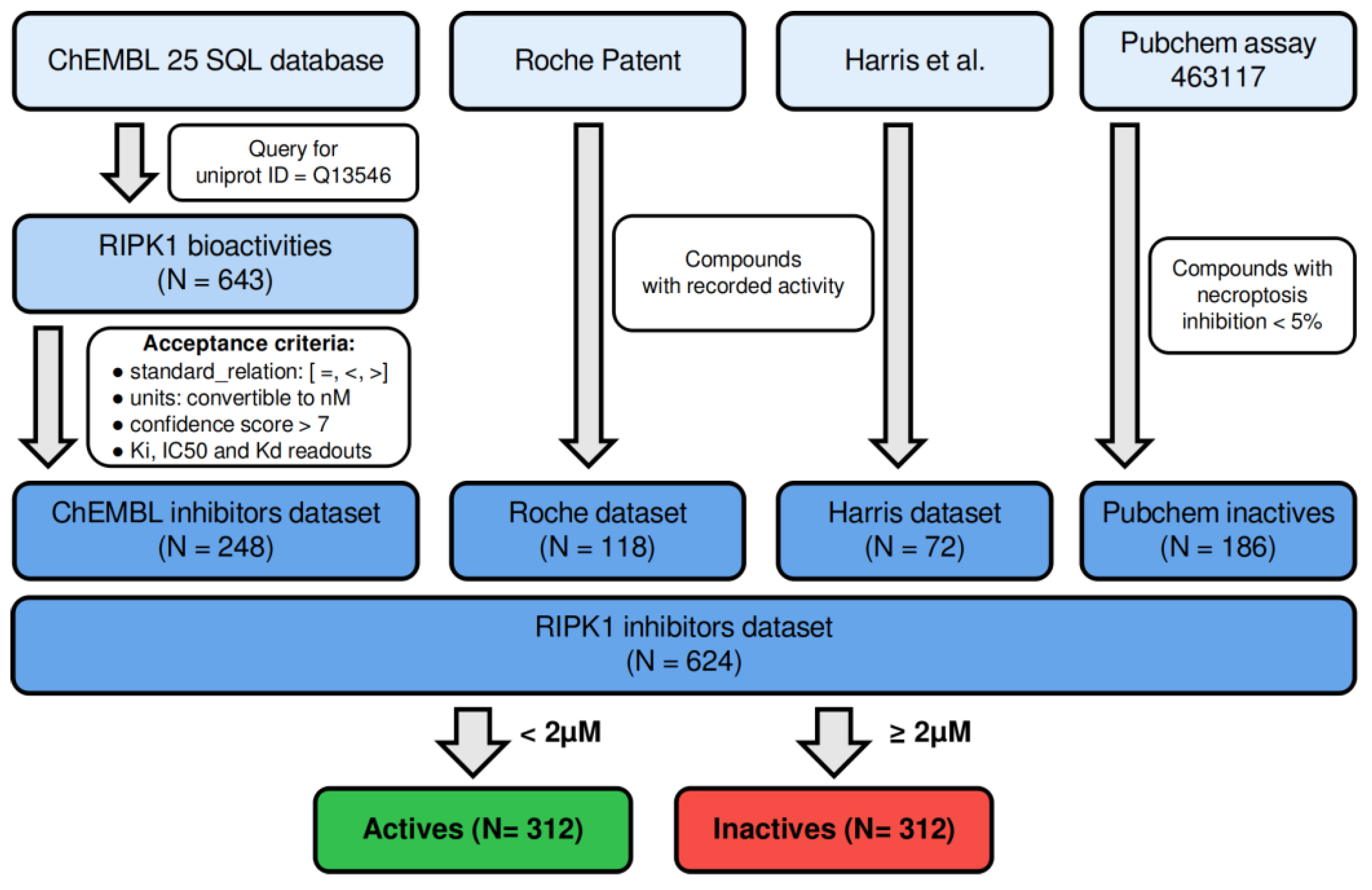
2.1. Assembly of the Dataset of RIPK1 Inhibitors
2.2. Preparation of Compound Structures for QSAR
2.3. Molecular Descriptors, Morgan Fingerprints, Protein-Ligand Interaction Fingerprints (PLIFs), and PLIFs Similarity
2.4. Molecular Docking Protocol Validation and Optimisation
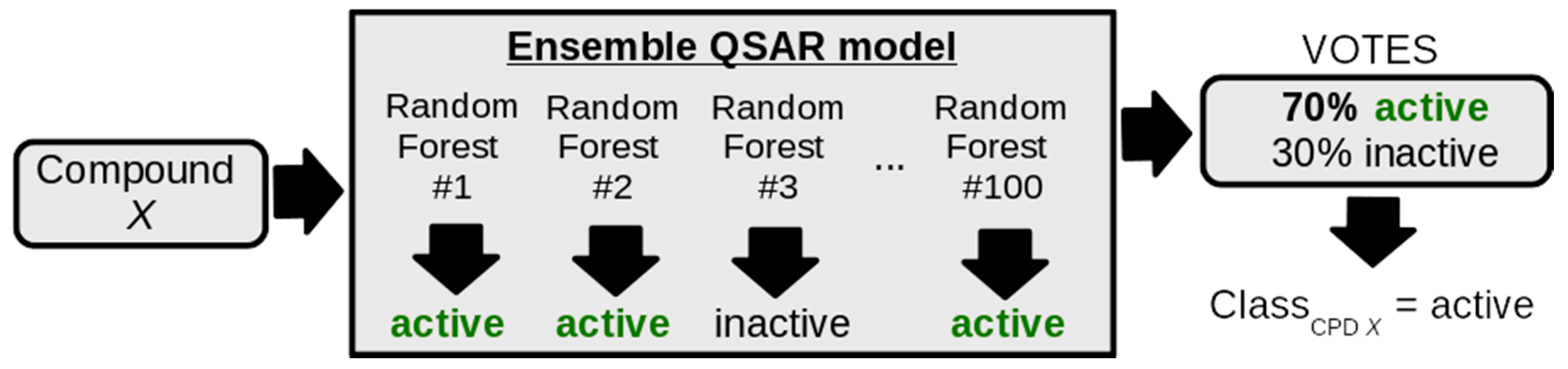
2.5. Development of a QSAR Model of RIPK1 Inhibition Using Machine Learning
2.6. Virtual Screening and Compound Selection
2.7. Phenotypic Assay of Necroptosis Inhibition
2.8. Biochemical Assay of RIPK1 Inhibition
2.9. Performance Evaluation
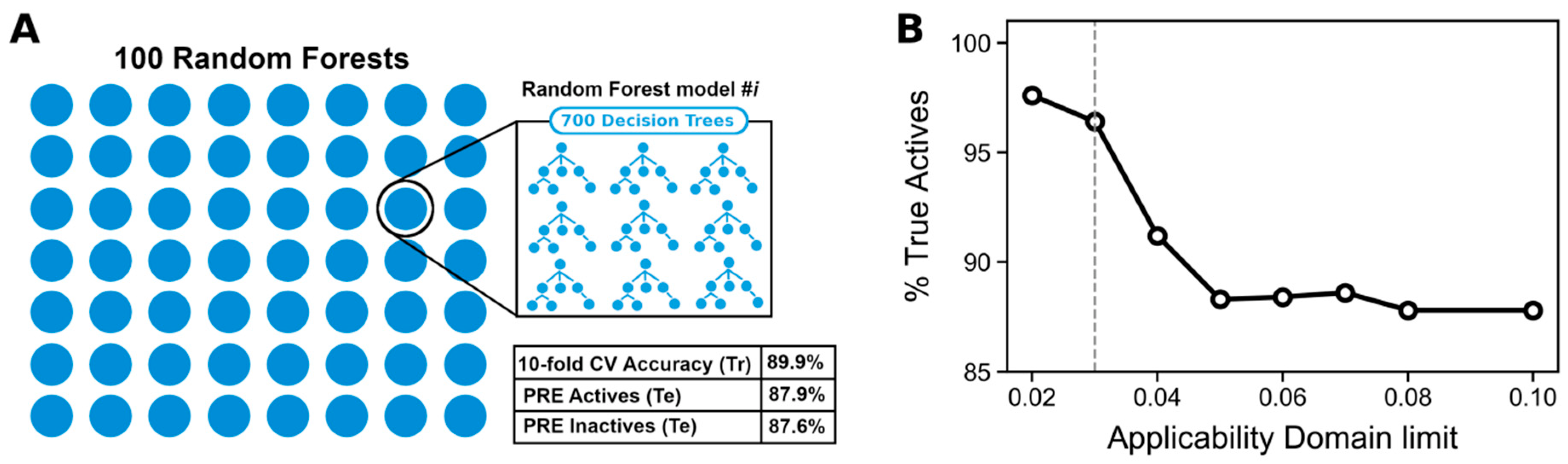
2.10. Applicability Domain
2.11. Analysis of Chemical Novelty for In Silico Hit Compounds
2.12. Data Analysis and Visualisation
3. Results and Discussion
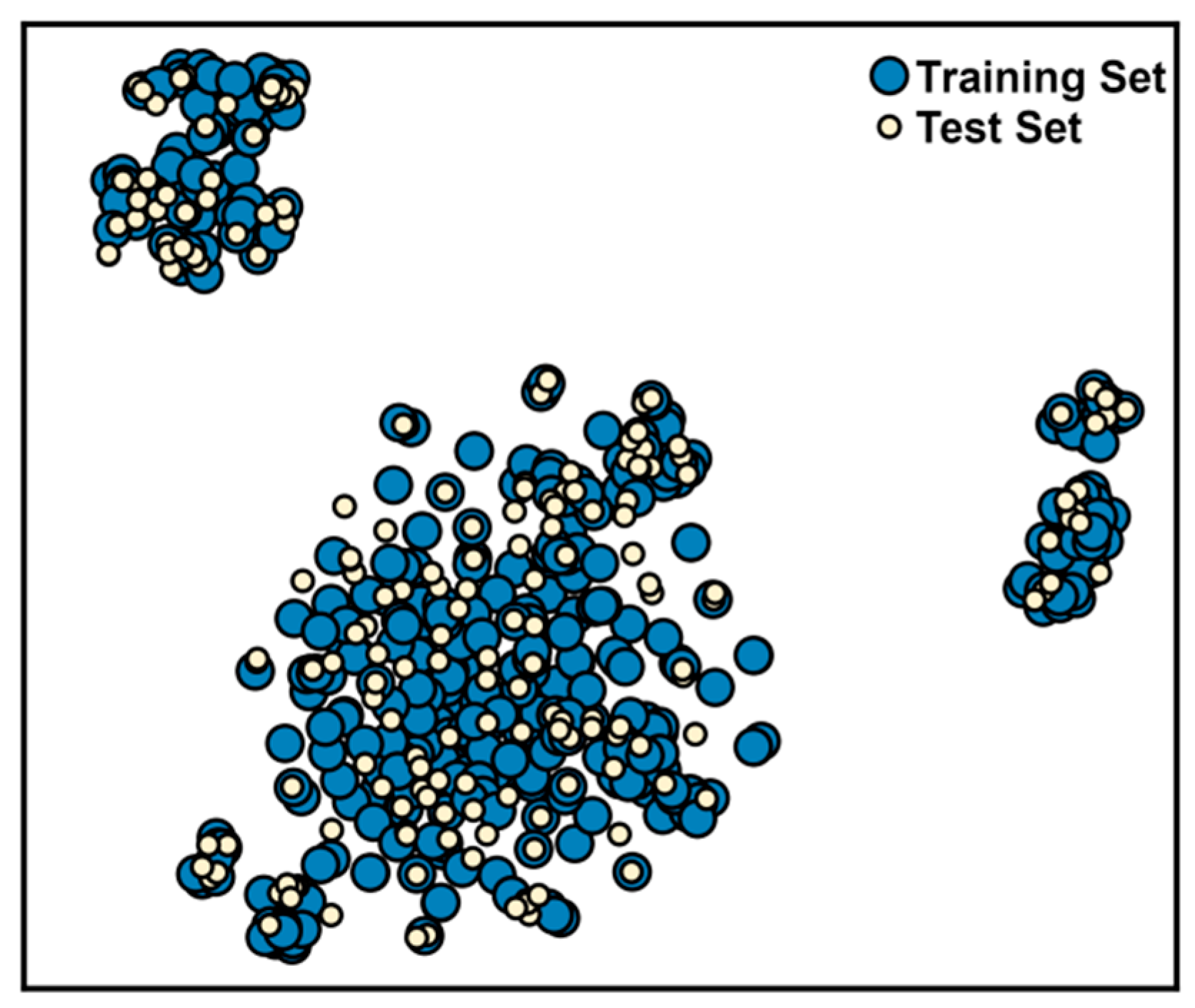
3.1. Chemical Space Analysis of RIPK1 Inhibitors
3.2. Validation of Molecular Docking Calculations
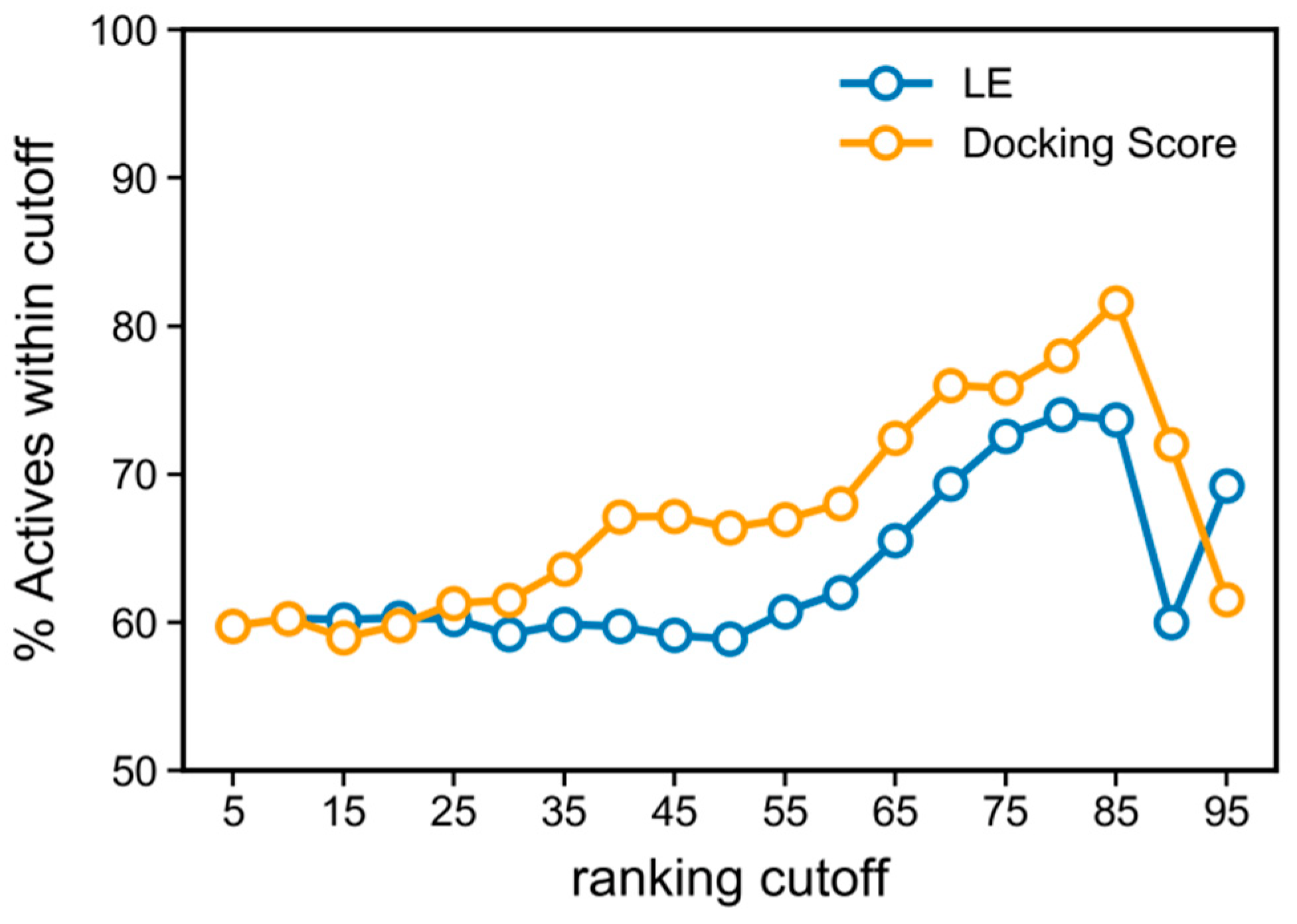
3.3. Docking Score and Ligand Efficiency Do Not Correlate with Activity
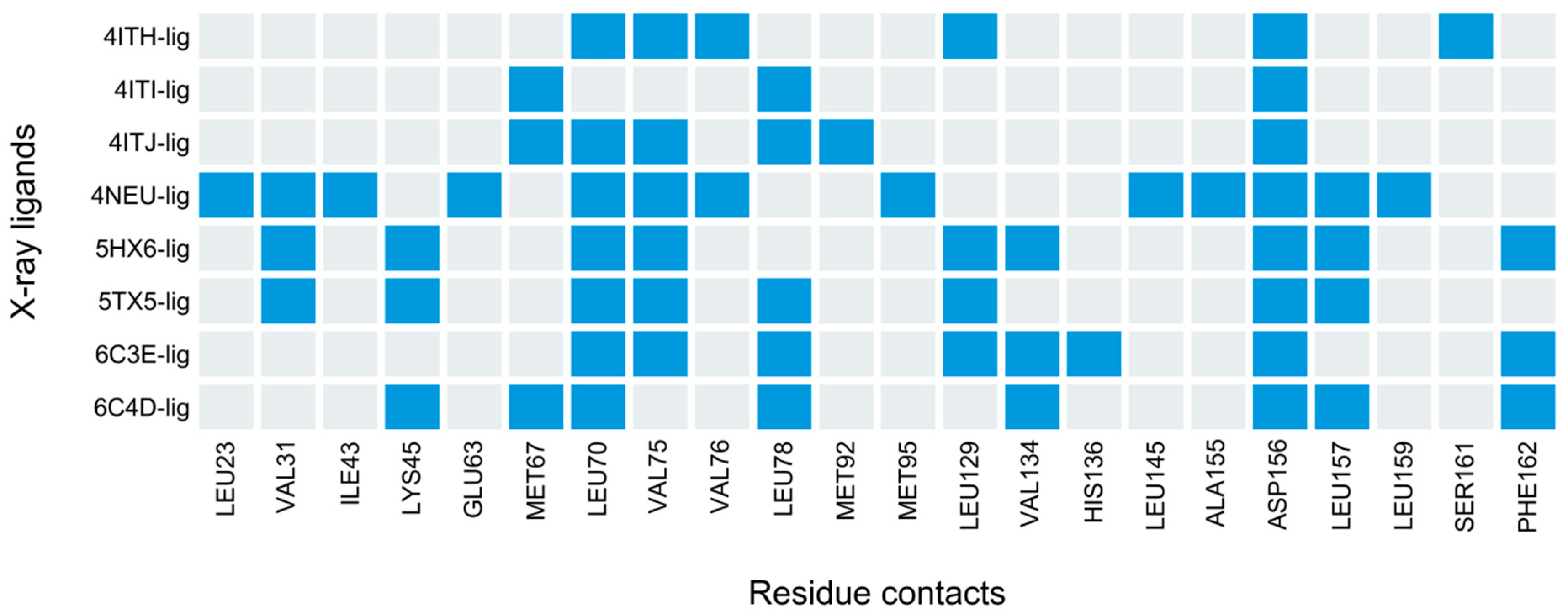
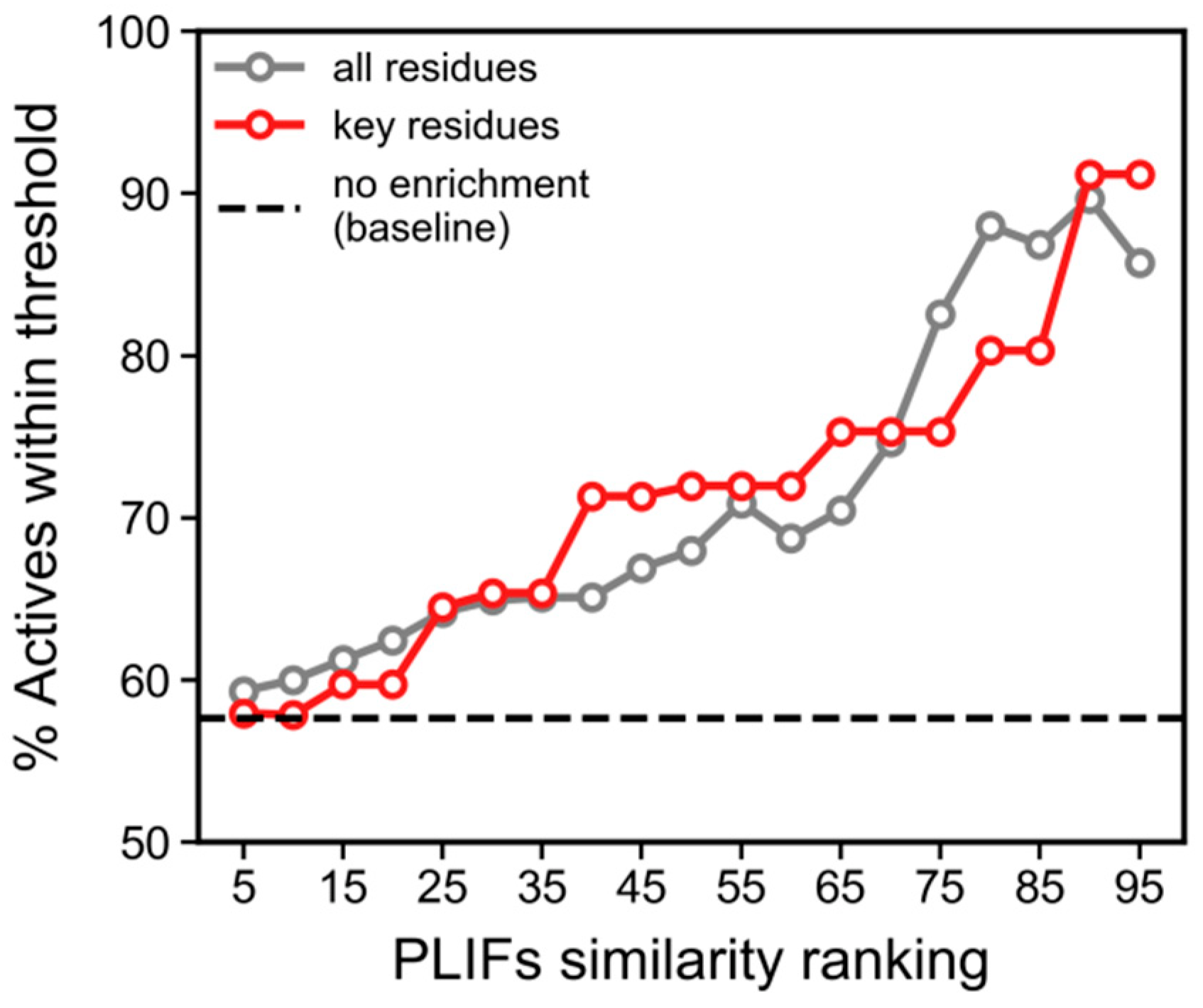
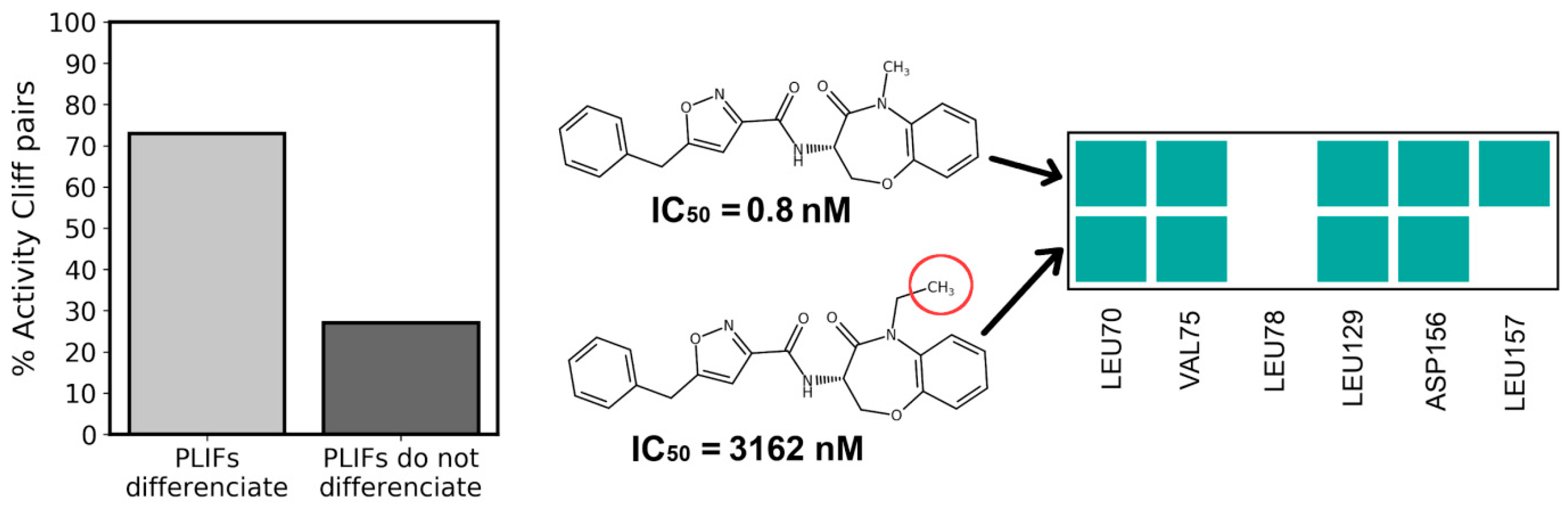
3.4. PLIFs Similarity as a New Guideline to Find RIPK1 Inhibitors
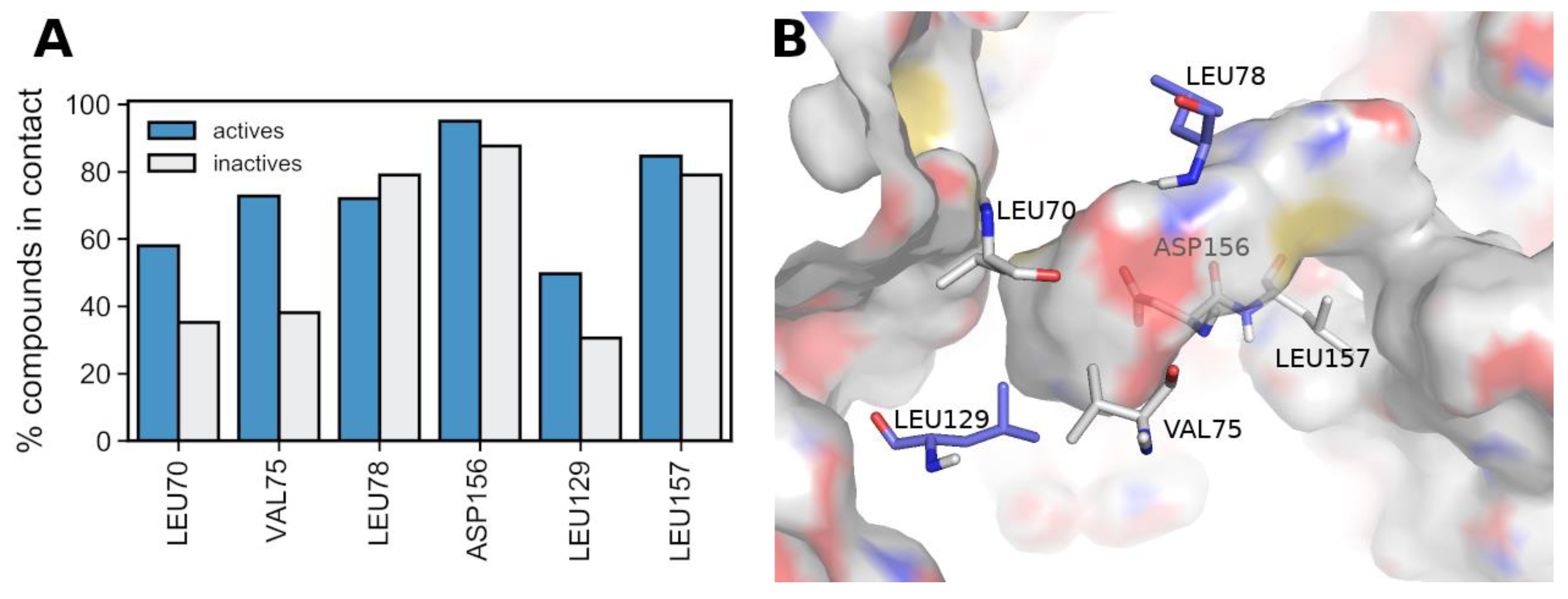
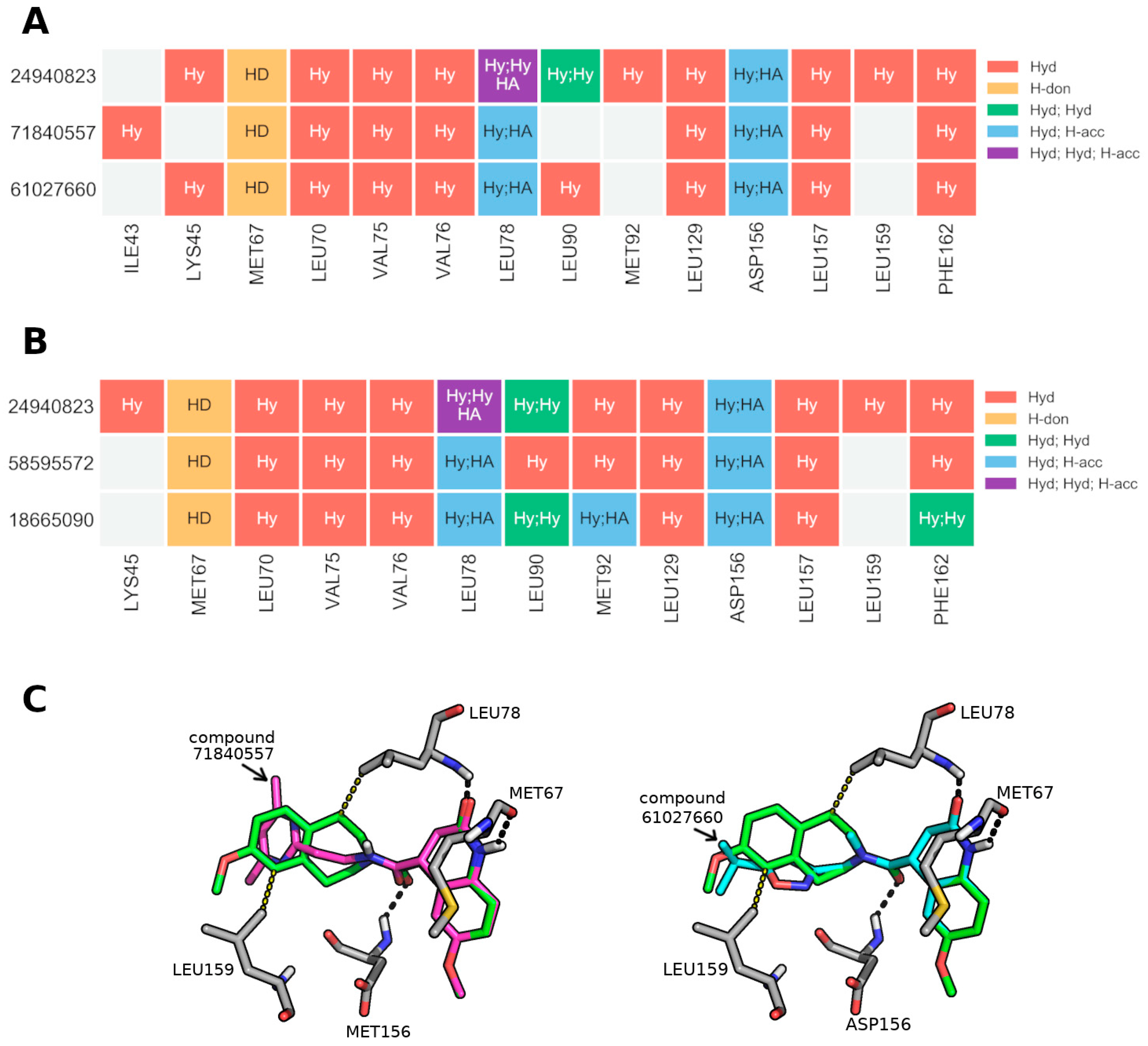
3.5. Deriving a New 4-Residue Rule to Predict RIPK1 Inhibitors
3.6. Building a Machine Learning Model to Predict Necroptosis Modulators
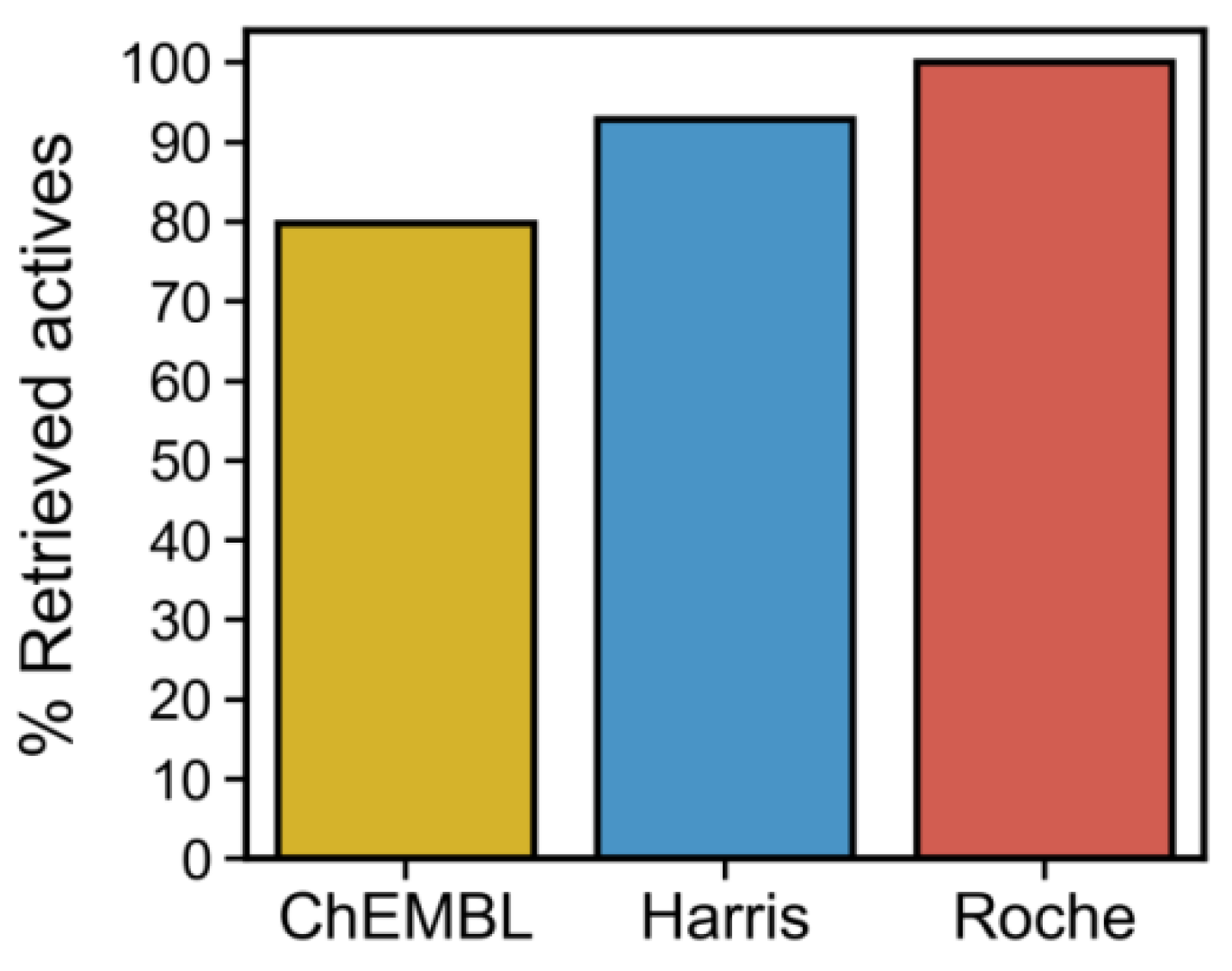
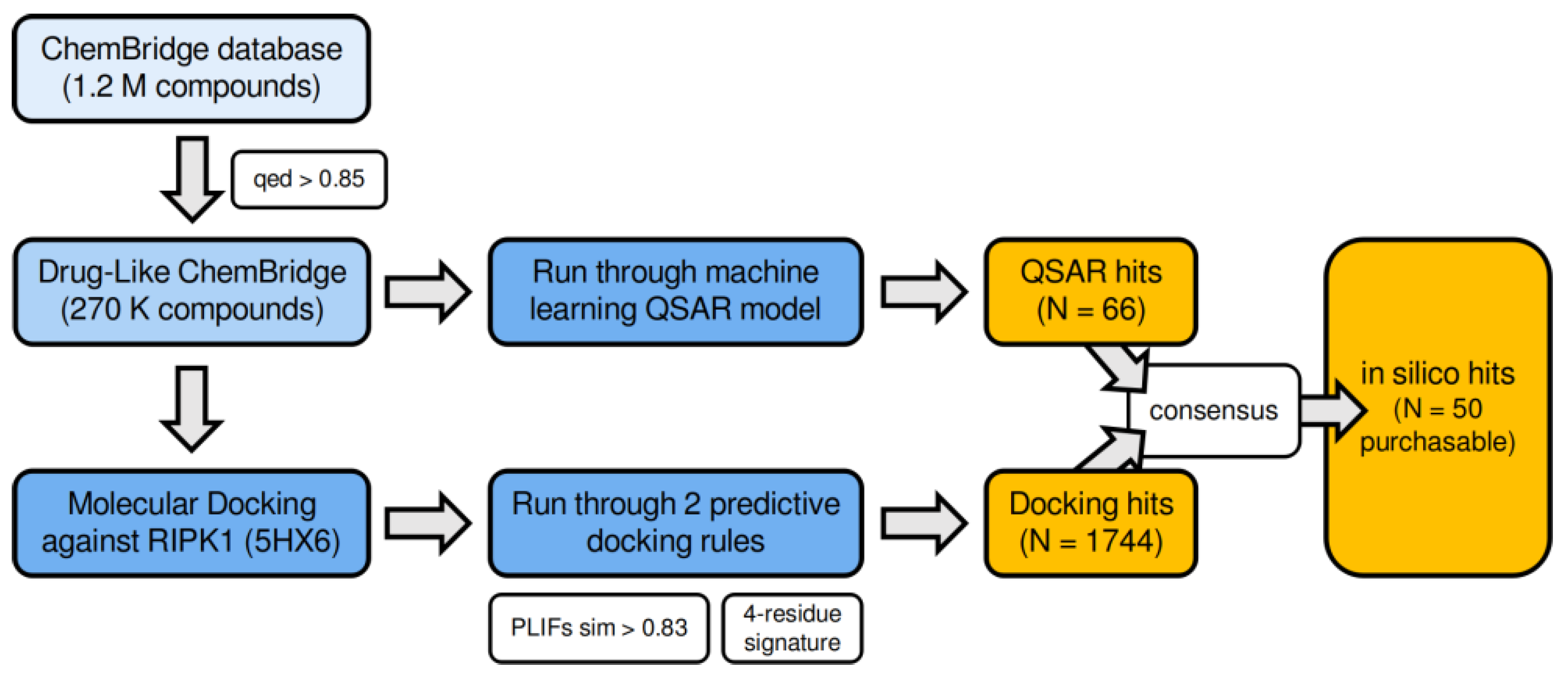
3.7. Virtual Screening of the ChemBridge Library Using the Docking-Based (4-Residue Signature and the PLIFs Similarity Filter) and the QSAR Models
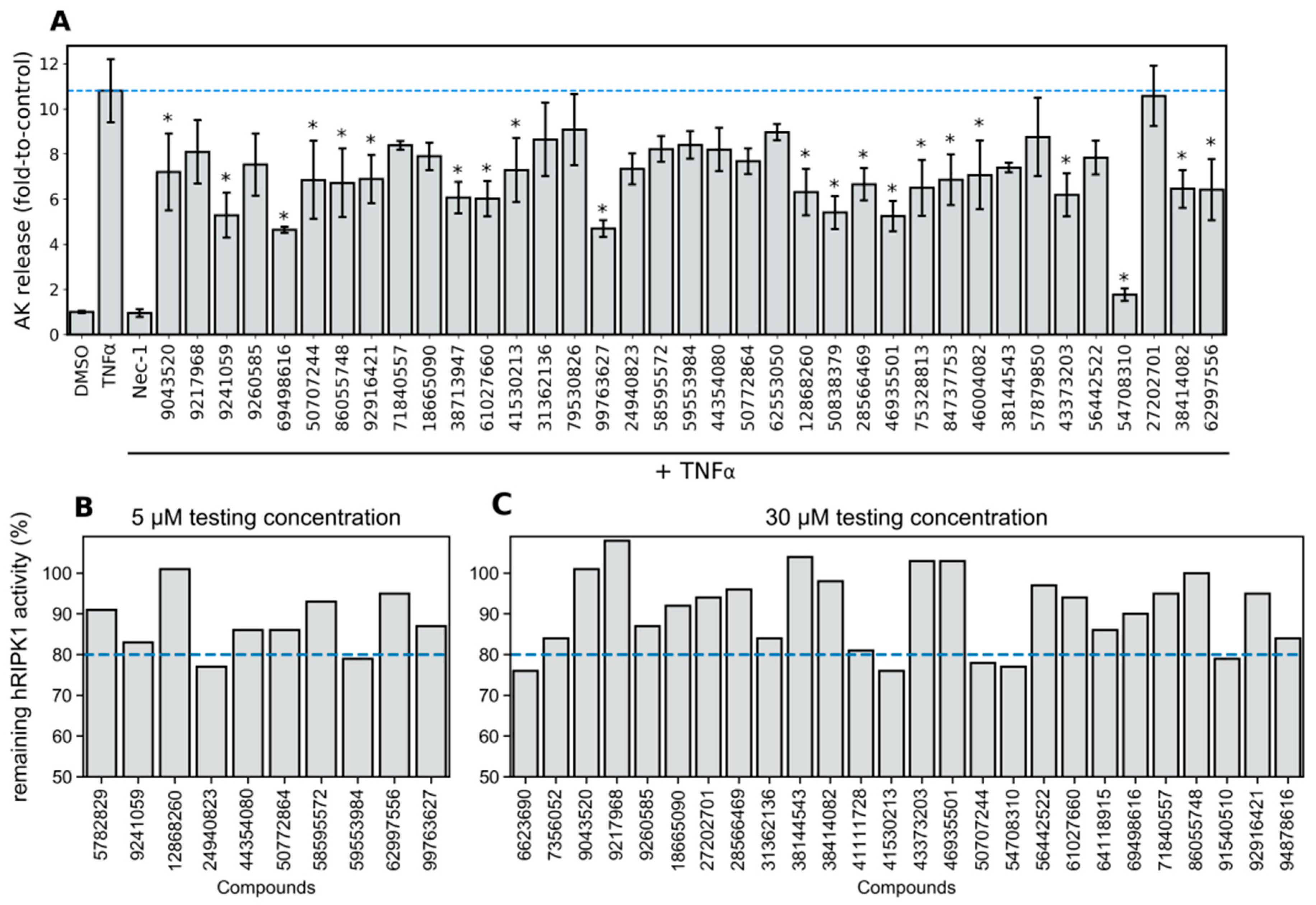
3.8. Phenotypic Necroptosis Inhibition Assay Reveals In Silico Hits with RIPK1 Inhibitory Activity
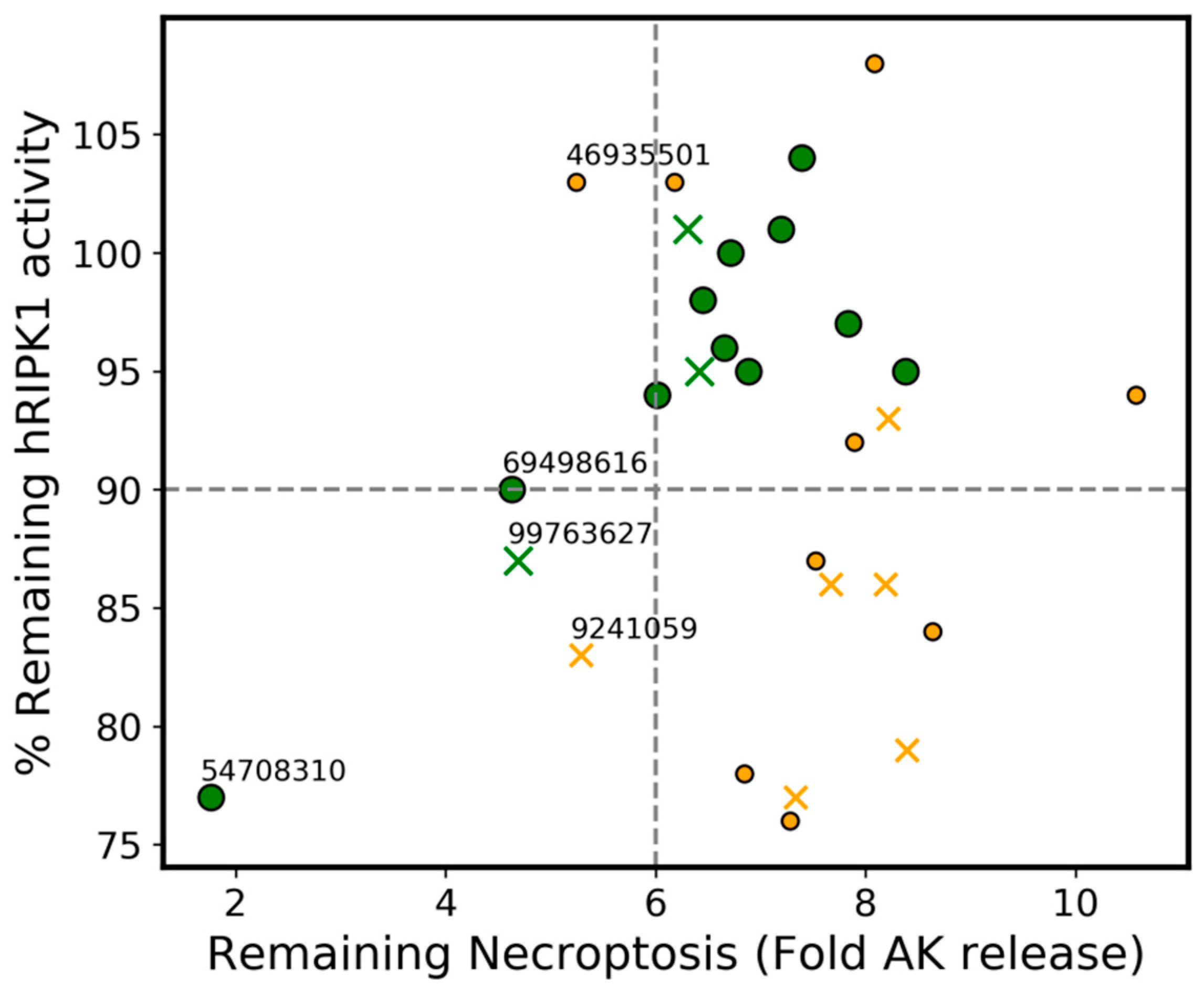
3.9. Agreement between the Phenotypic Assay of Necroptosis Inhibition in Murine L929 Cells and the Biochemical hRIPK1 Inhibition Assay
3.10. Extracting General Structure-Activity Relationships (SAR) in New RIPK1 Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Tang, M.-B.; Luo, H.-Y.; Shi, C.-H.; Xu, Y.-M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Webster, J.D.; Varfolomeev, E.; Kwon, Y.C.; Cheng, J.H.; Zhang, J.; Dugger, D.L.; Wickliffe, K.E.; Maltzman, A.; Sujatha-Bhaskar, S.; et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2019, 27, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’En, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Clinical Trials Database. Available online: https://clinicaltrials.gov (accessed on 1 December 2020).
- Hanson, S.M.; Georghiou, G.; Thakur, M.K.; Miller, W.T.; Rest, J.S.; Chodera, J.D.; Seeliger, M.A. What Makes a Kinase Promiscuous for Inhibitors? Cell Chem. Biol. 2019, 26, 390–399.e5. [Google Scholar] [CrossRef]
- Hou, J.; Ju, J.; Zhang, Z.; Zhao, C.; Li, Z.; Zheng, J.; Sheng, T.; Zhang, H.; Hu, L.; Yu, X.; et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 2019, 10, 493. [Google Scholar] [CrossRef] [Green Version]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico Methods for Design of Kinase Inhibitors as Anticancer Drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef] [Green Version]
- Kowalewski, J.; Ray, A. Predicting novel drugs for SARS-CoV-2 using machine learning from a >10 million chemical space. Heliyon 2020, 6, e04639. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; de Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Harris, P.A.; Faucher, N.; George, N.; Eidam, P.M.; King, B.W.; White, G.V.; Anderson, N.A.; Bandyopadhyay, D.; Beal, A.M.; Beneton, V.; et al. Discovery and Lead-Optimization of 4,5-Dihydropyrazoles as Mono-Kinase Selective, Orally Bioavailable and Efficacious Inhibitors of Receptor Interacting Protein 1 (RIP1) Kinase. J. Med. Chem. 2019, 62, 5096–5110. [Google Scholar] [CrossRef]
- Chen, H.; Hamilton, G.; Patel, S.; Zhao, G.; Daniels, B.; Stivala, C. Bicyclic Compounds For Use As Rip1 Kinase. Inhibitors. Patent WO2019/072942-A1, 2019. [Google Scholar]
- Lowe, D.M.; Corbett, P.T.; Murray-Rust, P.; Glen, R.C. Chemical Name to Structure: OPSIN, an Open Source Solution. J. Chem. Inf. Model. 2011, 51, 739–753. [Google Scholar] [CrossRef]
- Filippov, I.V.; Nicklaus, M. Optical Structure Recognition Software To Recover Chemical Information: OSRA, An Open Source Solution. J. Chem. Inf. Model. 2009, 49, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Mervin, L.H.; Afzal, A.M.; Drakakis, G.; Lewis, R.; Engkvist, O.; Bender, A. Target prediction utilising negative bioactivity data covering large chemical space. J. Cheminform. 2015, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Bosc, N.; Atkinson, F.; Felix, E.; Gaulton, A.; Hersey, A.; Leach, A.R. Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug Discovery. J. Cheminform. 2019, 11, 4. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE); Corporate Headquarters: Montreal, QC, Canada, 2019. [Google Scholar]
- RDKit. RDKit: Open-source Cheminformatics. Available online: https://www.rdkit.org/ (accessed on 14 July 2022).
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. Structural Basis of RIP1 Inhibition by Necrostatins. Structure 2013, 21, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.A.; Bandyopadhyay, D.; Berger, S.B.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Finger, J.N.; Hoffman, S.J.; Kahler, K.M.; et al. Discovery of Small Molecule RIP1 Kinase Inhibitors for the Treatment of Pathologies Associated with Necroptosis. ACS Med. Chem. Lett. 2013, 4, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.A.; King, B.W.; Bandyopadhyay, D.; Berger, S.B.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; Finger, J.N.; et al. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem. 2016, 59, 2163–2178. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Saitoh, M.; Katoh, T.; Seki, T.; Bigi, S.V.; Shimizu, Y.; Ishii, T.; Okai, T.; Kuno, M.; Hattori, H.; et al. Discovery of 7-Oxo-2,4,5,7-tetrahydro-6H-pyrazolo[3,4-c]pyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP1) Kinase Inhibitors: Analysis of Structure–Kinetic Relationships. J. Med. Chem. 2018, 61, 2384–2409. [Google Scholar] [CrossRef]
- Pedregosa, F.; Gael, V.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Duborg, V.; Vanderplas, J.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Aniceto, N.; Freitas, A.A.; Bender, A.; Ghafourian, T. A novel applicability domain technique for mapping predictive reliability across the chemical space of a QSAR: Reliability-density neighbourhood. J. Cheminform. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Kanji, G. 100 Statistical Tests; Sage Publications: London, UK, 2006. [Google Scholar]
- The PyMOL. Molecular Graphics System; Version 2.0; Schrödinger, LLC: New York, NY, USA, 2010. [Google Scholar]
- Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M. Estimating Electron Density Support for Individual Atoms and Molecular Fragments in X-ray Structures. J. Chem. Inf. Model. 2017, 57, 2437–2447. [Google Scholar] [CrossRef]
- Bietz, S.; Rarey, M. SIENA: Efficient Compilation of Selective Protein Binding Site Ensembles. J. Chem. Inf. Model. 2016, 56, 248–259. [Google Scholar] [CrossRef]
- Schöning-Stierand, K.; Diedrich, K.; Fährrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef] [Green Version]
- Van Der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, R.; Bajorath, J. Multitask Machine Learning for Classifying Highly and Weakly Potent Kinase Inhibitors. ACS Omega 2019, 4, 4367–4375. [Google Scholar] [CrossRef]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Neto, M.F.A.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef]
- Fischler, M.A.; Bolles, R.C. Random sample consensus: A Paradigm for Model Fitting with Applications to Image Analysis and Automated Cartography. Commun. ACM 1981, 24, 381–395. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, Q.; Phan, N.; Ren, J.; Yang, H.; Feldman, C.C.; Feltenberger, J.B.; Ye, Z.; Wildman, S.; Tang, W.; et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019, 10, 226. [Google Scholar] [CrossRef]
- Pearson, W. An Introduction to Sequence Similarity (“Homology”) Searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef]
- Turei, D.; Korcsmáros, T.; Saez-Rodriguez, D.T.J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aniceto, N.; Marques, V.; Amaral, J.D.; Serra, P.A.; Moreira, R.; Rodrigues, C.M.P.; Guedes, R.C. Harnessing Protein-Ligand Interaction Fingerprints to Predict New Scaffolds of RIPK1 Inhibitors. Molecules 2022, 27, 4718. https://doi.org/10.3390/molecules27154718
Aniceto N, Marques V, Amaral JD, Serra PA, Moreira R, Rodrigues CMP, Guedes RC. Harnessing Protein-Ligand Interaction Fingerprints to Predict New Scaffolds of RIPK1 Inhibitors. Molecules. 2022; 27(15):4718. https://doi.org/10.3390/molecules27154718
Chicago/Turabian StyleAniceto, Natália, Vanda Marques, Joana D. Amaral, Patrícia A. Serra, Rui Moreira, Cecília M. P. Rodrigues, and Rita C. Guedes. 2022. "Harnessing Protein-Ligand Interaction Fingerprints to Predict New Scaffolds of RIPK1 Inhibitors" Molecules 27, no. 15: 4718. https://doi.org/10.3390/molecules27154718
APA StyleAniceto, N., Marques, V., Amaral, J. D., Serra, P. A., Moreira, R., Rodrigues, C. M. P., & Guedes, R. C. (2022). Harnessing Protein-Ligand Interaction Fingerprints to Predict New Scaffolds of RIPK1 Inhibitors. Molecules, 27(15), 4718. https://doi.org/10.3390/molecules27154718